


Abstract
The role of protein kinase C and intracellular Ca2+ on amphetamine-mediated dopamine release through the norepinephrine plasmalemmal transporter in undifferentiated PC12 cells was investigated. The selective protein kinase C inhibitor chelerythrine completely inhibited endogenous dopamine release elicited by 1 μM amphetamine. Direct activation of protein kinase C increased dopamine release in a Ca2+-insensitive, imipramine-sensitive manner and the release was not additive with amphetamine. Exocytosis was not involved since these events were not altered by either deletion of extracellular Ca2+ or reserpine pretreatment. Down-regulation of protein kinase C activity by long-term phorbol ester treatment resulted in a dramatic decrease in amphetamine-mediated dopamine release with no apparent effect on [3H]dopamine uptake. To more completely examine a role for Ca2+, intracellular Ca2+ was chelated in the cells. Depletion of intracellular Ca2+ considerably decreased dopamine release in response to 1 μM amphetamine compared with vehicle-treated cells, but had no effect on the [3H]dopamine uptake. Thus, our results suggest that amphetamine-mediated dopamine release through the plasmalemmal norepinephrine transporter is highly dependent on protein kinase C activity and intracellular but not extracellular Ca2+. Furthermore, protein kinase C and intracellular Ca2+ appear to regulate [3H]dopamine inward transport and amphetamine-mediated outward transport of dopamine independently in PC12 cells.
Amphetamine (AMPH) exerts its physiological effects by enhancing the transport of monoamines into the synapse through a reversal of their respective plasmalemmal transporters. AMPH has a high affinity for the norepinephrine (NE) and dopamine (DA) plasmalemmal transporters, NET and DAT, respectively. DAT and NET have 78% sequence similarity and both contain consensus sequences for protein kinase C (PKC) and protein kinase A (Giros and Caron, 1993; Bruss et al., 1997). NET and DAT are postulated to function similarly to other Na+-dependent carriers, in a gated channel mechanism, dependent on the coordinate opening and closing of the inner and outer channels (Rudnick and Clark, 1993). Following binding of a substrate molecule from one compartment, the carrier reverses its orientation and the substrate is released on the other side. For net inward transport, the carrier resumes its initial orientation without bound substrate. When AMPH is the substrate, the inward-facing transporter now binds cytoplasmic DA and carries it to the outside. Until recently, the mechanism of exchange diffusion has been the most accepted mechanism for AMPH action (Fischer and Cho, 1979; Seiden et al., 1993). There are emerging reports concerning AMPH action that are inconsistent with a simple model of exchange diffusion (Langeloh et al., 1987; Sitte et al., 1998; Pifl et al., 1999).
Recent studies have demonstrated that phosphorylation, especially PKC-mediated activity, can regulate catecholamine transporter activity (Copeland et al., 1996; Huff et al., 1997; Zhang et al., 1997; Zhu et al., 1997; Pristupa et al., 1998; Apparsundaram et al., 1998; Daniels and Amara, 1999; Melikian and Buckley, 1999). Although these studies have demonstrated a down-regulation of transporter-mediated monoamine uptake in response to PKC activation, we (Kantor and Gnegy, 1998;Cowell et al., 2000) and others (Davis and Patrick, 1990; Giambalvo, 1992) have shown that PKC activation can also lead to an immediate increase in outward transport of the monoamine through the transporter. We found that AMPH-mediated DA release in rat striatum was blocked by PKC inhibitors in an action that did not involve synaptic vesicles (Kantor and Gnegy, 1998). In rat synaptosomes, 12-O-tetradecanoylphorbol 13-acetate (TPA) elicited a rapid release of DA that was independent of extracellular Ca2+, was blocked by cocaine and GBR12935, and was not additive with AMPH (Cowell et al., 2000). These effects were not dependent on extracellular Ca2+ and were not affected by reserpine pretreatment of the rats. Therefore, it appeared that the effect of PKC on amphetamine-mediated DA release was unrelated to exocytotic events.
It is unknown whether the apparent PKC regulation of AMPH action is unique for the DA transporter or is inherent for the mechanism of AMPH at any transporter for which it is a substrate. Therefore, we chose to examine the role of PKC in undifferentiated rat pheochromocytoma PC12 cells, which contain NET (Bonisch, 1984; Langeloh et al., 1987). Both DA and AMPH are excellent substrates for NET (Gu et al., 1994). In the undifferentiated PC12 cell, AMPH releases both NE and DA. In PC12 cells, DA can be released via exocytosis (Kittner et al., 1987) and/or through NET in response to AMPH (Sulzer et al., 1995). It was desirable to use cells in which endogenous DA would be released to obviate problems concerning preloading of [3H]DA into various pools or metabolism of [3H]DA. Our results demonstrate that PKC activation is required for AMPH to release DA through NET and that intracellular Ca2+, but not extracellular Ca2+, is required for the ability of AMPH to elicit outward transport of DA.
Materials and Methods
Drugs.
Chelerythrine, 1-[3-(amidinothio)propyl-1H-indoyl-3-yl]-3-(1-methyl-1H-indoyl-3-yl)maleimide; methane sulfonate (Ro-31-8220); 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetra(acetoxymethyl) ester (BAPTA-AM); and 12-O-tetradecanoylphorbol 13-acetate (TPA) were purchased from Calbiochem (La Jolla, CA). Imipramine was purchased from RBI/Sigma (Natick, MA). GBR-12935 dihydrochloride and nisoxetine dihydrochloride were purchased from Sigma (St. Louis, MO).
Cell Culture.
Undifferentiated PC12 cells were grown in monolayers in a 75-cm2 tissue culture flask in growth medium composed of Dulbecco's modified Eagle's medium (Sigma) supplemented with 5% (v/v) fetal bovine serum, 10% (v/v) heat-inactivated horse serum, 100 μg/ml of streptomycin, and 100 units/ml of penicillin and were incubated at 7.5% CO2 until they reached confluency. Cells were subcultured once a week and medium was changed every other day. In experiments involving reserpine, cells were treated for 3 h with 50 nM reserpine or vehicle. This treatment was shown to give nearly complete depletion of catecholamines in PC12 cells by 3 h (Drukarch et al., 1996). Reserpine was dissolved in dimethyl sulfoxide but diluted so that no more than 0.005% dimethyl sulfoxide was present in the cell culture.
Superfusion Assay.
The cells were harvested by washing them from flasks with Kreb's Ringer buffer (KRB) containing 125 mM NaCl, 2.7 mM KCl, 1.0 mM MgCl2, 1.2 mM CaCl2, 1.2 mM KH2PO4, 10 mM glucose, 24.9 mM NaHCO3, and 0.25 mM ascorbic acid, oxygenated by 95% O2 and 5% CO2 for 1 h. The cell suspension was centrifuged at 500g for 5 min, the supernatant was removed and cells were resuspended in KRB to achieve a protein concentration of ∼1.2 μg/μl. The cells were then placed in appropriate chambers and perfused with KRB or drug for at least 30 min until a stable baseline was attained.
Measurement of Endogenous Dopamine Release.
The PC12 cell suspension was transferred to Whatman GF/B glass filters (Maidstone, England) in the appropriate chambers of a Brandel superfusion apparatus (Brandel SF-12, Gaithersburg, MD). Superfusion chambers were maintained at 37°C and medium was perfused through the chambers at a rate of 100 μl/min. Samples were collected at 5-min intervals. After cells were perfused with KRB or drug (transporter blockers or PKC inhibitors) for 30 min, a 2.5-min bolus of 1 μM AMPH, 1 μM AMPH plus drug, drug alone, or 250 nM TPA was delivered. The stimulation was terminated by replacing AMPH with fresh KRB or drug. Collection continued for another 40 min. Results are not corrected for the time that it takes the perfused AMPH to reach the cells. The AMPH is added at fraction 7; calculating the time of delivery, the AMPH would reach the sample at fraction 9 and dopamine would elute at fraction 10. Samples were collected into vials containing 25 μl of internal standard solution (0.05 M HClO4, 4.55 mM dihydrobenzylamine, 1 M metabisulfate, and 0.1 M EDTA). Samples were stored at −70°C. The DA content in the perfusate was measured by HPLC with electrochemical detection using dihydrobenzylamine as an internal standard. Stock concentrations of chelerythrine (10 mM), Ro31-8220 (1.8 mM), TPA (10 mM), and BAPTA-AM (10 mM) in dimethyl sulfoxide were prepared and diluted in KRB to final concentration of 1 μM for chelerythrine and Ro31-8220, 250 nM for TPA and 50 μM for BAPTA. The final concentration of dimethyl sulfoxide in the perfusate was less than 0.1%.
Statistical analysis was performed on the values attained at the peak AMPH response, which is always fraction 10. Statistical significance was determined using one-way ANOVA with post-test Tukey-Kramer multiple comparison analysis or by Student's t test.
[3H]DA Uptake.
Since DA is taken up through NET with a greater affinity than NE (Gu et al., 1994), and we are measuring DA release from PC12 cells, we determined uptake through NET using [3H]DA instead of [3H]NE. Media was removed from cells and cells were washed twice with KRB. Cells are incubated at 37°C with 50 μM imipramine for 20 min followed by 10 min of incubation with either 30 nM [3H]DA (18.3 Ci/mmol) or 1 μM [3H]DA. After 10 min, the solution was removed, the cells were washed three times with ice-cold saline, lysed, and radioactivity determined by liquid scintillation spectrometry. All the samples were counted in ScintiVerse BD in a Beckman LS 5800 scintillation counter. Uptake experiments involving BAPTA-AM treatment were performed in cell suspension rather than with plated cells because BAPTA-AM treatment precluded adhesion of cells to plates. PC12 cells were harvested, collected, and resuspended in KRB as described above. Cell suspensions (200 μl) were treated with 50 μM BAPTA-AM for 30 min, whereas the control groups received KRB/dimethyl sulfoxide (0.1%) buffer. Following the preincubation, [3H]DA (18.3 Ci/mmol) was added at a concentration of 30 nM or 1 μM, and the incubation proceeded for 10 min. The reaction was terminated by filtering the cells through GF/B filters on a Hoeffer filtering apparatus (San Francisco, CA) and washing three times with ice-cold saline.
Immunoblots.
Undifferentiated PC12 cells were removed from the plates with trypsin (1:4) diluted with KRB. Cells were homogenized in a buffer containing 0.32 M sucrose, 40 mm Tris-HCl, pH 7.4, 10 μM leupeptin, 10 μM pepstatin, and 1 mM phenylmethylsulfonyl fluoride. The homogenate fractions were resolved by electrophoresis on 7.5% SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were blocked with 5% milk in Tris-buffered saline containing 0.1% Tween 20 and incubated in 1% bovine serum albumin in phosphate-buffered saline with rabbit polyclonal anti-DAT (Chemicon, Temecula, CA) (1:1000 dilution, 2 h), rabbit polyclonal anti-NET (Chemicon) (1:1000 dilution, 2 h), or with rabbit polyclonal anti-PKCα (Life Technologies, Grand Island, NY) (1:500 dilution, 2 h). After three washes with the Tween-containing blocking buffer, membranes were incubated with a biotinylated second antibody and avidin conjugated with alkaline phosphatase (Amersham Pharmacia Biotech, Piscataway, NJ). The blots were washed and developed with nitroblue tetrazolium and 5-bromo-4-chloro-3-indoyl phosphate (Sigma).
Measurement of PKC Activity.
Cells were grown and treated with 70 nM TPA or with vehicle in media for 24 h in T-75 flasks. After 24 h, the medium was removed and cells were washed twice with cold phosphate-buffered saline. Each well received 2 ml of cold lysis buffer (10 mM Tris-HCl, pH 7.4, 10 μM leupeptin, 10 μM pepstatin, and 1 mM phenylmethylsulfonyl fluoride). Cells were incubated for 2 min at room temperature prior to collection with a cell scraper and sonication for 10 s. Lysates were then centrifuged at 100,000g for 60 min. Pellets were brought to 1% Triton X-100 in the above-described buffer and incubated at least 30 min on ice prior to a 10-fold dilution into the assay. PKC activity was assayed, using myelin basic protein4-14 (Upstate Biotechnology, Lake Placid, NY) as a substrate in both supernatant and membrane fractions, as described (Goldsmith and Gnegy, 1999).
Calcium Measurement.
Intracellular free Ca2+ concentration was measured in fura-2-loaded PC12 cells using dual-wavelength spectrofluorometry according to Fisher et al. (1989). PC12 cells were harvested and resuspended in KRB buffer with and without 1.2 mM CaCl2 to a protein concentration of 3 mg/ml. Cells were incubated with 50 μM BAPTA-AM for 30 min at 37°C followed by centrifugation to remove BAPTA-AM. The pellet was resuspended in KRB with and without added Ca2+. The cells were then incubated with 2 μM fura-2/AM buffer for 15 min at 37°C, washed twice, and resuspended in KRB with or without 1.2 mM CaCl2 at a protein concentration of about 3 mg/ml. Fluorescence measurements were made on 1-ml aliquots of cells maintained at 37°C and constantly stirred. Changes in intracellular free Ca2+ concentration were monitored as variations in the fluorescence ratio of the 340/380-nm excitation wavelength in a Shimadzu RF-5000 spectrofluorimeter. Calcium concentrations were calculated as described (Fisher et al., 1989).
Results
Undifferentiated PC12 Cells Contain NET.
PC12 cells commonly contain the norepinephrine transporter NET (Bonisch, 1984). To ascertain that our PC12 cells contained NET and not DAT, we immunoblotted the cells for NET and DAT. As shown in Fig.1, NET was readily detected in the undifferentiated PC12 cells (lanes 2 and 3), whereas DAT was absent or in very small amounts (lanes 5 and 6). To further ascertain that AMPH was releasing DA through NET, the ability of a selective NET blocker, nisoxetine, or a selective DAT blocker, GBR 12935, to block AMPH-mediated DA release was measured. As shown in Fig.2, the ability of 1 μM AMPH to release DA was significantly inhibited by 300 nM nisoxetine but 50 nM GBR 12935 had no effect. Since the Ki of nisoxetine for DAT is approximately 2 μM (Buck and Amara, 1995), very little, if any, of the DAT would be blocked by 300 nM nisoxetine. On the other hand, since the Ki of GBR12935 for DAT is approximately 1 nM (Buck and Amara, 1995), 50 nM GBR12935 would totally block existing DAT. Similarly, the ability of a serotonin transporter/NET blocker, imipramine, or a DAT blocker, GBR12935, to affect [3H]DA uptake was measured. The percentage of control ([3H]DA only) uptake was 35.6 ± 3.6% in the presence of 50 μM imipramine but was 113 ± 7.0% in the presence of 500 nM GBR 12935 (p < 0.00005 compared with imipramine, n= 4). Although this concentration of imipramine would be large enough to block DAT, our results demonstrate that the transporter in our PC12 cells for which AMPH is a substrate is NET and not DAT.

Immunoblot of NET and DAT in undifferentiated PC12 cells. PC12 cells were homogenized and subjected to SDS-PAGE as described under Materials and Methods. All lanes, except for the molecular weight markers, contained 15 μg of protein. Lanes 1, 4, and 9 contained molecular weight markers. Lanes 2 and 3 are undifferentiated PC12 cells immunoblotted for NET; lanes 5 and 6 are undifferentiated PC12 cells immunoblotted for DAT; lanes 7 and 8 are rat striatum to serve as a control for DAT. The molecular weight markers in lanes 1 and 4 are phosphorylase b at 97.4 kDa and bovine serum albumin at 66 kDa. The molecular weight markers in lane 9 were included in the BenchMark protein ladder (Life Technologies). Both DAT and NET migrate at approximately 80 kDa.

Effect of NET and DAT blockade on AMPH-mediated DA release in undifferentiated PC12 cells. PC12 cells were pretreated during perfusion with either KRB, 50 nM GBR-12909, or 300 nM nisoxetine as described under Materials and Methods. AMPH (1 μM) was added for 2.5 min. DA collected in the peak fraction was measured by HPLC as described under Materials and Methods. Results are given in picomoles of DA released per milligram of protein ± S.E.M. n = 3. Baseline, ■; AMPH, ▨. In an ANOVA for the AMPH responses, p < 0.01. In post hoc Tukey-Kramer multiple comparisons test, values for nisoxetine differed from those for control and GBR-12909 atp < 0.05.
PKC Inhibitors Block AMPH-Mediated DA Release.
We chose to examine DA release through NET in the PC12 cells because DA is plentiful in those cells and can be released in response to either exocytotic stimuli (Kittner et al., 1987) or AMPH (Sulzer et al., 1995). Furthermore, both the affinity and maximal velocity of NET for DA is equal to or slightly greater than that for NE (Pacholczyk et al., 1991; Gu et al., 1994). Therefore, we felt it was physiologically relevant to examine DA release in this cell line. To examine the role of PKC in AMPH-mediated DA release through NET, the effect of two selective PKC inhibitors from different chemical classes was investigated. Chelerythrine or Ro31-8220, at 1 μM, effectively inhibited AMPH-mediated DA release (Fig.3, A and B) but had no effect on basal DA release. The PKC inhibitors had no effect on [3H]DA uptake after 30-min incubation at 1 μM concentration (data not shown). These results mimicked those obtained using DAT-containing rat striatum (Kantor and Gnegy, 1998).
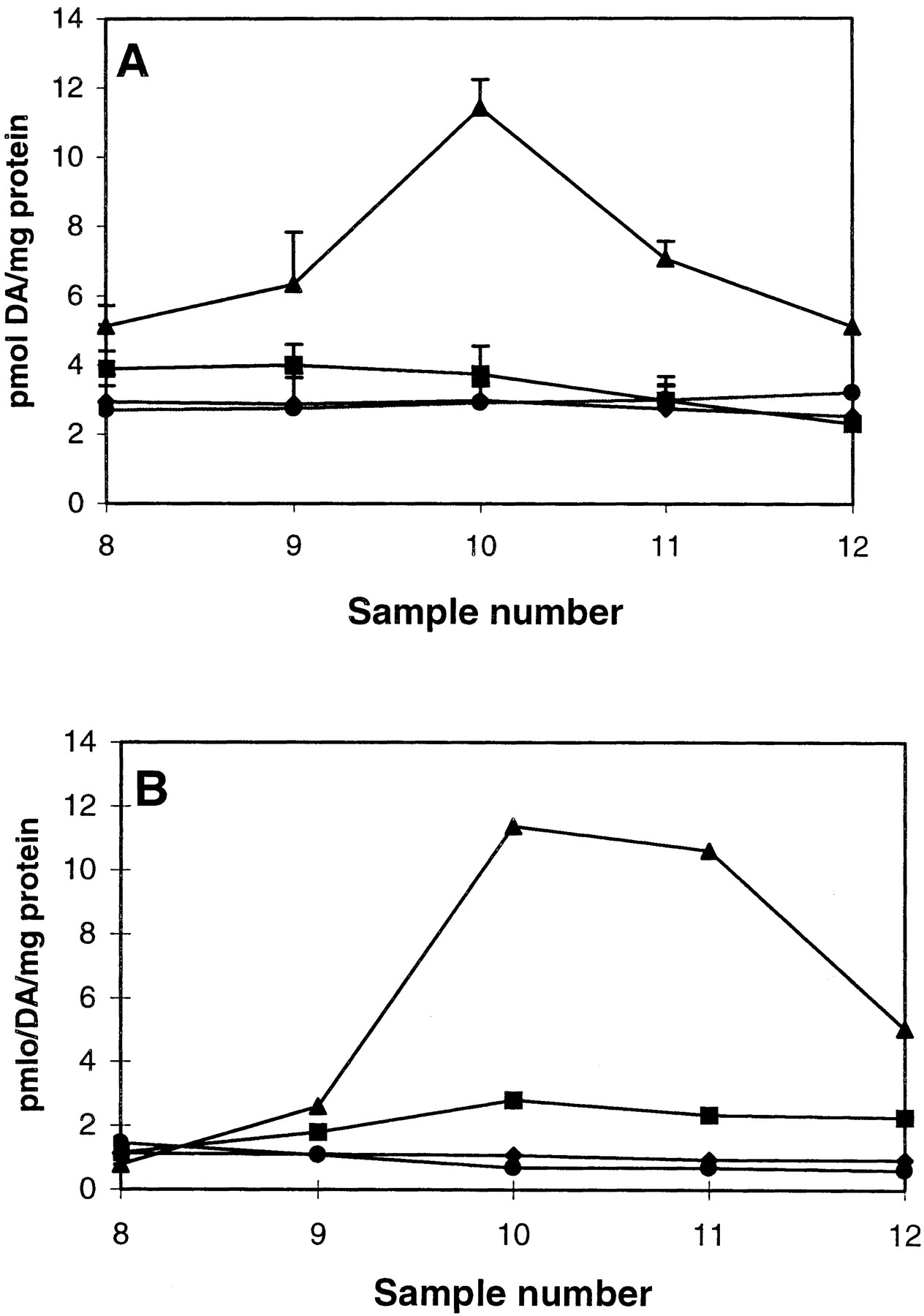
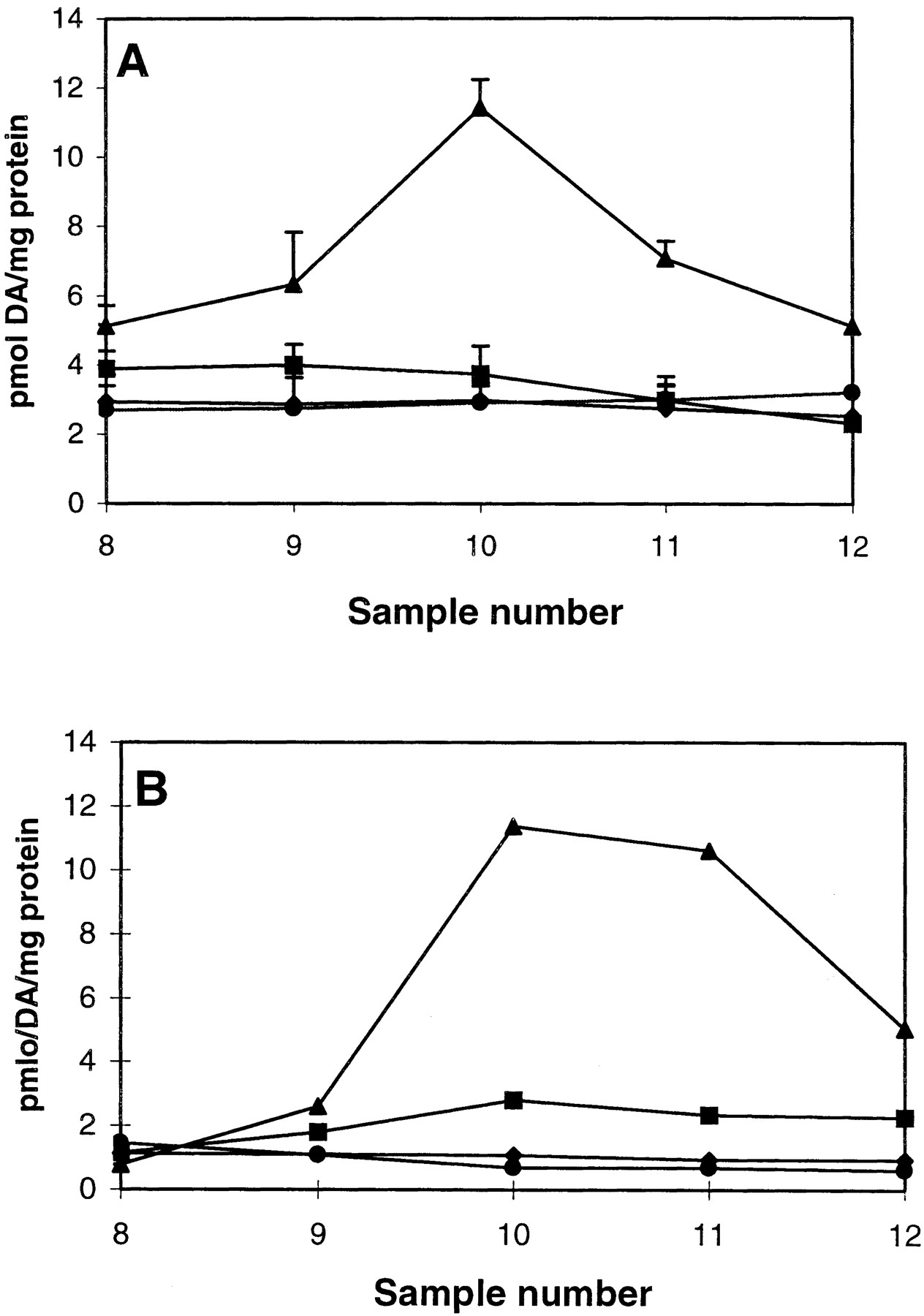
Effect of chelerythrine and Ro31-8220 on AMPH-mediated DA release. PC12 cells were perfused with 1 μM chelerythrine (CH) (A) or 1 μM Ro31-8220 (Ro) (B) for 30 min prior to a 2.5-min bolus of 1 μM AMPH as described under Materials and Methods. For A, ▴, AMPH; ▪, CH + AMPH; ♦, CH; and ●, control. For B, ▴, AMPH; ▪, Ro + AMPH; ♦, Ro; and ●, control. DA was measured by HPLC-electrochemical detection. Chelerythrine or Ro31-8220 alone did not alter the DA basal release. For chelerythrine, results are shown as picomoles of DA released per milligram of protein ± S.E.M. n = 3. In ANOVA for sample 10, p < 0.001. In the post hoc Tukey-Kramer comparison test, AMPH differed from all other groups atp < 0.01. For Ro31-8220, n = 2 with values not differing more than 5%. Note than in both experiments there was minimal AMPH-mediated DA release in the presence of Ro31-8220.
A PKC Activator Mimics AMPH-Mediated DA Release.
The blockade of AMPH-mediated DA release by PKC inhibitors suggests that PKC activity is involved in outward transport. We reported that direct activation of PKC by TPA released DA in rat striatum (Cowell et al., 2000). To test whether this was true in NET-containing cells, we examined the ability of the PKC activator TPA to directly elicit DA release in the PC12 cells. A bolus of 250 nM TPA, perfused in the same manner as 1 μM AMPH, produced DA release similar to that of AMPH (Fig. 4). When the drugs were given in combination (250 nM TPA + 1 μM AMPH), the DA release was not additive, suggesting that TPA shared the effect of AMPH. Both AMPH and TPA were releasing DA through NET, since the DA release elicited by both drugs was blocked by imipramine (Fig. 4). Although imipramine blocks both NET and DAT at high concentrations, our experiments show that AMPH is releasing DA through NET (Figs. 1 and 2).

TPA and AMPH release DA through NET and are not additive. Cells were perfused with a 2.5-min bolus of 250 nM TPA or 1 μM AMPH either individually or together (TPA + AMPH) as described under Materials and Methods. In some experiments, cells were perfused with 50 μM imipramine (IMI) for 30 min before addition of TPA (TPA + IMI) or AMPH (AMPH + IMI). Control cells received vehicle buffer only. ⋄, control; ■, AMPH; ▵, TPA; ○, AMPH + TPA; ♦, IMI; ●, IMI + AMPH; and ▴, IMI + TPA. Results are given in picomoles of DA per milligram of protein ± S.E.M.n = 3. ANOVA for fraction 10, p< 0.001, in post hoc Tukey-Kramer comparison tests, values for AMPH, TPA, and TPA + AMPH with imipramine differed from nonimipramine containing values at p < 0.01.
Extracellular Ca2+ and Vesicular Function Are Not Required for AMPH- or TPA-Mediated DA Release.
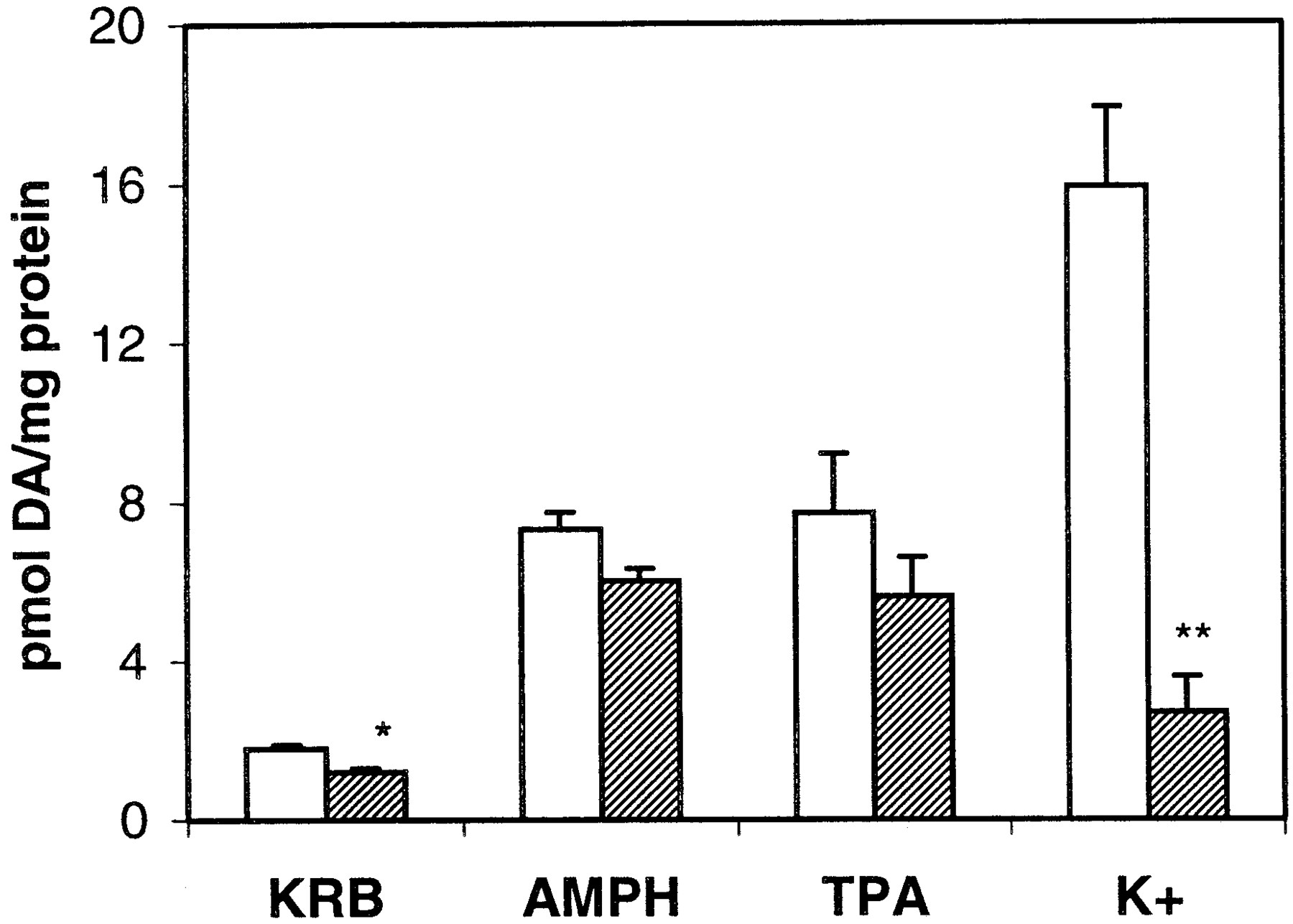
PKC activation can enhance extracellular Ca2+-dependent exocytotic neurotransmitter release through synaptic vesicles (for review, seeRobinson, 1991). To examine whether AMPH- and TPA-mediated DA release from PC12 cells was dependent on extracellular Ca2+, CaCl2 was deleted from the KRB superfusion buffer. As shown in Fig.5, DA release elicited by either 1 μM AMPH or 250 nM TPA was unaffected by the lack of extracellular Ca2+, whereas the depolarization-mediated DA release, elicited by 50 mM KCl, was abolished. The baseline release of DA was significantly reduced by the removal of extracellular Ca2+, such that the fold-stimulation by either AMPH or TPA over baseline was unaltered by the removal of Ca2+.

Absence of extracellular Ca2+ does not alter AMPH- or TPA-mediated DA release. PC12 cells were incubated in KRB prepared with or without 1.2 mM CaCl2. The amount of DA released in response to a 2.5-min bolus of 1 μM AMPH or 250 nM TPA was collected and measured by HPLC as described under Materials and Methods. Results are given as picomoles of DA released per milligram of protein ± S.E.M. n = 3. Only values for the baseline (p < 0.01) and K+ stimulation (p < 0.005) were altered by the absence of Ca2+ as assessed by Student'st test. The fold-stimulation over baseline by AMPH in the presence (■) and absence (▨) of extracellular Ca2+was 4.5 ± 0.5 and 5.1 ± 0.6, respectively. The fold-stimulation over baseline by TPA in the presence and absence of extracellular Ca2+ was 4.5 ± 1.0 and 4.3 ± 0.3, respectively.
To ascertain whether there is a role of exocytosis and synaptic vesicles in AMPH- and TPA-mediated DA release, the PC12 cells were pretreated with 50 nM reserpine for 3 h, which effectively depleted the cells of DA. The concentration of DA in vehicle- and reserpine-treated cells was 12 pmol/μg protein ± 4 (standard error of the mean, S.E.M.) and 0.1 pmol/μg protein ± 0.02, respectively, n = 4. As shown in Fig.6, reserpine pretreatment did not alter the ability of 1 μM AMPH or 250 nM TPA to release DA from PC12 cells. On the other hand, the reserpine pretreatment completely abolished the release of DA in response to depolarization with 50 mM KCl. Despite the reserpine pretreatment, chelerythrine could still inhibit AMPH- and TPA-mediated DA release (Fig. 6). However, chelerythrine had no effect on depolarization-mediated DA release.
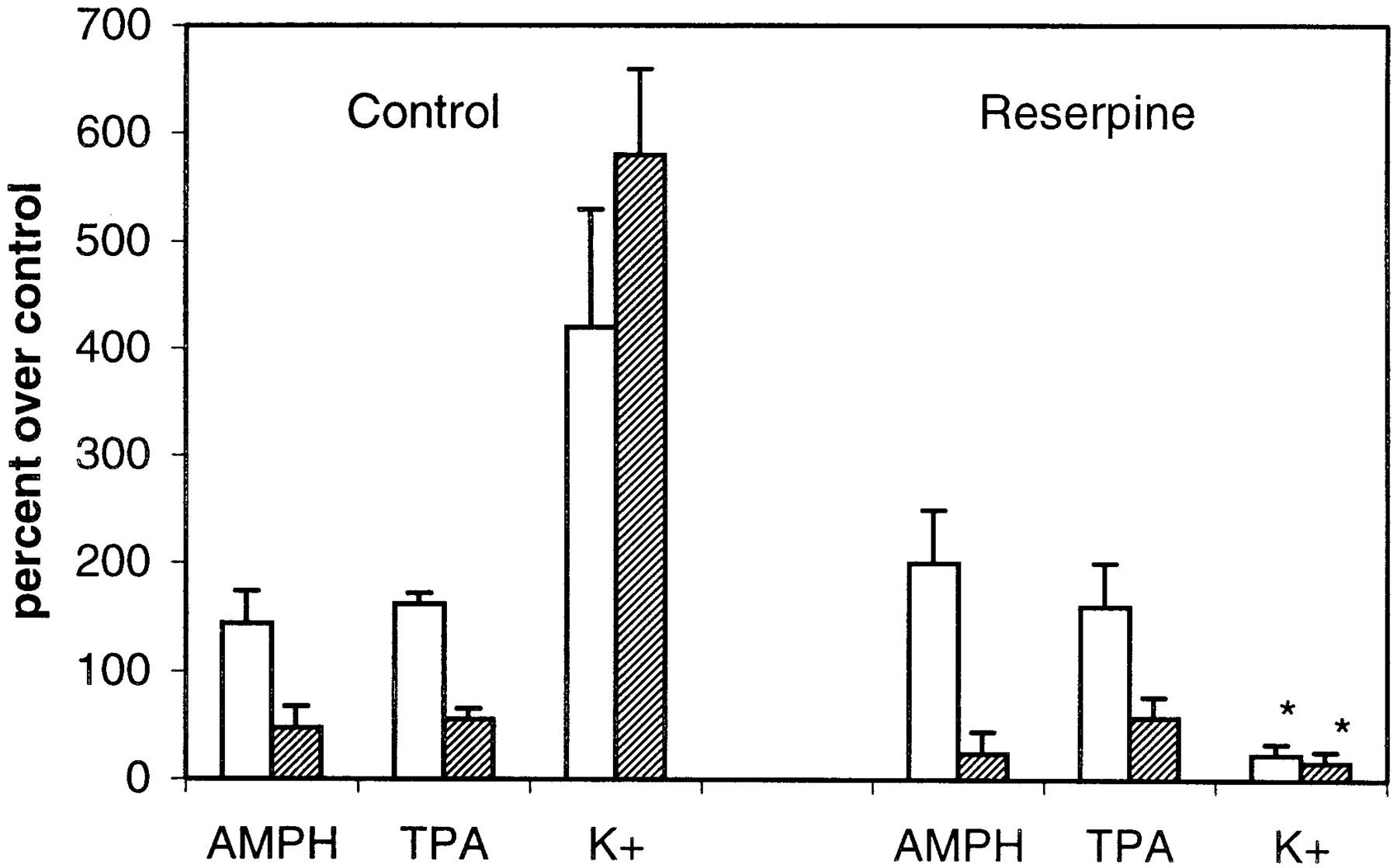
Reserpine pretreatment does not alter AMPH- or TPA-mediated DA release nor the inhibitory actions of chelerythrine (CH). PC12 cells were pretreated with vehicle or reserpine for 3 h as discussed under Materials and Methods. Following the incubation, cells were perfused with buffer containing KRB (■) or KRB with 1 μM chelerythrine (▨). DA release in response to a 2.5-min bolus of 1 μM AMPH, 250 nM TPA, or 50 mM KCl was measured. Results are given in percentage over control, where control is the DA released in the absence of AMPH, TPA, or KCl. Chelerythrine alone did not alter baseline. n = 3. For all groups, ANOVA gavep < 0.0001. In the post hoc Tukey-Kramer comparison test, values for K+ in control cells differed from those in reserpine-treated cells, in the presence or absence of chelerythrine, at p < 0.001. There is no significant difference in values for AMPH or TPA between control and reserpine groups in the presence or absence of chelerythrine.
Long-Term TPA Treatment Reduces DA Release but not Uptake.
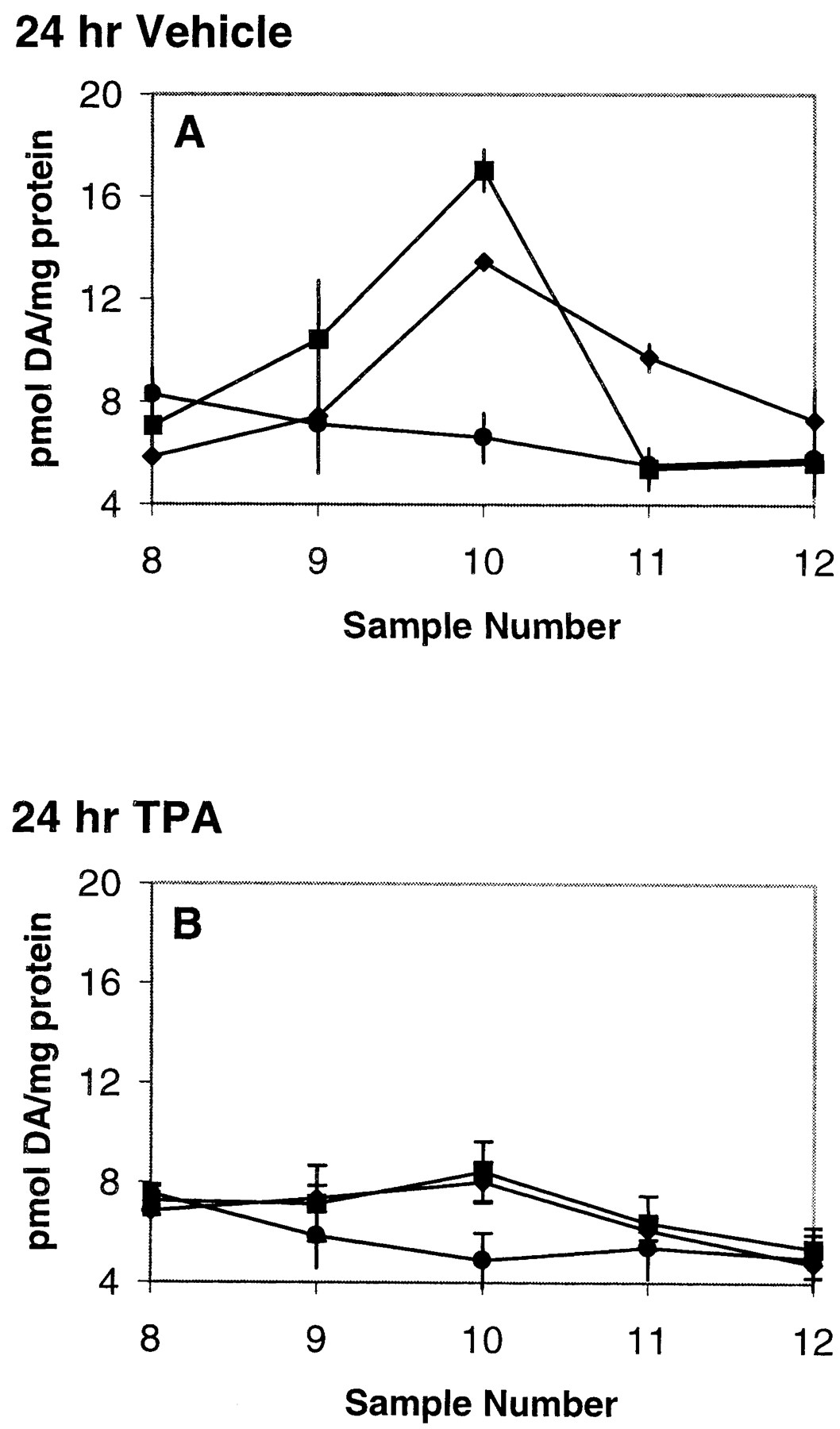
If PKC is essential for AMPH-mediated reverse transport, then down-regulation of the PKC should decrease the ability of AMPH to release DA. PC12 cells were treated with 70 nM TPA or vehicle for 24 h. The treatment was terminated by removing media containing TPA and washing the cells with phosphate-buffered saline. The cells were harvested, placed in the perfusion apparatus and perfused with KRB for 30 min. A bolus of 1 μM AMPH or 250 nM TPA was administered for 2.5 min. The results demonstrate that cells that were treated with vehicle for 24 h were able to release DA in response to AMPH or TPA (Fig. 7A), whereas those pretreated for 24 h with TPA were unresponsive to either agent (Fig. 7B). Since phorbol ester treatment has been demonstrated to increase internalization of DAT in many cell lines and reduce transporter function (Huff et al., 1997; Zhang et al., 1997; Zhu et al., 1997;Daniels and Amara, 1999; Pristupa et al., 1998) we measured uptake of [3H]DA in cells treated with vehicle or TPA for 24 h. The data of Fig. 8 reveal that long-term TPA treatment had no effect on [3H]DA uptake compared with vehicle-treated group. These experiments were performed using both 30 nM and 1 μM [3H]DA (data not shown) with identical results. When present, 50 μM imipramine inhibited [3H]DA uptake similarly in both treatment groups.

Long-term TPA incubation reduces AMPH- and TPA-mediated DA release. PC12 cells were incubated with vehicle (A) or 70 nM TPA (B) for 24 h. Following preincubation, cells were washed and then perfused with a 2.5-min bolus of 1 μM AMPH or 250 nM TPA as described under Materials and Methods. For A and B, ♦, TPA; ▪, AMPH; and ●, control. The long-term treatment with TPA did not effect basal DA release. Data are shown as picomoles per milligram of protein ± S.E.M. n = 3. For A, 24-h vehicle, ANOVA on values from fraction 10, p ≤ 0.0005. In post hoc Tukey-Kramer comparison test, control differed from values for TPA and AMPH at p < 0.01. TPA and AMPH values did not differ. For B, 24-h TPA, ANOVA on values from fraction 10, p ≤ 0.1, not significant.

Long-term TPA incubation does not alter [3H]DA uptake. PC12 cells were incubated with vehicle or 70 nM TPA for 24 h. Uptake of 30 nM [3H]DA was measured in cells as described under Materials and Methods in the absence and presence of 50 μM imipramine (IMI). The cells were incubated with 50 μM imipramine for 20 min followed by uptake of 30 nM [3H]DA with for 10 min at 37°C. Identical results were obtained using 1 μM [3H]DA. Values are shown as femtomoles per minute per milligram of protein ± S.E.M. n = 3. ANOVA,p < 0.0001. In the post hoc Tukey-Kramer comparison test, all values in the absence of imipramine differed from those in the presence of imipramine at p < 0.001. There was no significant difference in uptake of [3H]DA between vehicle- and TPA-treated cells values in the presence or absence of imipramine.
Long-Term TPA Treatment Down-Regulates PKCα and Decreases PKC Activity.
To demonstrate that PKC was down-regulated by 24 h TPA, the cells were immunoblotted for PKCα and PKC activity was measured in cytosol and membrane fractions. Long-term treatment with 70 nM TPA down-regulated the expression of PKCα compared with controls (Fig. 9) but did not reduce it entirely. We also performed immunoblots for PKCβ and ε, but the immunoblot signal was strongest and most measurable for PKCα in our PC12 cells. PKC activity decreased after long-term TPA treatment compared with control cells (Table 1). The reduction in the PKC activity was much greater in the supernatant than the membrane fraction, suggesting some PKC can still remain active in membranes. We found similar results following chronic TPA treatment in SH-SY5Y cells (Goldsmith and Gnegy, 1999).

Immunoblot for PKCα isozyme in vehicle (Veh)- and 24-h TPA-treated PC12 cells. PC12 cells were treated with vehicle or 70 nM TPA for 24 h. Cells were then washed, homogenized, and 50 μg of protein was subjected to SDS-PAGE and immunoblotted with antibody to PKCα as described under Materials and Methods. The leftmost lane contains rat striatum as a control. The experiment was repeated three times. All lanes contain 50 μg of protein. PKCα migrated at an apparent Mr of 80 kDa.
Effect of 24-h TPA treatment on PKC activity
Internal Ca2+ Is Required for AMPH-Mediated DA Release.
Extracellular Ca2+ is not required for AMPH-mediated DA release (Raiteri et al., 1979; Seiden et al., 1993) but the role of intracellular Ca2+ on monoamine transporter function has not been fully explored. To assess a role for intracellular Ca2+ in AMPH-mediated outward transport, cells were treated with 50 μM of the membrane permeable Ca2+chelator BAPTA-AM, in the presence of KRB containing no CaCl2. To ensure that BAPTA-AM significantly lowered intracellular Ca2+, intracellular Ca2 was measured using fura-2. The results in Table2 demonstrate that, in the absence of external Ca2+, BAPTA-AM effectively chelates the internal Ca2+ and abolishes the Ca2+ influx in response to 50 mM KCl. When Ca2+ is included in the media, BAPTA-AM reduces, but does not abolish the peak response to KCl, demonstrating a partial chelation of internal Ca2+. AMPH-mediated DA release was measured in PC12 cells preincubated with 50 μM BAPTA-AM in the absence of Ca2+ in the external media. The data of Fig. 10 show that BAPTA-AM abolished the ability of 1 μM AMPH to elicit release of DA from the cells. BAPTA-AM had no effect on basal DA release. Since BAPTA treatment could block uptake of DA, we measured [3H]DA uptake in cells treated with 50 μM BAPTA. Preincubation of the cells with BAPTA had no effect on uptake of 30 nM [3H]DA, nor in the ability of imipramine to block the uptake (Fig. 11). This experiment was repeated with 1 μM [3H]DA with the same result. These data strongly suggests that internal Ca2+ must be present to generate AMPH-mediated DA release.
Effect of BAPTA-AM on the intracellular calcium concentration

BAPTA-AM pretreatment reduces AMPH-mediated DA release. PC12 cells were placed into the perfusion apparatus and perfused with KRB that did not contain CaCl2 in the absence or presence of 50 μM BAPTA-AM. After 30 min, either the KRB or AMPH (1 μM) was given for 2.5 min in the appropriate KRB buffer. Following the bolus of AMPH, the cells were returned to the perfusion with vehicle or BAPTA-containing buffer. Incubation with BAPTA-AM did not affect basal DA release. Values are given as picomoles of DA per milligram of protein ± S.E.M., n = 5. In comparing maximal DA released in fraction 10, ANOVA p < 0.0001. In a post hoc comparison of individual values using the Tukey-Kramer multiple comparisons test, AMPH (▪) significantly differed from control (●), BAPTA (▴), and BAPTA + AMPH (♦) atp < 0.001. BAPTA + AMPH did not significantly differ from either control or BAPTA alone.
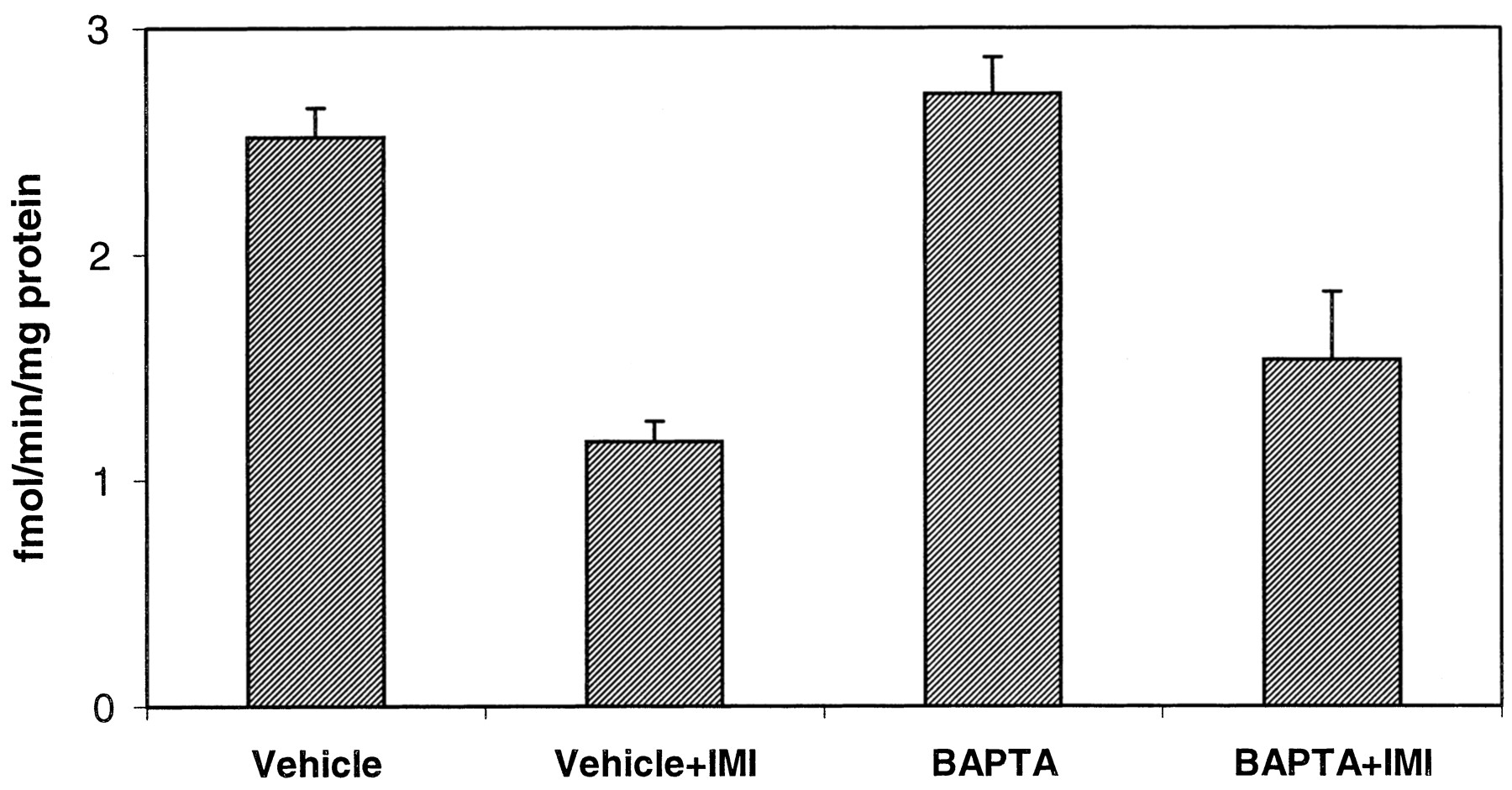
BAPTA-AM pretreatment does not alter [3H]DA uptake. PC12 cells scraped from the plate, washed, and preincubated for 20 min with 50 μM BAPTA-AM or with vehicle with or without 50 μM imipramine (IMI) in KRB without calcium. [3H]DA (30 nM) was added and incubated with cells for an additional 10 min at 37°C. Cells were filtered, washed, and uptake of 30 nM [3H]DA measured as described under Materials and Methods. Identical results were obtained using 1 μM [3H]DA. There is no significant difference between vehicle- and BAPTA-pretreated cells in either the presence or absence of imipramine. n = 3. ANOVA, p< 0.001. In the post hoc Tukey-Kramer comparison test, all values in the absence of imipramine differed from those in the presence of imipramine at p < 0.01.
Discussion
In this study, we found that AMPH requires an active PKC to release DA through reverse transport of NET in undifferentiated PC12 cells. This result was identical to that from our studies examining the role of PKC in AMPH-induced DA release in rat striatum through DAT (Kantor and Gnegy, 1998; Cowell et al., 2000). This indicates that the requirement for PKC activation for AMPH-mediated release is not a characteristic unique to a certain transporter, but instead is a characteristic of the substrate AMPH. We have also demonstrated that intracellular, but not extracellular, Ca2+ is required for AMPH to release DA through the transporter.
The requirement of PKC for AMPH action is demonstrated by the fact that inhibition of PKC activity, whether through the use of two different PKC inhibitors or TPA-induced down-regulation, blocks the DA releasing effects of AMPH. The role of PKC activity in reverse transport is also demonstrated by the ability of TPA to directly release DA. This release occurred through NET because it was independent of extracellular Ca2+ and was antagonized by imipramine, an uptake blocker. TPA-induced DA release from rat striatum was similarly independent of extracellular Ca2+, was blocked by cocaine and GBR12935, and was not additive with AMPH (Cowell et al., 2000). A PKC activator-induced release of DA that was independent of extracellular Ca2+ has been reported (Davis and Patrick, 1990). The substrate for the PKC is unknown. Both DAT (Giros and Caron, 1993) and NET (Bruss et al., 1997), however, contain consensus sequences for PKC.
AMPH can enhance releasable DA through blockade of VMAT (Sulzer et al., 1995) and is postulated to increase vesicular DA release through a release of intracellular Ca2+ (Mundorf et al., 1999). The vesicular monoamine transporter VMAT, however, is not required for AMPH-mediated catecholamine release (Pifl et al., 1995;Fon et al., 1997) although it plays an important role in the rate of the response to AMPH (Pifl et al., 1995; Jones et al., 1998). Our results do not support a role for DA-containing synaptic vesicles in either the releasing action of AMPH or the effect of PKC activation or inhibition. PKC activators, such as TPA, diacylglycerol, and arachidonic acid, enhance exocytotic neurotransmitter release and those effects require extracellular Ca2+ (Robinson, 1991). Neither the lack of extracellular Ca2+ nor reserpine pretreatment altered the ability of 1 μM AMPH or 250 nM TPA to release DA. The fact that imipramine blocked the effect of AMPH and TPA would suggest that both drugs are acting to reverse the action of NET. Similarly, PKC inhibitors have little or no effect on Ca2+-evoked release in the absence of supplementary PKC activation (see Discussion in Robinson, 1991). The ability of the PKC inhibitors to block AMPH-mediated DA release required no exogenous source of PKC activation, was not altered by reserpine pretreatment, and did not require extracellular Ca2+. In addition, our preliminary experiment in hDAT-transfected Cos-7 cells, which contain neither VMAT nor synaptic vesicles, demonstrated that the AMPH-induced DA release and requirement for PKC activation was independent of synaptic vesicles. From our studies, it seems unlikely that synaptic vesicles and exocytosis were contributing to the ability of AMPH and TPA to release DA. The ability of PKC inhibitors to block AMPH-mediated DA release in rat striatal slices and synaptosomes was unaffected by deletion of extracellular Ca2+ and reserpine pretreatment of the rat, showing that our results were not idiosyncratic for the PC12 cells (Kantor and Gnegy, 1998; Cowell et al., 2000).
Although there have been some reports of a requirement for extracellular Ca2+ in AMPH-mediated DA release (Crespi et al., 1997; Mundorf et al., 1999), most studies have found that acute AMPH-mediated release is independent of extracellular Ca2+ (Raiteri et al., 1976; Arnold et al., 1977;Takimoto et al., 1983; Carboni et al., 1989; Hurd and Ungerstedt, 1989). In some studies reporting a role for extracellular Ca2+, synaptosomes or slices were incubated for up to an hour with EGTA before measuring the AMPH effect (Crespi et al., 1997; Kramer et al., 1998). Ca2+ is lost from intracellular stores following incubation in millimolar EGTA (Fisher et al., 1989). This implies that intracellular Ca2+ could play a role in AMPH action and our studies have demonstrated this. The intracellular Ca2+ chelator BAPTA nearly completely inhibited the DA-releasing effect of AMPH. We found that pretreatment of rat striatal slices with BAPTA-AM also attenuated AMPH-mediated DA release (L. Kantor and M. E. Gnegy, unpublished observation). Mundorf et al. (1999) reported an inhibiting effect of BAPTA on AMPH-stimulated catecholamine release from bovine chromaffin cells.
The requirement for internal Ca2+ would be consistent with an activation of a Ca2+-dependent PKC activity, but at this point the molecular process requiring the Ca2+ is unknown. In this study we demonstrated that PKCα, a Ca2+-requiring isozyme, was down-regulated by long-term TPA treatment concomitant with the reduction in AMPH-mediated DA release. Although we did not measure all PKC isozymes in the PC12 cells, it should be noted that the prominent PKC isozyme in dopaminergic terminals in the striatum is the α-isozyme (Yoshihara et al., 1991). It is not known whether AMPH actively elicits an increase in PKC activity or whether an AMPH-mediated alteration of transporter conformation activates PKC. We (Iwata et al., 1997) and Giambalvo (Giambalvo, 1992) reported that AMPH treatment of synaptosomes can increase PKC activity. In addition,Kramer et al. (1998) demonstrated that 3,4-methylenedioxymethamphetamine, acting through the serotonin transporter in cerebral cortical synaptosomes, leads to an activation of PKC within the nerve terminal. Amphetamine has been reported to increase the release of intracellular Ca2+(Mundorf et al., 2000).
Our data further suggest that AMPH-mediated outward transport and the inward transport of [3H]DA can be separately regulated. Since NET and DAT function as gated channels (Rudnick and Clark, 1993), they would be expected to be fully reversible, such that an agent that affects uptake would affect release and the rate of uptake would equal the rate of release. Evidence suggests, however, that AMPH-induced outward transport is not a simple reversal of inward transport (Langeloh et al., 1987; Sitte et al., 1998; Kantor and Gnegy, 1998; Pifl et al., 1999). In PC12 cells, the relationship between rate of uptake and rate of release for releasing amine substrates is multifactorial, not linear (Langeloh et al., 1987). In addition, AMPH has higher release rates than those for other unlabeled amines (Sitte et al., 1998). The release rate paralleled an inward current, presumably a Na+ current, induced by these substrates in patch-clamp experiments. AMPH could elicit a rapid increase of a Ca2+-dependent PKC, which could, in turn, enhance a Na+ current permitting more binding of intracellular DA to an inward-facing transporter. Therefore, the action of AMPH in eliciting outward transport can involve a different or additional mechanism and mode of regulation than that of the uptake process.
Our data appear at variance with the reports that a PKC-mediated phosphorylation reduces monoamine inward transport and elicits internalization of the plasmalemmal transporters (Copeland et al., 1996; Huff et al., 1997; Zhang et al., 1997; Zhu et al., 1997;Apparsundaram et al., 1998; Pristupa et al., 1998; Daniels and Amara, 1999; Melikian and Buckley, 1999). Experiments suggest that the catecholamine transporters DAT and NET constitutively cycle between the plasma membrane and an intracellular endosomal pool through a clathrin- and dynamin-mediated pathway that seems to involve PKC activation (Daniels and Amara, 1999; Melikian and Buckley, 1999; Saunders et al., 2000). The lack of change in uptake of [3H]DA following 24 h of TPA could be accounted for by the cessation of internalization of the transporter due to down-regulation of PKC. AMPH itself has been demonstrated to induce internalization of DAT and thus can participate in the endosomal pathway (Saunders et al., 2000). AMPH was much more active in this activity than DA. It is possible that AMPH has two activities, one very short-term that increases reverse transport at the plasmalemma membrane and another, with slightly later activity, resulting in increased internalization. Therefore, AMPH on the very short term would enhance release of DA, but on a longer term would contribute to a compensatory down-regulation of transporter function.
In conclusion, we have demonstrated that AMPH-mediated DA release through NET in PC12 cells requires activation of PKC and the presence of intracellular calcium. The regulation of AMPH action through NET in PC12 cells by PKC parallels that of DAT in striatum. [3H]DA uptake and AMPH-mediated DA release were differentially affected by PKC inhibition or chelation of intracellular calcium, suggesting an asymmetry in their regulation. It is possible that AMPH elicits a rapid activation of a Ca2+-dependent PKC, which either enhances a Na+ current, causing greater binding of intracellular DA to the inward-facing transporter, or actually leads to a short-term enhancement of outward function at the plasmalemmal membrane.
Acknowledgments
We thank Dr. Richard Neubig and Masakatsu Nanamori for help in transfecting Cos-7 cells with the plasmid pcDNA3-hDAT1 plasmid. We also thank Dr. Z. Pristupa, University of Toronto, for donating the plasmid. We thank Jason Kurzer for help in measurement of AMPH-mediated DA release following long-term TPA treatment. We thank Dr. Stephen Fisher for helpful discussions and reading of the manuscript.
Footnotes
-
Send reprint requests to: Margaret E. Gnegy, Department of Pharmacology, 2220 MSRB III, University of Michigan School of Medicine, Ann Arbor, MI 48109-0632. E-mail: pgnegy{at}umich.edu
-
This work was supported by National Research Service Award Grant 5F32DA05912 (to L.K.), Basic Science Research Partnership Grant, University of Michigan Medical School, and Grant DA11697 from the National Institutes of Health.
- Abbreviations:
- AMPH
- amphetamine
- NE
- norepinephrine
- DA
- dopamine
- NET
- norepinephrine transporter
- DAT
- dopamine transporter
- PKC
- protein kinase C
- TPA
- 12-O-tetradecanoylphorbol 13-acetate
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetra(acetoxymethyl) ester
- KRB
- Krebs-Ringer buffer
- HPLC
- high-performance liquid chromatography
- PAGE
- polyacrylamide gel electrophoresis
- VMAT
- vesicular monoamine transporter
- ANOVA
- analysis of variance
- Received December 1, 2000.
- Accepted January 31, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}