


Abstract
Nociceptin is an endogenous peptide that produces its biological effects by binding to the opioid receptor-like (ORL1) receptor. It has been shown that activation of ORL1receptor leads to inhibition of the adenylyl cyclase activity, but stimulation of the extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases. In this report, we demonstrate that activation of the G protein-coupled ORL1receptor in transfected COS-7 cells leads to stimulation of the JNK subgroup of mitogen-activated protein kinases in a Ras/Rac-dependent manner, and it was insensitive to wortmannin. This increased JNK activity was mainly mediated by PTX-sensitive Gi proteins, and partially contributed by a PTX-insensitive component. Among all known PTX-insensitive G proteins, Gz, G12, G14, and G16 seemed to have functional coupling with the ORL1 receptor in terms of JNK activation. Stimulation of the endogenous ORL1 receptor in NG108-15 cells also led to activation of a PTX-sensitive JNK activity in a wortmannin-insensitive manner. The induced JNK activation is accompanied by the active phosphorylation of c-Jun and activating transcription factor-2. This is the first report that demonstrates the stimulatory effect of ORL1 receptor on JNK, and the subsequent activation of c-Jun and activating transcription factor-2.
Nociceptin/orphanin FQ (N/OFQ) is the endogenous ligand for the G protein-coupled opioid receptor-like (ORL1) receptor (Meunier et al., 1995; Reinscheid et al., 1995). The ORL1 receptor has high sequence homology to different opioid receptor subtypes but does not exhibit high-affinity binding for opioid ligands. The mRNA of ORL1 and the peptide precursor of N/OFQ are widely distributed in the central nervous system (Mollereau et al., 1994; Nothacker et al., 1996). The biological functions associated with the ORL1 receptor appear to be somewhat opposite to those of the opioid receptors. ORL1receptors expressed in the central nervous system mediate pain regulation, and intraventricular injection of N/OFQ into the mouse brain resulted in enhanced painful reactivity (Meunier et al., 1995).
The ORL1 receptor produces its biological effects through different members of the G protein superfamily. Like opioid receptors, the ORL1 receptor is known to inhibit adenylyl cyclases (Mollereau et al., 1994) and mediate ion channel activities (Connor et al., 1996) by coupling to the pertussis toxin (PTX)-sensitive Gi and Goproteins, respectively. We have demonstrated that the ORL1 receptor is also capable of coupling to PTX-insensitive Gz, G14,G16, and, to a lesser extent, G12 to regulate the activities of different second messenger systems (Chan et al., 1998; Yung et al., 1999). The ability of the ORL1 receptor to interact with multiple G proteins may in part explain the diverse physiological effects of N/OFQ, ranging from modulation of nociceptive function to locomotor activity (Reinscheid et al., 1995) and stress adaptation (Koster et al., 1999).
In less than a decade, we have come to the realization that many G protein-coupled receptors regulate cell proliferation and differentiation through the mitogen-activated protein kinases (MAPKs). MAPKs are serine/threonine protein kinases capable of phosphorylating several transcription factors (Price et al., 1996), and hence, regulate subsequent transcriptional events. There are at least three subtypes of MAPK, the extracellular signal-regulated kinases (ERKs) are mainly stimulated by growth factors (Boulton et al., 1991), whereas c-Jun NH2-terminal kinases (JNKs) and p38 MAPK are more responsive to cellular stress (Han et al., 1994; Kyriakis et al., 1994). Recent studies suggest that the signaling of ORL1 receptors involves the activation of ERK and p38 MAPK (Lou et al., 1998; Zhang et al., 1999) to modulate the activities of the downstream transcription factors Elk-1 and Sap1a (Bevan et al., 1998). However, the capability of the ORL1 receptor to activate JNK remains unknown.
JNK modulates the activities of several transcription factors. Depending on the JNK isoforms involved, c-Jun, ATF-2, and Elk-1 can be actively phosphorylated upon JNK activation (Gupta et al., 1996), whereas the phosphorylation of NFAT4 by JNK prevents its translocation to the nucleus (Chow et al., 1997). Therefore, activation of JNK may regulate gene transcription in both directions to up-regulate the expression of certain genes and to down-regulate others. There is evidence to suggest that ORL1receptor-mediated MAPK stimulation and the subsequent modulation of transcriptional events may have some physiological significance. Chronic activation of the ORL1 receptor leads to supersensitization of adenylyl cyclase (Chan and Wong, 1999), and this adaptive response may involve gene transcription as in the case of immediate-early genes transcription associated with supersensitization of dopamine receptor functions (LaHoste et al., 1993). In this report, we used COS-7 cells transiently expressing the ORL1 receptor, as well as neuroblastom × glioma hybrid NG108-15 cells, which endogenously express the receptor, to investigate the characteristics of N/OFQ-induced JNK activation. Our results clearly demonstrated the capability of ORL1 receptor to stimulate JNK activity in terms of active phosphorylation of c-Jun and ATF-2.
Materials and Methods
Reagents.
The cDNAs encoding ORL1receptor and JNK-HA were kindly provided by Dr. Gang Pei (Shanghai Institute of Cell Biology, Shanghai, China) and Dr. Tatyana A. Voyno-Yasenetskaya (University of Illinois, Chicago, IL), respectively. The cDNAs of the dominant-negative mutants of Ras (RasS17N) and Rac (RacT17N) were generous gifts from Dr. Eric J. Stanbridge (University of California, Irvine, CA). [γ-32P]ATP was purchased from DuPont NEN (Boston, MA). Anti-phospho-JNK, anti-JNK, anti-phospho-c-Jun, and anti-phospho-ATF-2 antibodies were obtained from New England BioLabs (Beverly, MA). PTX and 12CA5 (anti-HA) antibody were purchased from List Biological Laboratories (Campbell, CA) and Roche Molecular Biochemicals (Indianapolis, IN), respectively. N/OFQ was obtained from Research Biochemicals International (Natick, MA). Cell culture reagents, including LipofectAMINE PLUS were obtained from Life Technologies (Gaithersburg, MD) and all other chemicals were purchased from Sigma (St. Louis, MO).
Cell Culture and Transfection.
COS-7 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal calf serum, 50 units/ml penicillin and 50 μg/ml streptomycin, and grown at 37°C in an environment of 5% CO2. In the case of NG108-15 cells, fetal calf serum was reduced to 5%. Transfection was performed on 3 × 106 COS-7 cells in a 10-cm plate by means of LipofectAMINE PLUS reagents following the supplier's instructions.
In Vitro JNK Assay.
COS-7 cells were transferred to six-well plates at 3 × 105 cells/well 12 h after transfection, and then kept in the growth medium for 36 h. The cells were then serum starved for 18 h in the presence or absence of PTX before drug treatment. In case of the wortmannin sensitivity assay, an additional treatment of wortmannin (100 nM, 15 min) was applied to the starved cells. The assay used was basically similar to that previously described (Berestetskaya et al., 1998). Transfected COS-7 cells in six-well plates were treated with the assay medium (Dulbecco's modified Eagle's medium with 20 mM HEPES) in the presence or absence of N/OFQ for 30 min at 37°C. Reactions were terminated by washing the cells with ice-cold phosphate-buffered saline, followed by addition of 500 μl of lysis buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 5 mM EDTA, 40 mM NaP2O7, 1% Triton X-100, 1 mM dithiothreitol, 200 μM Na3VO4, 100 μM phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, 4 μg/ml aprotinin, and 0.7 μg/ml pepstatin) and then gently shaken on ice for 30 min. A supernatant was collected for each sample by centrifugation at 16,000g for 5 min. Fifty microliters of each supernatant was used for the detection of JNK-HA expression, and the remaining was incubated for 1 h at 4°C with anti-HA antibody (2 μg/sample), followed by incubation with 30 μl of protein A-agarose (50% slurry) at 4°C for 1 h. The resulting immunoprecipitates were washed twice with lysis buffer and twice with kinase assay buffer [40 mM HEPES, pH 8.0, 5 mM Mg(C2H3O2)2, 1 mM EGTA, 1 mM dithiothreitol, 200 μM Na3VO4]. Washed immunoprecipitates were resuspended in 40 μl of kinase assay buffer containing 5 μg of GST-c-Jun per reaction, and the kinase reactions were initiated by the addition of 10 μl of ATP buffer (50 μM ATP containing 2 μCi of [γ-32P]ATP per sample). After a 30-min incubation at 30°C with occasional shaking, the reactions were terminated by 10 μl of 6× sample buffer, and the samples were resolved by 12% SDS-polyacrylamide gel electrophoresis. The radioactivity incorporated into GST-c-Jun was detected by autoradiogram, and the signal intensity was quantified by PhosphorImager (Molecular Dynamics 445 SI).
Western Blot.
NG108-15 cells were seeded on six-well plates at a density of 1.5 × 105 cells/well and were kept in the growth medium overnight. The cells were then serum starved for 18 h either in the presence or absence of PTX, followed with a wortmannin treatment (100 nM, 15 min) if necessary. The cells were stimulated with N/OFQ (100 nM) for 30 min and then lysed in 500 μl of lysis buffer. Supernatants were collected by centrifugation at 16,000g for 5 min. Eighty microliters of each supernatant was resolved by 12% SDS-polyacrylamide gel electrophoresis, and then transferred to nitrocellulose membranes. The presence of actively phosphorylated JNK, c-Jun, and ATF-2 were detected by phospho-specific antibodies as mentioned under Reagents, followed with horseradish peroxidase-conjugated secondary antibody. The blot was developed in the presence of enhanced chemiluminescence reagents, and the images detected in X-ray films were quantified by densitometric scanning using the Eagle Eye II still video system (Stratagene, La Jolla, CA).
Results
ORL1 Receptor Stimulates JNK Activity in COS-7 Cells.
It has been established that the ORL1receptor inhibits adenylyl cyclase by activating the Gi proteins (Mollereau et al., 1994). Because Gi-coupled receptors can activate JNK (Coso et al., 1996), we examined whether the functional coupling between ORL1 receptor and Giproteins has any effect on JNK activity. Agonist treatment on COS-7 cells transfected with JNK-HA alone had no observable changes in the kinase activity upon application of 100 nM N/OFQ (Fig.1A). However, COS-7 cells cotransfected with JNK-HA and ORL1 receptor exhibited increased JNK activity upon stimulation with N/OFQ (Fig. 1A). These results indicate that the COS-7 cells probably do not express the ORL1 receptor, or the receptor is only present at an extremely low level and does not affect our functional assay. The agonist-induced kinase activity was characterized by a dose dependence on the concentration of N/OFQ, reaching a maximum activity at 100 nM (Fig. 1B). When the transfected cells were stimulated with 100 nM N/OFQ for different durations, the JNK activity increased gradually and became saturated around 15 to 30 min of drug treatment (Fig. 1C). To examine whether the N/OFQ-induced stimulation of JNK required Gi proteins, we pretreated the transfectants with PTX before stimulation with N/OFQ. As shown in Fig. 1A, PTX treatment significantly reduced the N/OFQ-induced activation of JNK. This result indicated that the ORL1 receptor-mediated stimulation of JNK was primarily via endogenous PTX-sensitive Gi proteins. However, because the increased kinase activity could not be completely abolished by PTX (Fig. 1A), we could not exclude the possibility that the ORL1receptor may also stimulate the kinase activity through other pathways, for example, via endogenous PTX-insensitive G proteins.

Stimulation of JNK by the ORL1receptor in transfected COS-7 cells. COS7-cells were transfected with the cDNAs of JNK-HA alone, or cotransfected with the ORL1 receptor. JNK assay was performed as described under Materials and Methods. The JNK activity was determined at 30 min (A and B) in the absence (basal) or presence of 100 nM N/OFQ with or without PTX pretreatment (100 ng/ml, 18 h), or stimulated for the indicated periods (C). Values shown represent the mean ± S.E. from three separate experiments. The phosphorylation of GST-c-Jun and the expression of JNK-HA in the cell lysates are shown on the right-hand side. ∗, N/OFQ significantly stimulated the JNK activities in the transfected cells (Bonferroni t test,P < .05). ∗∗, PTX treatment significantly inhibited the N/OFQ-induced JNK stimulation (Bonferroni ttest, P < .05). In B, the JNK activity was significantly higher than the basal values when the N/OFQ concentration was >1 nM (Bonferroni t test, P < .05).
ORL1 Receptor-Mediated JNK Activity Is Dependent on the Low-Molecular-Weight GTPases and Is Insensitive to Wortmannin Pretreatment.
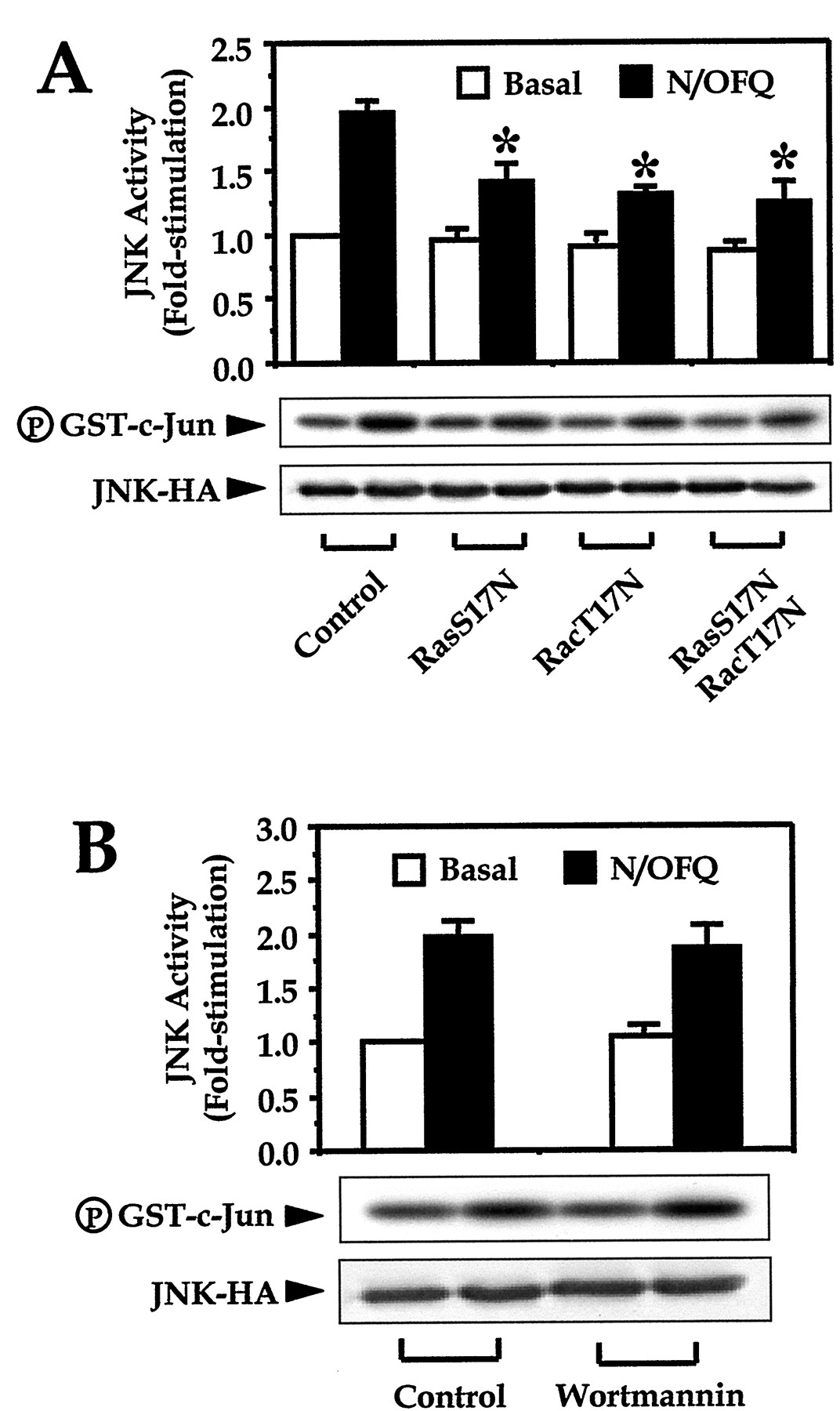
Low-molecular-weight GTPases have been shown to play an important role in the activation of MAPK activities (Thomas et al., 1992; Minden et al., 1995). The involvement of small GTPases such as Ras and Rac is often demonstrated with the use of dominant-negative mutants. By transfecting the dominant negative mutants of Ras (RasS17N) and Rac (RacT17N), we found that the ORL1receptor-mediated JNK activity was significantly inhibited (Fig.2A). Coexpression of the two dominant-negative mutants did not produce additive inhibition of the kinase activity (Fig. 2A). On the other hand, the role of phosphatidylinositol-3 kinase (PI3K) in modulating JNK activity has been studied by several groups, and both up-regulation (Lopez-Ilasaca et al., 1998) and down-regulation (Kwon et al., 2000) of JNK activity have been proposed. By pretreating the transfected cells with the specific PI3K inhibitor wortmannin, we observed no significant effect on the induced JNK activation (Fig. 2B). These results suggested that the low-molecular-weight GTPases, but not PI3K, served as signaling intermediates in the ORL1 receptor-mediated JNK activation.

ORL1 receptor-mediated JNK activity is dependent on the low-molecular-weight GTPases and is insensitive to wortmannin pretreatment. COS-7 cells were transfected with the cDNAs of JNK-HA and ORL1 receptor in the absence (B and the control in A) or presence of RasS17N and/or RacT17N (A). JNK activity is shown at 30 min in the absence (basal) or presence of 100 nM N/OFQ with or without wortmannin pretreatment (100 nM, 15 min) as indicated. Values shown represent the mean ± S.E. from three independent experiments. The phosphorylation of GST-c-Jun and the expression of JNK-HA in the cell lysates are shown below the graphs. ∗, N/OFQ-induced JNK activation was significantly inhibited in the presence of RasS17N and RacT17N (Bonferroni t test,P < .05).
N/OFQ-Induced Activation of JNK via PTX-Insensitive G Proteins.
Because the ORL1 receptor-mediated JNK activity was not completely abolished by PTX (Fig. 1A), PTX-insensitive G proteins endogenously expressed in COS-7 cells may link ORL1 receptors to the stimulation of JNK. The activated mutants of PTX-insensitive G12, G13, Gq/11, and G16 are capable of stimulating the JNK activity (Heasley et al., 1996; Voyno-Yasenetskaya et al., 1996). Our previous reports demonstrated that the ORL1 receptor is incapable of activating Gq, but it is functionally coupled to Gz, G14, G16, and to a lesser extent to G12 (Chan et al., 1998; Yung et al., 1999). Whether these functional couplings are linked to activation of JNK remains unknown. We coexpressed the ORL1receptor, JNK-HA, and the α-subunit of different PTX-insensitive G proteins in COS-7 cells, and examined the ORL1receptor-mediated JNK activation in the presence of PTX. Functional coupling of the ORL1 receptor to the coexpressed PTX-insensitive G protein should enhance the PTX-resistant JNK activity. No enhancement of N/OFQ-induced JNK activity was associated with cells coexpressing either PTX-insensitive Gαs or Gα13 compared with the control cells (Fig. 3). In contrast, COS-7 cells transfected with Gα12 was associated with a larger increase of JNK activity upon N/OFQ treatment, and this enhancement was even greater when PTX-insensitive Gαz, Gα14, or Gα16 was coexpressed instead (Fig. 3). These results indicated that the ORL1 receptor is capable of activating JNK through the PTX-insensitive Gz, G12, G14, and G16. However, it should be noted that the expression of Gα16 is restricted to hematopoietic cells (Amatruda et al., 1991), and COS-7 cells do not express endogenous Gαz (our unpublished data). Hence, the residual N/OFQ-induced JNK activity after PTX pretreatment was probably mediated through Gα12 and Gα14, which are ubiquitously expressed in different tissues (Strathmann and Simon, 1991) and found in kidney cells (Nakamura et al., 1991), respectively.

Functional coupling of ORL1receptor with PTX-insensitive G proteins leads to JNK activation. COS-7 cells were transfected with the cDNAs of JNK-HA, ORL1 receptor, in the absence (control) or presence of a Gα-subunit (Gαs, Gαz, Gα12, Gα13, Gα14, or Gα16). Transfected cells were pretreated with PTX (100 ng/ml, 18h). JNK activity is shown at 30 min after the addition of 100 nM N/OFQ. Values shown represent the mean ± S.E. from three separate experiments. The expression of JNK-HA and the phosphorylation of GST-c-Jun are also illustrated. ∗, coexpression of the Gα-subunit (Gαz, Gα12, Gα14, and Gα16) significantly enhanced the N/OFQ-induced kinase activity compared with the control (Bonferroni ttest, P < .05).
Stimulation of Endogenous ORL1 Receptor in NG108-15 Cells Resulted in Enhanced JNK Activity.
To verify that the ORL1 receptor-mediated JNK activation can occur in vivo, we applied N/OFQ treatment on NG108-15 cells that endogenously express ORL1 receptors, followed by subsequent detection of actively phosphorylated JNK by specific antibodies. Two immunoreactive bands, presumably representing JNK1 and JNK2, were detected by the JNK-specific antibodies. Treatment of N/OFQ induced the activation of JNK in a PTX-sensitive manner (Fig.4A). Densitometric quantification revealed a doubling of the JNK phosphorylation (JNK1 and JNK2) upon agonist treatment. Similar to the results obtained in the kinase assay of transfected COS-7 cells (Fig. 1A), the ORL1-mediated JNK activation in NG108-15 cells was also associated with a PTX-insensitive component (Fig. 4A). The N/OFQ-induced kinase activity is insensitive to wortmannin (Fig. 4B). Interestingly, the basal JNK activity was enhanced upon wortmannin pretreatment (Fig. 4B).

Stimulation of endogenous ORL1receptor in NG108-15 cells resulted in enhanced JNK activity in a PTX-sensitive but wortmannin-insensitive manner. NG108-15 cells were serum starved for 18 h in the absence or presence of PTX (100 ng/ml) before stimulation with 100 nM N/OFQ for 30 min (A), or accompanied by an additional wortmannin pretreatment (100 nM, 15 min) before the application of N/OFQ (B). Phosphorylated JNKs were detected by phospho-specific antibodies against JNKs and quantified by densitometry. Values shown represent the mean ± S.E. from three separate experiments. ∗, N/OFQ induced a significant increase of JNK phosphorylation in the control, PTX-treated (A) and wortmannin-treated (B) cells (Bonferroni paired t test, P < .05). ∗∗, PTX significantly inhibited N/OFQ-induced JNK phosphorylation (A), whereas wortmannin treatment (B) enhanced the basal JNK phosphorylation (Bonferroni t test,P < .05).
A previous study showed that JNK isoforms are capable of phosphorylating, and hence activating transcription factors such as c-Jun and ATF-2 (Gupta et al., 1996). To investigate whether the N/OFQ-induced JNK activation is accompanied with the active phosphorylation of these two transcription factors, we stimulated NG108-15 cells with N/OFQ for different periods of time, and detected the amount of activated JNK as well as the phosphorylated c-Jun and ATF-2. We found that treatment of NG108-15 cells with N/OFQ induced a time-dependent activation of the JNK (Fig.5A), accompanied with the phosphorylation of c-Jun and ATF-2 (Fig. 5B). JNK activity was slightly elevated with ≤15 min of N/OFQ treatment and no phosphorylation of c-Jun or ATF-2 was observed at those time points. At 30 or 45 min of agonist treatment, the JNK activity was doubled and phosphorylation of c-Jun and ATF-2 became apparent. Phosphorylation of c-Jun, but not of ATF-2, appeared to lag behind that of JNK (Fig. 5). However, the maximum JNK activity was delayed compared with the result obtained in the kinase assay of transfected COS-7 cells (Fig. 1C), and did not appear to be fully saturated even when the cells were stimulated by N/OFQ for 30 min.
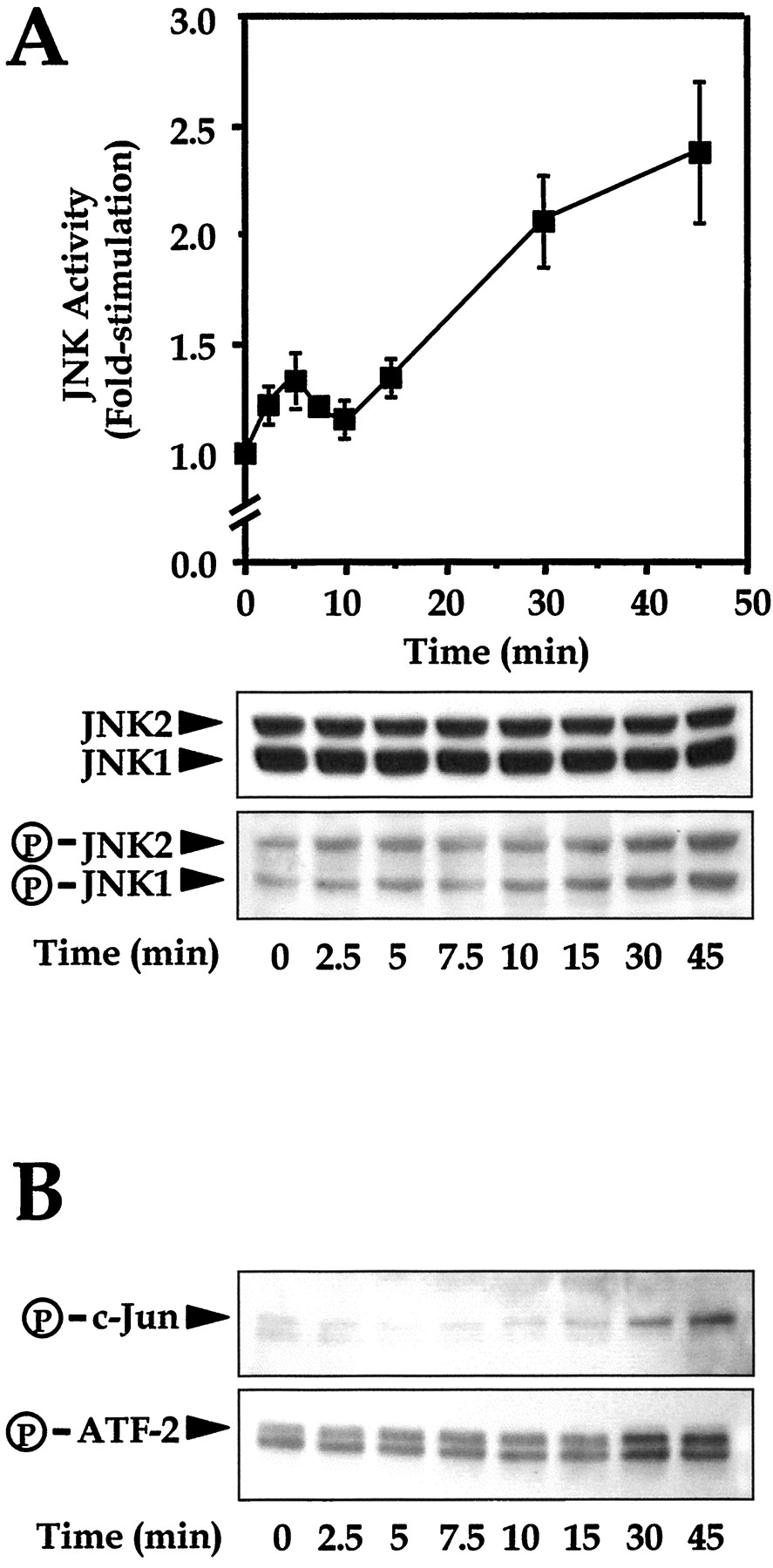
ORL1 receptor mediates JNK activation and the phosphorylation of c-Jun and ATF-2 in a time-dependent manner. Serum-starved NG108-15 cells were stimulated by 100 nM N/OFQ for different durations. Phosphorylated JNKs (A), and c-Jun and ATF-2 (B) were detected by phospho-specific antibodies, followed with subsequent quantification by densitometry. Values shown represent the mean ± S.E. from three separate experiments.
Discussion
The ORL1 receptor bears high resemblance to the opioid receptors in terms of its signal transduction properties. Given that the opioid receptors regulate neural development and synaptic plasticity by modulating neuronal survival and translational control, it is important to establish whether the ORL1 receptor can regulate similar events. The present study demonstrated that the functional coupling of ORL1 receptor with PTX-sensitive Gi proteins was associated with an increased activity of JNK. Because free Gβγ-subunits are more effective regulators than Gαi subunits for the stimulation of JNK activity (Coso et al., 1996; Yamauchi et al., 2000), the observed JNK activation might be mediated mainly through Gβγ-subunits released during the activation of Gi proteins by receptor stimulation. Indeed, the α-subunits of Go do not possess the ability to activate JNK (Yamauchi et al., 2000). Interestingly, PTX-insensitive G proteins such as Gz, G12, G14, and G16 can also link the ORL1 receptor to the activation of JNK in heterologous expression systems. Further analysis revealed that the N/OFQ-induced JNK activity was dependent on Ras and Rac but not on PI3K. N/OFQ-induced JNK signaling as well as the phosphorylation of c-Jun and ATF-2 were observed in NG108-15 cells that endogenously express the ORL1 receptor.
The mechanism by which G protein-coupled receptors regulate JNK activity is rather complicated. It has been suggested that stimulation of JNK by Gi-coupled receptors is Gβγ dependent, and the Gβγ-induced JNK activation involves PI3Kγ for signal transduction and is therefore wortmannin sensitive (Lopez-Ilasaca et al., 1998). In contrast, wortmannin-insensitive JNK activation mediated by Gβγ has also been described (Yamauchi et al., 1999). The Gβγ-mediated pathway probably involves nonreceptor tyrosine kinases, which act on specific guanine nucleotide exchange factors to activate low-molecular-weight GTPases such as Ras and Rac (Kiyono et al., 1999). A different perspective on the functional role of PI3K signaling is illustrated by the observations that Akt inhibits Rac-GTP binding, whereas wortmannin stimulates JNK activity (Kwon et al., 2000). The wortmannin insensitivity of our experimental results did not support the activating role of PI3K signaling on the ORL1 receptor-mediated JNK activation. Elevation of basal JNK activity in wortmannin-treated NG108-15 cells, in fact, suggested the presence of a PI3K inhibitory pathway in these cells. The characteristics of communication between PI3K and G protein-coupled receptor-mediated JNK signaling pathway may differ from one receptor to another, and cell-type specific.
It is generally believed that selective activation of the JNK signaling cascade is associated with the Rho subfamily of GTPases, particularly with Rac and Cdc42 (Minden et al., 1995). MEKK1 is a ubiquitously expressed MAPK kinase kinase that binds Rac1 in a GTP-dependent manner, and both Rac1 and MEKK1 can activate the JNK pathway (Fanger et al., 1997). The substantial decrease of N/OFQ-induced JNK activation in the presence of a dominant-negative Rac indicated the Rac dependence of the ORL1 receptor-mediated JNK activation. However, we also demonstrated that Ras might also contribute to the N/OFQ-induced JNK activation. For some G protein-coupled receptors the activation of ERK is mediated via Gβγ-subunits and is Ras dependent (Crespo et al., 1994). Direct interaction between Ras and MEKK1 may result in the latter being stimulated (Russell et al., 1995). It has recently been shown that EPS8, E3B1, and SOS-1 form a tri-complex with Rac-specific guanine nucleotide exchange factor activity, and probably transmit intracellular signals from Ras to Rac (Scita et al., 1999). Our results showed that coexpression of dominant-negative mutants of both Ras and Rac did not produce additive inhibitory effect on the N/OFQ-induced JNK activity. Thus, the ORL1receptor might transmit its activating signaling from Ras to Rac, and then through MEKK1 to stimulate JNK.
Inhibitory effects of ORL1 receptor on adenylyl cyclases through PTX-sensitive Gi proteins are well established (Mollereau et al., 1994). However, previous reports have shown that ORL1 receptor may also act through PTX-insensitive pathways to regulate the activity of adenylyl cyclase as well as phospholipase Cβ (Chan et al., 1998; Yung et al., 1999). Because the N/OFQ-induced JNK activation could not be completely inhibited by PTX in both transfected COS-7 cells and NG108-15 cells, PTX-insensitive G proteins may actually participate in the ORL1 receptor-mediated JNK activation. Many PTX-insensitive G proteins are characterized by differential tissue distribution, for example, Gz is mainly expressed in neuronal cells; G14 is found in spleen, pancreatic islets (Zigman et al., 1994), kidney, and early myeloid cells (Nakamura et al., 1991); and G16 is predominantly expressed in hematopoietic cells (Amatruda et al., 1991). However, G12 is a ubiquitously expressed G protein, and a strong activator for the Rho-dependent biological activities, including differentiation and apoptosis (Berestetskaya et al., 1998). The finding of ORL1 receptor expression in neuronal cells and lymphocytic cells implied that coexpression of the ORL1 receptor with these PTX-insensitive G proteins is likely. Our results clearly demonstrated a PTX-insensitive component of the ORL1receptor-mediated JNK activation in NG108-15 cells that endogenously express Gz. Further studies are needed to explore the physiological relevance of the functional coupling between the ORL1 receptor and PTX-insensitive G proteins.
As a subgroup of MAPK, JNK phosphorylates and activates the activator protein-1 transcription factor component; c-Jun, therefore induces activator protein-1 transcriptional activity (Van Dam et al., 1993). Other transcription factors such as ATF-2 and Elk-1 can also be activated by different isoforms of the JNK family (Gupta et al., 1996). Stimulation of ORL1 receptor has been shown to activate both ERK and p38 MAPK (Zhang et al., 1999), and the activating effects of these two subtypes of MAPK on different transcriptional factors have been extensively studied (Price et al., 1996). The ability of the ORL1 receptor to activate different subtypes of MAPK indicated the importance of N/OFQ signaling at the nuclear level. In fact, growth, differentiation, and even apoptotic events of neuronal cells are highly dependent on the activities of MAPK, and the effects of N/OFQ on neuronal differentiation have been proposed (Saito et al., 1997). We have recently demonstrated that chronic activation of ORL1 receptor induces supersensitization of adenylyl cyclases (Chan and Wong, 1999). On the other hand, supersensitivity of dopamine receptor functions is tightly associated with the transcription of immediate-early genes (LaHoste et al., 1993). Hence, the capability of ORL1receptor to stimulate both MAPK and transcription factors may imply that the resulting transcriptional events of immediate-early genes (e.g., c-Jun and ATF-2) could be critically important for N/OFQ signaling. In this report, we demonstrated the stimulatory effect of ORL1 receptor on JNK via PTX-sensitive and PTX-insensitive G proteins, and suggested the involvement of Ras and Rac as important intermediates in the signaling. The participation of c-Jun and ATF-2 in the ORL1 receptor-mediated pathway was also proposed. Further studies on the co-operativity of MAPK subtypes in the ORL1 receptor-mediated neuronal cell activity will provide us with a refined picture for the N/OFQ signaling, and reveal the inter-relationship between the N/OFQ-induced MAPK activities and the resulting physiological consequences.
Acknowledgments
We are indebted to the following individuals for the generous donations of cDNAs: G. Pei for the human ORL1receptor, T. Voyno-Yasenetskaya for the JNK-HA, E. Stanbridge for RasS17N and RacT17N, M. I. Simon for human Gα16, T. Nukada for bovine Gα14, Y. Kaziro for rat Gαz, and H. R. Bourne for various G protein subunits. We also thank Rico K. H. Lo for technical assistance.
Footnotes
-
Send reprint requests to: Dr. Yung H. Wong, Department of Biochemistry and the Biotechnology Research Institute, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: boyung{at}ust.hk
-
↵1 This study was supported in part by grants from the Research Grants Council of Hong Kong (HKUST 653/96 M, 6176/97 M, and 2/99C), the Hong Kong Jockey Club Biotechnology Research Institute (BRI-96-I-3), and the Gunnar Nillson Cancer Research Trust Fund to Y.H.W.
- Abbreviations:
- N/OFQ
- nociceptin/orphanin FQ
- ORL
- opioid receptor-like
- PTX
- pertussis toxin
- MAPK
- mitogen-activated protein kinase
- ERK
- extracellular signal-regulated protein kinase
- HA
- hemagglutinin
- JNK
- c-Jun N-terminal kinase
- ATF-2
- activating transcription factor-2
- GST
- glutathioneS-transferase
- PI3K
- phosphatidylinositol-3 kinase
- MEKK1
- mitogen-activated protein kinase kinase kinase
- Received June 15, 2000.
- Accepted August 24, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}