


Abstract
This study examined the high-affinity, selective dopamine D4 receptor antagonist, L-745,870 (3-{[4-(4-chlorophenyl)piperazin-1-yl]methyl}-1H-pyrrolo[2,3-b]pyridine) in rodent behavioral models used to predict antipsychotic potential and side-effect liabilities in humans. In contrast to the classical neuroleptic, haloperidol, and the atypical neuroleptic, clozapine, L-745,870 failed to antagonize amphetamine-induced hyperactivity in mice or impair conditioned avoidance responding in the rat at doses selectively blocking D4 receptors. Furthermore, L-745,870 failed to reverse the deficit in prepulse inhibition of acoustic startle responding induced by the nonselective dopamine D2/3/4 receptor agonist apomorphine, an effect which was abolished in rats pretreated with the D2/3 receptor antagonist, raclopride (0.2 mg/kg s.c.). L-745,870 had no effect on apomorphine-induced stereotypy in the rat but did induce catalepsy in the mouse, albeit at a high dose of 100 mg/kg, which is likely to occupy dopamine D2 receptors in vivo. High doses also impaired motor performance; in rats L-745,870 significantly reduced spontaneous locomotor activity (minimum effective dose = 30 mg/kg) and in mice, L-745,870 reduced the time spent on a rotarod revolving at 15 rpm (minimum effective dose = 100 mg/kg). Altogether these results suggest that dopamine D4 receptor antagonism is not responsible for the ability of clozapine to attenuate amphetamine-induced hyperactivity and conditioned avoidance responding in rodents. Furthermore, the lack of effect of L-745,870 in these behavioral tests is consistent with the inability of the compound to alleviate psychotic symptoms in humans.
It is now well established that the atypical neuroleptic, clozapine, exhibits a unique clinical profile in humans. Thus in contrast to classical neuroleptic agents such as haloperidol, clozapine can improve both the positive and negative symptoms of schizophrenia, is effective in some treatment-resistant patients, has a lower propensity to induce extrapyramidal side effects and is not associated with increased prolactin release (Kane et al., 1988; Fitton and Heel, 1990;Baldessarini and Frankenburg, 1991). Unfortunately, despite improved efficacy at treating psychotic symptoms, the 1 to 2% risk of developing agranulocytosis has restricted the use of this compound. Nevertheless clozapine unequivocally shows that antipsychotic activity can be maintained or even improved upon without inducing the major side-effect liabilities associated with classical neuroleptic agents, and considerable effort has been spent on trying to identify the pharmacological mechanism(s) involved.
Radioligand binding studies have shown that clozapine has high affinity for several neurotransmitter receptors including serotonergic 5HT2 receptors (Ki= 25 nM), alpha-1 adrenoceptors (Ki = 9 nM), histamine H1 receptors (Ki = 3 nM) and muscarinic receptors (Ki = 12 nM) (Richelson and Nelson, 1984; Canton et al., 1990). Although all may contribute toward its clinical profile, recent attention has focused on the role of the dopamine D4 receptor, cloned by Van Tol and colleagues in 1991, and for which clozapine also exhibits a high affinity (Ki = 9 nM). Interest was further fueled by studies comparing D2 and D4 receptor affinity with plasma concentrations of neuroleptic agents showing that, at clinically relevant concentrations, clozapine could preferentially occupy dopamine D4 receptors in the brain (Seeman, 1992). Additional studies have also reported increased levels of a dopamine D4-like receptor in the striatum of schizophrenic brain (Seeman et al., 1993, 1995), although others have not replicated these findings (Reynolds and Mason, 1994,1995) and it remains to be proven that the pharmacology of this site is consistent with that exhibited by the dopamine D4receptor.
In view of the suggestion that D4 receptor antagonism might contribute toward the antipsychotic efficacy of clozapine, the present study examined the behavioral profile of L-745,870 in rodent models generally used to predict antipsychotic potential (i.e., inhibition of amphetamine-induced hyperactivity and disruption of conditioned avoidance responding) and side-effect liability (i.e., blockade of apomorphine-induced stereotypy and induction of catalepsy). L-745,870 was recently identified as a potent and highly selective dopamine D4 receptor antagonist and displaces [3H]spiperone binding from HEK or CHO cell lines expressing the cloned human dopamine D2(CHO), D3 (HEK) or D4 (HEK) receptor with Ki values of 960, 2300 and 0.43 nM, respectively (Kulagowski et al., 1996; Patelet al., 1996). In hD4 receptor expressing cell lines, L-745,870 reversed the dopamine (1 μM)-mediated inhibition of adenylate cyclase activity, stimulation of [35S]GTPγS binding and stimulation of extracellular acidification rate consistent with antagonist activity (Patel et al., 1997). L-745,870 did not exhibit any intrinsic activity in these assays. High affinity and selectivity was also demonstrated for rat D4 receptors (Ki = 1.5 nM and 1600 nM for rat D4 and D2 receptors respectively) and pharmacokinetic studies showed the compound had excellent oral bioavailability (66% in rat) and readily penetrated into the brain (Patel et al., 1997). L-745,870 is thus an ideal tool for examining whether selective dopamine D4 receptor antagonists exhibit an atypical, clozapine-like profile in rodent tests.
Methods
Animals.
Experiments were carried out on male BKTO mice (20–35 g), male Sprague-Dawley rats (200–300 g), male Wistar rats (270–430 g) and male hooded Lister rats (330–450 g) obtained from Bantin and Kingman, Hull, UK. Animals were housed in groups of 3 (hooded Lister and Wistar rats) or 5 and maintained on a 12-hr light/dark cycle (lights on at 7:00 a.m.) with food and water freely available. All procedures were carried out in accordance with the U.K. Home Office Animals (Scientific Procedures) Act 1986. All rats were deprived of food overnight before given oral doses of test compounds.
Mescaline-induced head twitches in rodents.
Mice or Sprague-Dawley rats were given either L-745,870 (3–30 mg/kg p.o.) or vehicle (10 ml/kg p.o. or 3 ml/kg p.o., respectively) 30 min before injection of mescaline (25 mg/kg s.c. in mice or 20 mg/kg i.v. in rat). Ten minutes later, mice were placed in individual perspex observation boxes (230 × 280 × 210 mm) and the number of head twitches were recorded for the next 10 min. For rat studies, animals were placed in individual perspex observation boxes (315 × 215 × 335 mm) immediately after mescaline injection and the number of head twitches were recorded for 15 min.
Locomotor activity in mice.
Mice were habituated for at least 2 hr to individual photocell activity cages (230 × 280 × 210 mm) equipped with two parallel infrared beams, one at each end of the base of the cage to record cage crossings, i.e.,consecutive beam breaks. Animals were then given either L-745,870 (0.03–1 mg/kg p.o. or 10–100 mg/kg p.o.), clozapine (1.25–10 mg/kg p.o.), haloperidol (0.03–1 mg/kg p.o.) or appropriate vehicle (10 ml/kg p.o.) 30 min before subcutaneous injection of either saline (10 ml/kg) or amphetamine (5 mg/kg), and the number of cage crosses were recorded in 10-min intervals for 2 hr.
Prepulse inhibition of acoustic startle responding.
Startle responses in Wistar rats were recorded using 8 SR-LAB (San Diego Instruments, CA) stabilimeter chambers and measured as the mean displacement of a piezoelectric cartridge during a 100-msec period beginning at the onset of the tone. Before each experiment, all animals were tested for their startle responses to 10 × 120 db tones (40-msec duration) given at 30-sec intervals. Mean startle responses were calculated for each rat and used to assign animals to different treatment groups such that each group had a comparable mean ± S.E.M. startle response.
On experimental days, rats were housed in an adjacent holding room and exposed to white noise (65 db) for at least 1 hr before testing. Animals were then given either L-745,870 (0.001–1 mg/kg p.o.) or vehicle (3 ml/kg p.o.), placed in individual startle chambers 30 min later and given a 5-min acclimation period during which background noise (65 db) was present. At the conclusion of the acclimation period, 5 × 120 db tones (40-msec duration) were presented at 30-sec intervals to partially habituate the animals to the startle eliciting stimulus (see Davis, 1988). This was followed by the test session itself which comprised 12 repetitions of the following seven different trial types: 1) a 40-msec, 120-db tone (pulse-alone); 2 to 4) a 120-db tone preceded 100 msec before by a 5-msec 70-, 75- or 80-db prepulse (prepulse + pulse); 5 to 6) a 5-msec 75- or 80-db prepulse alone; and 7) a period when no stimulus was presented. In a separate experiment, the effects of L-745,870 on apomorphine-induced disruption of prepulse inhibition was examined. Animals were pretreated with either L-745,870 (0.01–1 mg/kg p.o.), raclopride (0.2 mg/kg s.c.) or vehicle (3 ml/kg p.o.) 30 min before saline (1 ml/kg s.c.) or apomorphine (0.6 mg/kg s.c.) injection. Ten minutes later, rats were placed in individual startle boxes and tested as described previously.
Conditioned avoidance responding.
Conditioned avoidance training was carried out in 32 hooded Lister rats with six shuttle boxes divided into two equal compartments each equipped with a light source and connected via an open doorway. Each box had an electrified grid floor which operated on a tilt mechanism to detect the location of the rat within the apparatus. Sessions commenced in darkness and consisted of 15 trials (intertrial interval = 25–40 sec) during which a 10-sec illumination of the compartment signaled the delivery of a 0.4 mA shock applied for a maximum of 10 sec. Active avoidance, i.e., crossing into the opposite compartment during the 10-sec conditioning stimulus (light) prevented shock delivery. Thereafter, crossing compartments after initiation of the shock terminated shock delivery and was recorded as an escape response. Training was carried out 5 days/week until the criterion of the avoidance of 10 or more shocks on 3 consecutive days was achieved after which animals were pretreated with either L-745,870 (0.01–1 mg/kg i.p.), clozapine (0.5–3 mg/kg s.c.), haloperidol (0.01–0.05 mg/kg s.c.) or appropriate vehicle (1 ml/kg). Conditioned avoidance responding was then tested 30 min later as described previously. A minimum of 3 days was allowed between successive drug tests during which rats were trained as usual.
Stereotyped behavior in rat.
Rats were habituated to individual perspex observation boxes for 30 min and then given either L-745,870 (0.01–1 mg/kg p.o.), clozapine (30–100 mg/kg p.o.), haloperidol (0.05–1 mg/kg p.o.) or appropriate vehicle [1 or 3 ml/kg p.o.(L-745,870)]. All animals received apomorphine (1 mg/kg s.c.) 20 min (L-745,870) or 60 min later and the time spent sniffing and licking/biting recorded 20 to 25 min after apomorphine injection.
Catalepsy in rodents.
Mice were given either L-745,870 (1–100 mg/kg p.o.), haloperidol (3 mg/kg p.o.) or vehicle (10 ml/kg p.o.). Thirty minutes later, the forepaws of each animal were placed over a 0.5 cm diameter bar positioned horizontally 3 cm above the bench, and the length of time that the animal was able to maintain this abnormal posture was noted to a maximum of 120 sec.
Motor impairment in rodents.
The effect of L-745,870 on motor performance was examined in both rats and mice. To assess impairment of spontaneous locomotor activity, rats were given either L-745,870 (3–30 mg/kg p.o.) or vehicle (3 ml/kg p.o.) and placed in novel photocell activity cages 30 min later. Photocell beam breaks were then recorded in 10-min intervals for 1 hr. To assess effects on motor coordination, mice were first trained to remain on a rotarod revolving at 15 rpm for 120 sec. Animals were then given L-745,870 (10–300 mg/kg p.o.) or vehicle (10 ml/kg p.o.) and the latency to fall from the rotarod was determined at various time intervals after dosing.
Statistical analysis.
All data were analyzed by analysis of variance (with repeated measures if appropriate) followed by Dunnett’st test to compare drug-induced effects with vehicle-pretreated animals. Doses inhibiting responses by 50% were calculated by Allfit analysis (DeLean et al., 1978) and reported as the ED50 values ± S.E. of the estimated ED50 dose. For prepulse inhibition experiments, the mean startle response to each stimulus type was calculated for each subject and analyzed as above. However, in addition, a repeated measures analysis of variance was performed on pulse and prepulse + pulse startle amplitudes by use of drug as a between-subjects variable and stimulus as a within-subject variable. These data were further examined by a simple main effects analysis of variance to compare pulse and prepulse + pulse responses at each dose tested. Finally, the level of prepulse inhibition in each rat was calculated by expressing the prepulse + pulse startle amplitude as a percentage decrease from the response to the pulse alone,i.e., %PPI = [pulse − (prepulse + pulse)]/pulse × 100. These data were also analyzed by analysis of variance followed by Dunnett’s t test.
Drugs.
Apomorphine HCl, (+)-amphetamine sulfate, mescaline hemisulfate (Sigma Chemical Co., St Louis, MO), clozapine (Sandoz Pharmaceuticals, Hanover, NJ) and haloperidol (Haldol; Janssen Pharmaceutica, Beerse, Belgium) were dissolved in 0.9% saline with the addition of 0.1% ascorbic acid for apomorphine. L-745,870 was synthesized at Merck, Sharp and Dohme Research Laboratories (Harlow, Essex, U.K.) and administered as a solution in 25% polyethylene glycol (molecular weight, 300) and water, pH 5.
Results
Inhibition of mescaline-induced head twitches.
Oral administration of L-745,870 dose-dependently attenuated the head twitch response induced by mescaline in rodents (rat: ED50 = 7 ± 2.6 mg/kg; mice: ED50 = 7.1 ± 1.3 mg/kg; fig.1). Similar effects were also observed in mice after i.p. injection of L-745,870 (ED50 = 6.7 ± 0.03 mg/kg; data not shown).

Inhibition of mescaline-induced head twitches in rodents. Animals were dosed orally with either L-745,870 (hatched columns) or vehicle (Veh; open columns) 30 min before mescaline injection (25 mg/kg s.c. in mice or 20 mg/kg i.v. in rats). Results are expressed as the mean ± S.E.M. number of head twitches recorded 10 to 20 min (mice) or 0 to 15 min (rat) later (n = 7–8/group). Data were analyzed by analysis of variance followed by Dunnett’s t test, *P < .05 compared with vehicle-pretreated animals.
Inhibition of amphetamine-induced hyperactivity.
In habituated mice, subcutaneous injection of amphetamine (2.5–10 mg/kg) dose-dependently increased the number of cage crosses recorded compared with animals given saline (data not shown). The lowest dose to induce a robust and reliable hyperactivity was 5 mg/kg and this response was significantly attenuated after oral administration of clozapine (ED50 = 2 ± 0.03 mg/kg; fig.2) and haloperidol (ED50 = 0.15 ± 0.04 mg/kg; fig. 2). In contrast, pretreatment with L-745,870, at doses up to 1 mg/kg, did not alter amphetamine-induced hyperlocomotion in mice (fig. 2). In subsequent studies examining a higher dose range (10–100 mg/kg p.o.), L-745,870 did significantly attenuate amphetamine-induced hyperactivity in mice [ED50 = 22 ± 5 mg/kg p.o.; mean ± S.E.M. total number of cage crosses recorded in 2 hr (n= 7–8 mice/group): vehicle/vehicle = 22 ± 9*; vehicle/amphetamine = 1039 ± 166; L-745,870 (10 mg/kg)/amphetamine = 997 ± 192; L-745,870 (30 mg/kg)/amphetamine = 257 ± 89*; L-745,870 (100 mg/kg)/amphetamine = 117 ± 55*; (*P < .05 compared with vehicle/amphetamine-treated mice, analysis of variance followed by Dunnett’s t test)].
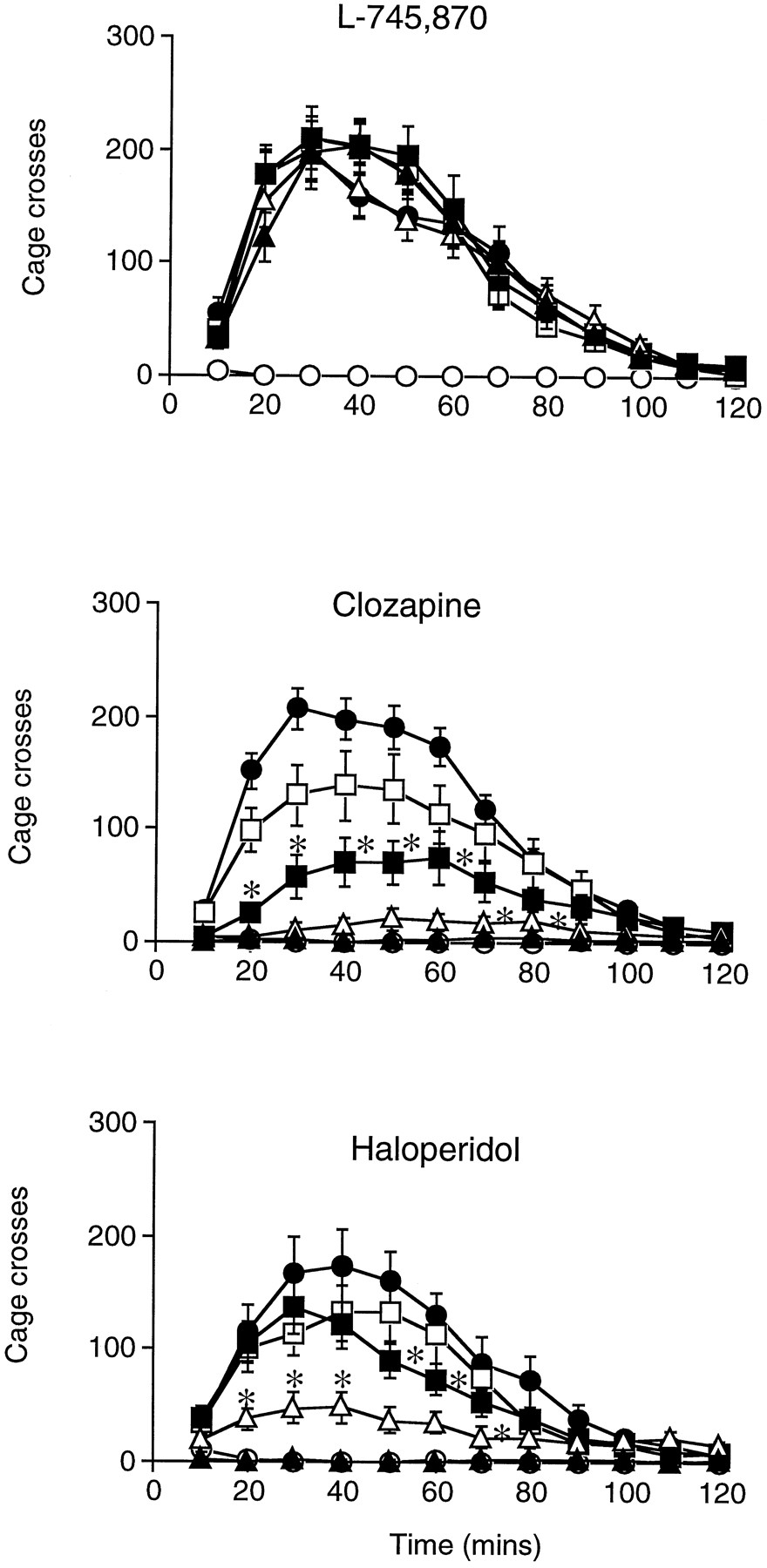
Inhibition of amphetamine-induced hyperactivity in mice. Animals were dosed orally with either L-745,870 [0.03 mg/kg (open squares), 0.1 mg/kg (closed squares), 0.3 mg/kg (open triangles) or 1 mg/kg (closed triangles)], clozapine [1.25 mg/kg (open squares), 2.5 mg/kg (closed squares), 5 mg/kg (open triangles) or 10 mg/kg (closed triangles)] or haloperidol [0.03 mg/kg (open squares), 0.1 mg/kg (closed squares), 0.3 mg/kg (open triangles) or 1 mg/kg (closed triangles)] 30 min before injection of amphetamine (5 mg/kg s.c.). Open and closed circles show data for vehicle/saline- and vehicle/amphetamine-treated mice, respectively. Results are expressed as the mean ± S.E.M number of cage crosses recorded in 10-min intervals for 2 hr (n = 6–9/group). Data were analyzed by analysis of variance followed by Dunnett’st test, *P < .05 compared with vehicle/amphetamine-treated mice. For clarity, only the minimum effective dose significantly attenuating amphetamine hyperactivity responses is denoted.
Inhibition of apomorphine-induced deficits in PPI.
When a 40-msec, 120-db stimulus was preceded by a 5-msec, 70-, 75- or 80-db prepulse, the amplitude of the startle response was significantly attenuated in a prepulse-dependent manner (fig.3). L-745,870 itself had no effect on any startle parameters or prepulse inhibition in the rat (table1). In contrast, subcutaneous injection of apomorphine (0.6 mg/kg) significantly reduced the % PPI, an effect which was observed at all prepulse intensities used (fig. 3). Analysis of individual trial types showed that the decrease in % PPI reflected increases in the startle amplitude to the prepulse + pulse stimuli rather than a reduction in startle amplitude to the pulse alone. Pretreatment with L-745,870 did not reverse the deficit in % PPI induced by apomorphine (fig. 3). In contrast, the dopamine D2/3 receptor antagonist, raclopride (0.2 mg/kg s.c.) significantly attenuated the apomorphine-induced disruption of PPI (fig. 3).

Effect of L-745,870 or raclopride (Rac) on the disruption in prepulse inhibition induced by apomorphine (Apo) in the rat. Animals were exposed to a 120-db tone (pulse-alone; stippled columns) or the same pulse preceded by either a 70-, 75- or 80-db prepulse (hatched columns). Mean startle responses to 12 repetitions of each stimulus type were calculated for each rat and the % PPI determined (% inhibition = [pulse − (prepulse + pulse)]/pulse × 100). Results are expressed as the group mean ± S.E.M. startle response or % PPI (n = 14–15 rats/dose). Data were analyzed by analysis of variance followed by Dunnett’s t test, *P < .05 compared with vehicle-pretreated rats (startle amplitudes) or apomorphine-treated rats (% PPI). In addition a simple main effects analysis of variance was used to compare pulse and prepulse + pulse startle responses at each dose, † P < .05 compared with pulse alone.
Effect of L-745,870 on prepulse inhibition of acoustic startle responding in the rat
Inhibition of conditioned avoidance responding in the rat.
Stable conditioned avoidance responding was acheived after 48 training sessions with rats actively avoiding on average 13 of 15, 0.4 mA shocks within 4 to 5 sec after presentation of the conditioning stimulus (table 2). Pretreatment with haloperidol or clozapine dose-dependently and significantly attenuated the number of “avoids” recorded but did not prevent the animal from escaping after the initiation of the shock (table 2). Thus, the number of escape responses was significantly increased in haloperidol- or clozapine-treated rats. The latency to either avoid or escape from an electric shock was not altered significantly. In contrast, L-745,870 did not alter active avoidance responding at doses up to 1 mg/kg (table2).
Conditioned avoidance responding in the rat
Inhibition of apomorphine-induced stereotypy.
Pretreatment with haloperidol dose-dependently antagonized the intense stereotyped sniffing and licking/biting induced by apomorphine in the rat (ED50 = 0.4 ± 0.08 mg/kg p.o.; fig.4). In contrast, only a modest decrease in sniffing and licking/biting was seen in rats given a high dose of 100 mg/kg clozapine. Oral administration of L-745,870, at doses up to 1 mg/kg, failed to alter apomorphine-induced stereotypy in the rat (fig.4).

Inhibition of apomorphine-induced stereotypy in the rat. Animals were dosed orally with either L-745,870, clozapine, haloperidol or vehicle (Veh) 20 min (L-745,870) or 60 min before injection of apomorpine (1 mg/kg s.c.). Results are expressed as the mean ± S.E.M duration (sec) of stereotyped sniffing (open columns) and licking/biting (hatched columns) recorded 20 to 25 min later (n = 8/group). Data were analyzed by analysis of variance followed by Dunnett’s t test, *P < .05 compared with vehicle-treated rats.
Induction of catalepsy in the mouse.
When tested for cataleptic responses, mice given L-745,870 (0.1–10 mg/kg p.o.) did not show a significant increase in their ability to maintain an abnormal posture produced by placing both forepaws over a circular bar [mean ± S.E.M. time spent in a cataleptic position (sec): vehicle = 2.2 ± 0.5; L-745,870: 0.1 mg/kg = 2.5 ± 1, 1 mg/kg = 5.7 ± 1.6, 10 mg/kg = 7.9 ± 1.9). However, mice given a high dose of 100 mg/kg did show a significant cataleptic response which was similar to that induced by haloperidol [100 mg/kg L-745,870 = 34 ± 13 sec*; 3 mg/kg haloperidol = 47 ± 11 sec* (*P < .05 compared to vehicle-treated mice)].
Motor impairment in rodents.
When rodents are placed in novel photocell activity cages, exploratory activity is initially high and then rapidly declines as they habituate to their new surroundings. Oral administration of L-745,870 dose-dependently and significantly attenuated the number of cage crossings recorded compared with vehicle-pretreated rats with a modest reduction in spontaneous locomotor activity observed at 30 mg/kg (fig.5). Similarly in mice, high doses of L-745,870 (100 and 300 mg/kg) significantly attenuated the latency to remain on a rotarod revolving at 15 rpm, a deficit which was still apparent 6 hr after dosing (fig. 5).

(a) Changes in spontaneous locomotor activity in the rat. Animals were dosed orally with either vehicle (open circles) or L-745,870 [3 mg/kg (closed circles), 10 mg/kg (open squares) or 30 mg/kg (closed squares)] and placed in novel photocell activity cages 30 min later. Results are expressed as the mean ± S.E.M. number of cage crosses recorded in 10-min intervals for 1 hr (n = 8/group). (b) Impairment of rotarod performance in mice. Animals were dosed orally with either vehicle (open circles) or L-745,870 [10 mg/kg (closed circles), 30 mg/kg (open squares), 100 mg/kg, (closed squares) or 300 mg/kg (open triangles)], and the ability to remain on a rotarod revolving at 15 rpm was tested up to 6 hr later. Results are expressed as the mean ± S.E.M. time spent on the rotarod (sec). Data were analyzed by analysis of variance followed by Dunnett’s t test, *P < .05 compared with vehicle-treated rats.
Discussion
L-745,870 was recently identified as a potent and highly selective antagonist for the dopamine D4 receptor subtype (Kulagowski et al., 1996; Patel et al., 1996;Patel et al., 1997). Given that considerable interest has focused on whether D4 receptor antagonism explains the unique clinical profile seen with clozapine in man, the present studies were designed to evaluate the compound in rodent models generally used to detect novel antipsychotic agents.
Before initiating these experiments, the absence of a measurable neurochemical or behavioral response known unambiguously to be mediated through D4 receptor activation led us to look for an alternative means of estimating the doses at which L-745,870 would selectively block D4 receptors in vivo. Although extensive counterscreening showed that L-745,870 had negligible affinity for a wide range of mammalian receptors (Patelet al., 1997), modest affinity for the sigmabinding site (Ki = 130 nM) and the 5HT2 receptor (Ki = 200 nM) was observed. In mouse brain, in vivo[3H]SKF 10,047 binding to the sigmarecognition site was dose-dependently displaced by L-745,870 (ED50 = 3 mg/kg p.o.; Patel et al., 1997). Given that the affinity of L-745,870 is 87-fold higher at the rat D4 receptor (Ki = 1.5 nM compared to 130 nM at the sigma binding site), extrapolation from the ED50 dose for displacement of sigma binding suggests that 35 μg/kg L-745,870 would occupy 50% of D4 receptors in vivo. In contrast, given that L-745,870 is 12-fold weaker at the rat D2 receptor (Ki = 1600 nM), then a predicted dose of 36 mg/kg would occupy 50% of D2 receptors. Extrapolations from thesigma binding assay also predicted that greater than 90% of brain D4 receptors would be blocked at a dose of 1 mg/kg L-745,870 (Patel et al., 1997). Such calculations seem reasonable given that brain concentrations of L-745,870 increase proportionally with dose (Patel et al., 1997).
Although receptor occupancy might not accurately predict functional potency, several observations appear consistent with the conclusions drawn from the in vivo sigma binding assay. First, at dose ranges predicted to occupy dopamine D2 receptors, L-745,870 1) reduced amphetamine-induced hyperactivity in mice (present study), 2) induced catalepsy in mice (present study) and 3) produced parkinsonian-like symptoms in squirrel monkeys (Patel et al., 1997). Second, clozapine and haloperidol also displaced in vivo sigma binding and the predicted doses occupying D2 receptors (50% receptor occupancy at 0.3 mg/kg and 0.004 mg/kg s.c., respectively) were in reasonable agreement with those blocking amphetamine-induced hyperactivity [ED50 = 0.3 mg/kg and 0.02 mg/kg s.c. (L. J. Bristow, unpublished observations)]. Finally, pretreatment with L-745,870 also antagonized mescaline-induced head twitches, a well-characterized behavioral response resulting from activation of 5HT2A receptors in rodents (Schreiber et al., 1995). Again, given the relative receptor affinities of L-745,870 for the rat D4 and 5HT2A receptor (Ki = 200 nM), extrapolation from the ED50 dose for blockade of head twitches (7 mg/kg) suggests that a dose of 52 μg/kg L-745,870 would block 50% of D4 receptors in rodent brain. These estimates are in excellent agreement with those derived from the in vivo sigma binding assay.
Although it has been assumed that L-745,870 reduces mescaline-induced head twitches via direct blockade of 5HT2A receptors, other pharmacological actions could explain this effect. In particular, D2receptor antagonists such as haloperidol and sigma ligands such as DuP 734 and BMY 14802 also block head twitches induced by activation of 5HT2 receptors (Schreiber et al., 1995; Bristow et al., 1991; Tam et al., 1992). Although it’s difficult to completely dismiss a role for these receptors, the effects seen with haloperidol occur at doses similar to, if not greater than, those blocking amphetamine-induced hyperactivity in contrast to the reverse profile seen with L-745,870. In addition, the role of the sigma site is unclear given that DuP 734 also has high affinity for 5HT2A receptors and that the effects of BMY 14802 have been attributed to 5HT1A receptor agonism (Bristow et al., 1991). Finally, the rank order in potency in the in vivo assays, i.e., ED50versus sigma binding < head twitch < amphetamine hyperactivity, reflects the rank order in the affinity of L-745,870 for these receptors. Thus, it’s clear that L-745,870 can have multiple pharmacological actions and that effects seen at doses greater than 1 mg/kg can not be solely attributed to antagonism of D4 receptors. This is particularly important because, in addition to the well-established effects of D2 receptor antagonists, evidence suggests that 5HT2A antagonists (Schmidt et al., 1995; Kehne et al., 1996) and sigma ligands (Largent et al., 1988) might also have antipsychotic potential.
The present study shows that, at doses predicted to block > 90% of brain D4 receptors, L-745,870 fails to exhibit an antipsychotic-like profile in rodents. Thus, in contrast to both haloperidol and clozapine, L-745,870 did not attenuate amphetamine-induced hyperactivity or impair conditioned avoidance responding at doses ≤1 mg/kg. The compound also failed to attenuate apomorphine-induced stereotypy, an effect thought to be predictive of extrapyramidal side effects in man (Costall and Naylor, 1973; Burkiet al., 1977) and which is dose-dependently blocked by classical neuroleptic agents like haloperidol but not by clozapine. Catalepsy, however, was observed in the mouse, albeit at high doses which are likely to be blocking dopamine D2receptors. Thus although L-745,870 shows a relatively benign side-effect profile, its lack of effect in models thought to be predictive of antipsychotic activity suggests that D4 receptor antagonism alone does not explain the behavioral profile of clozapine in these tests.
Our results with L-745,870 are similar to those recently reported for U-101,387 (S(−)-4-{4-[2-(isochroman-1-yl)ethyl]piperazin-1yl}benzenesulfonamide), a potent D4 receptor antagonist (Ki = 10 nM) which also failed to block amphetamine-induced hyperactivity in rodents (Merchant et al., 1996). In contrast, the observation that the D4 antagonist NGD 94–1 reversed apomorphine-induced disruption in prepulse inhibition of acoustic startle responding (Hoffman et al., 1995) led us to evaluate L-745,870 in this test. L-745,870 clearly had no effect on any startle parameters or prepulse inhibition per se and failed to attenuate the disruption induced by apomorphine. In contrast, the nonselective D2/3 receptor antagonist, raclopride, abolished this effect. In as much as the doses of L-745,870 were restricted to those selective for D4receptor blockade, these results suggest that D4receptors do not contribute toward this effect of apomorphine. In the absence of similar information for NGD 94–1 it is unclear whether the effects reported in prepulse inhibition relate directly to D4 receptor antagonism or perhaps to some other pharmacological property of the compound.
The behavioral profile of dopamine D4 receptor antagonists differs dramatically from those reported by use of the antisense approach to “knock out” brain D4receptors. Thus, Zhang et al. (1996) recently reported that intraventricular infusion of D4 antisense oligodeoxynucleotide for 3 days significantly attenuated amphetamine-induced hyperactivity in the rat. This effect appeared selective in that infusion of D2 or D3 antisense had no effect, which suggests that D4 receptors play an essential role in mediating this response. In addition, both D3 and D4 antisense-treated rats also showed a significant reduction in amphetamine-induced stereotyped behaviors; surprisingly, this response was not affected by D2 antisense infusion. These observations clearly contradict those reported with high-affinity D4receptor antagonists like L-745,870 and U-101,387. Indeed, these data should be interpreted with caution, because in the absence of a radioligand suitable for labeling D4 receptors in the brain, the behavioral effects seen with the antisense approach cannot be attributed unambiguously to a selective down-regulation of D4 receptors.
Although the present study suggests that L-745,870 is unlikely to be antipsychotic in humans, it could be argued that the behavioral tests used to explore its antipsychotic potential are inappropriate for evaluating D4 receptor antagonists. This is especially the case given that hyperactivity and stereotypy reflect activation of mesolimbic and striatal dopamine systems (Kelly et al., 1975; Pijnenburg et al., 1975; Costall and Naylor, 1973) which terminate in regions densely populated with D2 and D3 receptors but where dopamine D4 receptor mRNA is low or undetectable (reviewed in Reynolds, 1996). In contrast, D4 mRNA appears to be mainly expressed in regions such as the frontal cortex (Van Tol et al., 1991; Matsumotoet al., 1995) which is also implicated in the pathophysiology of schizophrenia (Roberts, 1991). Indeed, based on such arguments, L-745,870 recently entered phase II clinical trials in hospitalized, schizophrenic patients who were treated with either placebo or L-745,870 (15 mg/day) for 4 weeks. However, although the compound was well tolerated and the plasma levels achieved were consistent with a high level of D4 receptor occupancy in the brain, psychotic symptoms were not improved, suggesting that selective D4 receptor antagonism is not important in the clinical action of clozapine (Kramer et al., 1997).
In conclusion, L-745,870 was recently identified as a high-affinity, selective dopamine D4 receptor antagonist with an excellent pharmacokinetic profile in rodents and good tolerability in humans. Although both preclinical and clinical evidence argues against a utility in the treatment of schizophrenia, L-745,870 should prove a valuable tool for exploring both the functional relevance of the D4 receptor and the clinical potential of antagonists selective for the dopamine D4receptor subtype.
Footnotes
-
Send reprint requests to: Dr. Linda J. Bristow, Merck, Sharp and Dohme Research Laboratories, Neuroscience Research Centre, Terlings Park, Eastwick Rd., Harlow, Essex, CM20 2QR U.K.
- Abbreviations:
- L-745
- 870, 3-{[4-(4-chlorophenyl)piperazin-1-yl]methyl}-1H-pyrrolo[2,3-b]pyridine
- PPI
- prepulse inhibition
- %PPI
- percentage prepulse inhibition
- CHO
- Chinese hamster ovary
- HEK
- human embryonic kidney
- 5-HT
- 5-hydroxytryptamine
- Received April 21, 1997.
- Accepted August 25, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}