


Abstract
The finding that ascending cholinergic systems are severely degenerated in Alzheimer’s disease has driven the search for a cholinomimetic therapy. Adverse effects observed with cholinesterase inhibitors and high-efficacy muscarinic agonists led us to design compounds with an improved profile. SB 202026 (R-(Z)-(+)-α-(methoxyimino)-1-azabicyclo[2.2.2] octane-3-acetonitrile) displaced [3H]-oxotremorine-M from muscarinic receptors in the rat brain with high affinity (IC50 = 14 nM), a potency similar to that of oxotremorine-M itself (IC50 = 13 nM), but exhibited low affinity for cholinergic nicotinic receptors and other neuroreceptors. In studies using cloned human muscarinic receptors, SB 202026 possessed approximately equal affinity in displacing [3H]-quinuclidinyl benzilate from all muscarinic receptor subtypes. In functional models in vitro, SB 202026 caused maximal depolarization of the rat superior cervical ganglion at low concentrations (300 nM) (M1-mediated effect), while producing a lower maximal effect than the high-efficacy agonists oxotremorine-M and carbachol on M2-mediated release of ACh and M3-mediated smooth muscle contraction (guinea pig ileum), respectively. The functional selectivity and partial agonist profile seen in vitro were reflected in vivothrough potent cognition-related activity (M1-induced increase in hippocampal EEG power) combined with low efficacy, compared with arecoline or oxotremorine, on induction of bradycardia (M2-mediated response), hypotension (viaM3-mediated vasorelaxation) and tremor (thought to be mediated by M3 receptors). The foregoing profile of SB 202026 predicted that cognition-enhancing activity would be achieved at doses below those that initiate undesirable side effects, and this has subsequently been demonstrated in rodents, marmosets and humans.
The degeneration of cholinergic neuronal systems, in particular those projecting from the basal forebrain to the hippocampus and cerebral cortex, is a consistent feature in the neuropathology of AD (Whitehouseet al., 1982; Bowen, 1983; Mash et al., 1985;Lehericy et al., 1993). These systems play an intrinsic role in learning and memory processes (Hagan and Morris, 1988; Durkin, 1989;Dunnett and Fibiger, 1993), and the degree of cholinergic degeneration has been shown to correlate with the loss of cognitive function (Lehericy et al., 1993; Terry et al., 1991). Although the total number of muscarinic receptors in the brain is largely unchanged in the disease, differences have been found in receptor numbers in discrete brain regions. Thus postsynaptic receptors in the cortex and hippocampus (Levey et al., 1991; Pearce and Potter, 1991) remain largely intact, whereas receptors located presynaptically on the nerve terminals are lost as the presynaptic fibers degenerate (Mash et al., 1985; Araujo et al., 1988). Indeed, it has been postulated that the postsynaptic receptors may be up-regulated in AD (Probst et al., 1988;Harrison et al., 1991).
Replacement of the lost cholinergic function is an attractive proposition for the treatment of AD, but the usefulness of cholinomimetics developed to date has been compromised by low bioavailability and a lack of separation between cognition enhancement and side effects. ChEIs, for example, by enhancing endogenous ACh, can exert actions at both muscarinic and nicotinic sites and would be expected, therefore, to induce a number of nonspecific, undesirable effects mediated via both muscarinic and nicotinic receptors in the CNS and periphery. First-generation ChEIs, although they possess some efficacy, have also been found to cause significant side effects (Becker and Giacobini, 1988). It was believed that muscarinic agonists that act directly at postsynaptic muscarinic receptors, and are inherently devoid of nicotinic activity, would have an inherently better therapeutic ratio than ChEIs (Tariot et al., 1988). However, early muscarinic agonists tested in the clinic were found to have poor bioavailability (Penn et al., 1988) or a narrow therapeutic window as a result of undesirable side effects mediated by actions on muscarinic receptors throughout the body, including nausea, vomiting, GI disturbances, sweating and salivation (Spiegel, 1984;Wettstein and Spiegel, 1984; Sunderland et al., 1988;Hollander et al., 1987). The potential for effects on the cardiovascular system is also an important consideration, because cardiovascular function can be significantly modulated by stimulation of muscarinic receptors located in the CNS and periphery, resulting in a range of outcomes that depend on the location of the receptors stimulated (Brezenoff and Giuliano, 1982; Wilffert et al., 1983; Sundaram et al., 1988; Eglen and Whiting, 1990) and the influence of homeostatic reflex mechanisms (Baum, 1990). The objective was therefore to design a compound that was able to enhance cognition but was without undesirable side effects at therapeutic doses.
Significant advances have been made in recent years in the discovery and characterization of muscarinic receptor subtypes. To date, five subtypes have been identified through receptor cloning (Bonner et al., 1987, 1988), and three have been functionally described (Doods et al., 1987; Eglen and Watson, 1996; Michel and Whiting, 1987). Distribution studies have shown that M1sites predominate postsynaptically in the cerebral cortex and hippocampus (Cortes et al., 1987; Waelbroeck et al., 1990; Levey et al., 1991; Flynn and Mash, 1993) and that M2 and M3 receptors are more abundant in the periphery (Levey, 1993) where they can mediate a range of undesirable effects. These findings led to the hypothesis that agonists that demonstrate selectivity for the M1 subtype could have therapeutic utility in AD (Cortes et al., 1987) by selectively stimulating postsynaptic receptors in the cortex and hippocampus while having little or no effect at other sites; this would result in an improved therapeutic ratio over early, nonselective compounds. However, because of the high conservation of the agonist binding site across the five subtypes (Hulme et al., 1990), it has proved difficult to synthesize compounds that demonstrate M1 subtype selectivity per se in cloned receptor models (Traub and Freedman, 1992). As a consequence, we explored an approach based on work first described by Ringdahl et al.(1987; Ringdahl et al., 1987): using partial agonists to confer functional selectivity.
Ringdahl demonstrated that high-efficacy agonists induced a wider range of responses mediated by muscarinic receptors than lower-efficacy compounds and that this difference was related to the intrinsic efficacy of compounds rather than to differences in their affinity for receptors at the various sites of action (Ringdahl et al., 1987; Ringdahl, 1987). The tissue response to an agonist is a function of drug factors (affinity and intrinsic efficacy) and tissue factors (number of receptors on the tissue and efficiency of the coupling mechanism between the receptor stimulus and the response), and these factors can determine whether a compound with low intrinsic efficacy behaves as a full agonist, partial agonist or antagonist when tested in a given tissue (Kenakin, 1986, 1990; Ringdahl et al., 1987;Ringdahl, 1987). Partial agonists may therefore exhibit functional selectivity because they can elicit a response in one tissue (high receptor reserve) but are less effective in another (low receptor reserve). Ringdahl (1987) concluded that compounds that had high affinity for muscarinic receptors and low intrinsic efficacy were the most likely to exert selectivity by this mechanism. He further concluded that in AD, where the availability of ACh is diminished, a partial agonist would act on a virtually empty and presumably supersensitive postsynaptic receptor system and would be expected to behave as a full agonist.
This hypothesis provided another way of achieving selectivity (an alternative to synthesizing M1 selective agonists), and this approach has also been pursued by others (Hargreaves et al., 1992; Sedman et al., 1995; Shannon et al., 1994; Ensinger et al., 1993), who have shown that low-efficacy muscarinic agonists can demonstrate functional selectivity in in vitro and in vivo models.
The relationship between displacement affinity against antagonist (QNB)vs. agonist (OXO-M) muscarinic ligands and intrinsic efficacy has been described in the past (Freedman et al., 1988; Brown et al., 1988a; Hawkins et al., 1992) and is related to the existence of different-affinity binding sites for agonists and antagonists (Freedman et al., 1988). Thus it has been shown that the intrinsic efficacy of a muscarinic compound can be estimated with some accuracy from its ligand binding profile (Freedman et al., 1988). Such binding studies were used as a screen to identify muscarinic partial agonists that, because of the potential for functional selectivity and reduced side effects (Ringdahlet al., 1987), could have utility in the treatment of AD (Orlek et al., 1991). SB 202026 (fig.1) was the culmination of a significant research effort focused on the synthesis of potent muscarinic partial agonists and was selected from a large number of compounds for further study.

Structure of SB 202026.
This paper describes a range of studies that characterize the pharmacology of SB 202026. These include in vitro ligand binding studies using rat cortical tissue and human cloned receptors,in vitro functional studies that investigate efficacy in tissues rich in particular receptor subtypes and in vivostudies designed to assess the therapeutic ratio between desirable central actions and undesirable side effects.
Materials and Methods
Materials
The following drugs and chemicals were used in this study: arecoline, carbachol, muscarine, oxotremorine, scopolamine, pirenzepine, PBZ (Sigma Chemical Co., Poole, Dorset, U.K.), OXO-M, R-(−)-QNB, methoctramine, fHHSiDF, hemicholinium-3 (RBI, Semat Technical Ltd., St. Albans, U.K.), AF-DX 116 (Dr. Karl Thomae, GmbH, Germany) [3H]-QNB, [3H]-choline chloride, [3H]-CGP12177, [3H]-prazosin, [3H]-rauwolcine, [3H]-muscimol, [3H]-mesulergine, [3H]-spiperone (Amersham International, Little Chalfont, U.K.), [3H]-OXO-M, [3H]-SCH23390, [3H]-nicotine, [3H]-serotonin, [3H]-ketanserin, [3H]-granisetron, [35H]-TBPS (NEN-Dupont, Stevenage, U.K.). AF 102B was synthesized at Beecham Pharmaceuticals, Harlow, U.K. SB 202026 was synthesized at SmithKline Beecham, Harlow, UK, as either the oxalate or the hydrochloride salt. Testing of both salt forms showed there was no detectable difference in activity between the salts when doses were calculated in terms of free base.
Ligand Binding Studies In Vitro
[3H]-OXO-M and [3H]-QNB binding assays.
The ligand binding assay methods used to determine affinity for muscarinic agonist and antagonist sites were based on those of Bevan (1984a) and Yamamura and Snyder (1974), respectively. Cerebral cortex from male Hooded Lister rats (Olac, Bicester, UK) was homogenized in 2.5 vol (compared with wet weight) of ice-cold 50 mM Tris buffer, pH 7.7. After centrifugation at 24,000 ×g for 15 min at 4°C, the pellet was resuspended in 2.5 vol of fresh, cold buffer and washed twice more. Incubations for [3H]-OXO-M binding were in a total volume of 2 ml of ice-cold 50 mM Tris containing 2 mM magnesium chloride. [3H]-OXO-M acetate (specific activity 87 Ci/mmol) was added to a concentration of 1.88 nM. Cortex homogenate was diluted to 300 vol based on the original wet weight (equivalent to 0.145 mg protein/ml). Nonspecific binding was defined using 10 μM oxotremorine sesquifumarate. After equilibration at 37°C for 30 min, samples were filtered through Whatman GF/B filters presoaked for 30 min in a 0.05% aqueous solution of polyethylenimine to prevent adsorption of [3H]-OXO-M onto the glass fiber. [3H]-QNB (specific activity 44 Ci/mmol, final concentration 0.27 nM) binding was carried out in a similar manner, except that the magnesium chloride was omitted and the dilution of the homogenate was increased to 1500 vol (7.8 μg protein/ml). Nonspecific binding was defined with 1 μM atropine sulphate.
Muscarinic cloned receptors.
CHO cells expressing M1, M2, M3 and M4 receptors were obtained from N.I.M.H. (Bethesda, MD) (Bonneret al., 1987). Cells were grown in Alpha MEM containing nucleosides and deoxynucleotides (Life Technologies, Paisley, UK), supplemented with 10% fetal bovine serum. The initial cell suspension was 105 viable cells/ml. Cells were collected from log-phase cultures that did not exceed 9 × 105 viable cells/ml. The culture was centrifuged at 1000 to 2000 ×g for 10 to 15 min. The supernatant was discarded and the cell pellets resuspended with ice-cold PBS (Dulbecco ‘A’, Life Technologies), using approximately 200 ml for each liter of starting culture. This was then repeated with approximately 50 ml of PBS for each liter of starting culture. After a final spin, the supernatant was discarded and the pellet stored at −40°C.
Homogenates of CHO cells were prepared in Tris buffer, pH 7.7, at 25°C, as described for the cortex. The densities of cells for the clones in the first homogenization were as follows: HM1, 1.2 × 108 cells/ml; HM2, 7 × 107 cells/ml; HM3, 2 × 108cells/ml; HM4, 1 × 108 cells/ml. The membranes were washed as described for the cortex and stored as 1-ml aliquots, having been resuspended at 1 to 3 × 108cells/ml. Binding assays were performed as described for the cortex with 0.1 nM [3H]-QNB.
Binding to other neurotransmitter receptors.
The methods used in ligand binding studies against nicotinic; dopamine D1; dopamine D2; 5-HT2C; 5-HT1B/1D; 5-HT2; 5-HT3; and GABAergic receptors are cited in table 1.
Affinity of SB 202026 for neurotransmitter receptors
Functional Studies In Vitro
Rat SCG (M1).
Male Hooded Lister rats (Olac, UK, 200–300 g) were killed with an overdose of urethane anesthetic (2 g/kg i.p., 25% w/v). Both SCG with their attached cervical sympathetic and internal carotid nerves were removed and mounted as previously described by Newberry and Priestley (1987). The effect of muscarinic agonists at M1 receptors (depolarization of the ganglion) was evaluated using a contact time of 2 min and a cumulative dosing strategy. The buffer contained AF-DX 116 (400 nM), which has been shown previously to block the M2-mediated hyperpolarization while either not affecting or increasing the depolarizing response (Newberry and Gilbert, 1989). A dose-response curve for carbachol was obtained, and the dose that produced the maximal effect was determined (1 μM). The results obtained with SB 202026 were expressed as log concentration-response curves (percentage of response to 1 μM carbachol). The geometric mean and standard error of the mean were obtained for each concentration.
ACh release from rat cortical slices (M2).
Cerebral cortex was dissected from male Hooded Lister rats (Olac, Bicester, UK, 250–350 g) and placed on ice-cold Petri dishes. A strip of cortex was dissected and submerged in 5 ml of bicarbonate buffer of the following composition (in mM of ANALAR grade reagents): NaCl 124, NaHCO3 25.6, KCl 3.1, KH2PO4 1.3, CaCl 1.3, MgSO4 1.3, glucose 10, equilibrated with 95% O2/5% CO2. Slices were prepared (350 μm × 350 μm), washed and incubated for 30 min in 5 ml of bicarbonate buffer containing 20 μCi of [3H]choline chloride (specific activity 75.9 Ci/μmol, Amersham, UK). Slices were washed again with bicarbonate buffer containing 10 μM hemicholinium-3, and 50-μl aliquots were pipetted into each of eight release chambers (volume 0.5 ml). Stainless steel mesh electrodes at the top and bottom of each chamber allowed for electrical stimulation of the slices. The chambers were superfused with bicarbonate buffer at 0.625 ml/min for an equilibration period of 48 min. At 12 min (S1) and 40 min (S2) after the end of equilibration, the slices were stimulated for 2 min with rectangular electrical pulses 2 msec in duration at 3 Hz and an amplitude of 20 mA, using a constant current stimulator (Leeds University, Pharmacology Department). Agonists were given in the superfusate for a period of 16 min before S2. At the end of the collection period, 7.5 ml of scintillation fluid (Packard Ultima Gold) was added to each sample before counting (Packard 2500 TR liquid scintillation analyzer).
Contraction of guinea pig ileum (M3).
Male guinea pigs (Harlan, Porcellus, UK, 250–300 g) were sacrificed by cervical dislocation. Strips of longitudinal muscle (2–3 cm) were mounted in 10-ml tissue baths containing bicarbonate buffer (NaCl 121.5, NaHCO3 25, KCl 4.7, KH2PO41.8, CaCl2 2.5, MgSO4 1.2, glucose 5.6, equilibrated with 95% O2/5% CO2) at 37°C with a tension of 500 mg. After an equilibration period of 30 min, agonist dosing commenced. The dosing regimen was a contact time of 40 sec and a cycle time of 10 min. All compounds were dissolved in bicarbonate buffer. Carbachol (Sigma, Poole, UK) at 1 μmol was regularly given until consistent responses were obtained. Successive doubling concentration-response sequences were performed for carbachol and SB 202026. Receptor inactivation was then performed by adding the alkylating agent PBZ, 1 μM, for 15 min. After a further equilibration period of 45 min with regular washing (10-min intervals), the concentration-response sequences for carbachol and the test compound were repeated.
Functional Studies In Vivo
Body temperature, tremor and salivation in mice.
Male mice (CD1, Charles River, Margate, UK, 25–35 g) were used, and test compounds were administered s.c. Body temperature was measured using a rectal probe (Comark Electronic Thermometer Type 9001). Induction of salivation and tremor was assessed subjectively, using a scoring system: 0 = absent, 1 = mild, 2 = moderate, 3 = severe. Each parameter was assessed 30 min before and immediately before dosing and 15, 30, 45, 60, 90 and 120 min after dosing. The minimal effective dose for salivation was determined as the lowest dose where salivation was observed. The dose that caused a mean fall in temperature of 3°C was calculated by linear regression analysis of the maximal fall in temperature of each animal, using BBN RS/1 software. The ED50 for tremor was calculated as the dose of test compound that caused a mean tremor score of 1.5 (50% of the maximal mean score) at any of the timepoints.
Induction of hippocampal RSA.
The experimental procedure used was based on that of Bevan (1984b). Male rats (Hooded Lister, Olac, Bicester, UK, 300–380 g) were anesthetized with urethane (25% w/v, 0.6 ml/kg i.p.). Body temperature was maintained at 37°C by a thermostatically controlled heated blanket. The femoral vein was cannulated for drug administration in all animals. Animals were placed in a Kopf stereotaxic frame, and the dorsal skull was exposed. A small burr hole was drilled in the skull, and a bipolar electrode (Clark Electromedical, Reading, UK, model NE-100) was positioned in the CA1 region of the right hippocampus, using predetermined coordinates calculated from a stereotaxic atlas (Paxinos and Watson, 1982). These were (mm, relative to the interaural line): AP +4.6; L −2.3; V +6.6. All rats were pretreated with atropine methyl nitrate (0.1 mg/kg i.v.), a muscarinic antagonist that does not cross the blood-brain barrier, to prevent peripherally mediated muscarinic effects.
Hippocampal EEG was monitored continuously using an oscilloscope. The signal was amplified and filtered using Neurolog System modules (NL100AK headstage, NL103 AC preamplifier, NL106 AC-DC amplifier, NL125 filters; Digitimer Ltd., Welwyn Garden City, UK). Vehicle or test compound was administered i.v. Raw data were collected, digitized and analyzed using a CED1401 Laboratory Interface and a commercial software package (EEG Analysis System, Cambridge Electronic Design Ltd, Cambridge, UK). Arecoline (0.32 mg/kg i.v.) was used as a standard in each experiment. Drug effects were analyzed quantitatively as changes in mean power (μV) in the θ bandwidth of the frequency spectrum (3–7 Hz). The maximal effects obtained with each compound were compared using Student’s t test.
Cardiovascular effects in the anesthetized rat.
Male rats (Hooded Lister, Olac, Bicester, UK, 300–380 g) were used. General anesthesia was induced by urethane (25% w/v, 0.6 ml/kg i.p.). Body temperature was maintained at 37°C using a thermostatically controlled heated blanket. The femoral vein and carotid artery were cannulated for drug administration and monitoring of BP, respectively. BP was recorded via a Druck pressure transducer connected to a polygraph (Lectromed Multitrace 4). HR was derived electronically using a ratemeter triggered by the BP signal (BRL Instrument Services, Harlow, UK). Tracheae were intubated to assist respiration. Increasing doses of test compound were administered i.v. at 45-min intervals. BP and HR were recorded continuously throughout the experiment. Transient changes in both BP (mm Hg) and HR (beats/min) were recorded after i.v. administration, and the peak effect occurred within 1 min of i.v. administration. Peak effects were expressed as a percentage of predose values. Comparisons of the percentage change in BP or HR with each compound were made using Student’s t test.
Results
Ligand Binding Studies
Table 1 summarizes the binding affinity of SB 202026 for a range of neurotransmitter receptors. SB 202026 had high affinity for the muscarinic agonist binding site, as measured by displacement of the agonist ligand [3H]-OXO-M. In contrast, it had low affinity for all other receptors tested, including cholinergic nicotinic receptors. Table 2 summarizes the binding profile of SB 202026 to muscarinic receptors in rat cortex homogenates and to cloned muscarinic receptor subtypes compared with selected muscarinic agonists and antagonists. SB 202026 had a very high affinity for muscarinic receptors in rat cortex (IC50 = 14 nM), a potency similar to that of OXO-M itself (IC50 = 13 nM). The antagonist/agonist ratio has previously been shown to predict intrinsic efficacy (Freedman et al., 1988; Brown et al., 1988a; Hawkins et al., 1992): ratios near to or greater than 100 predict full agonism, those close to unity predict antagonism and intermediate values predict partial agonism. SB 202026 had a ratio of 22, indicative of low intrinsic efficacy.
Binding profile of a range of muscarinic ligands in rat cerebral cortex and cloned human muscarinic receptor subtypes
In studies using cloned human muscarinic receptors, the relatively selective antagonists pirenzepine (M1), AF-DX 116 (M2) and fHHSiDF (M3) exhibited affinity profiles for the cloned receptors consistent with their classification on the basis of previous data (Buckley et al., 1989). SB 202026 possessed high affinity for all subtypes and did not display selectivity for one receptor subtype.
Functional Studies In Vitro
Rat SCG.
Carbachol and SB 202026 induced depolarizations of the rat SCG with similar maxima (100% response of approximately 800 μV) and with parallel dose-response curves, which suggests that SB 202026 shares carbachol’s high efficacy on this system but is approximately 3 times more potent. Results for both compounds are shown in figure 2 and are expressed as percentage of response to the 1 μM carbachol dose. It has been previously reported that this depolarizing response is mediated by M1 receptors (Newberry et al., 1985; Newberry and Priestley, 1987).

Depolarization of rat SCG in response to carbachol or SB 202026. •-• SB 202026; □-□ carbachol. Each point represents the mean of three experiments. Results are expressed as a percentage of the maximal response to carbachol (1 μM) ± S.E. The maximal depolarization to carbachol (100%) was approximately 800 μV.
ACh release from rat cortical slices.
Release of ACh in the cortex and hippocampus is controlled by a negative feedback mechanism through stimulation of muscarinic M2 autoreceptors located presynaptically (Richards, 1990). OXO-M (0.01–3.0 μM) produced a concentration-dependent inhibition of electrically evoked ACh release with 80% inhibition at 1 μM (fig. 3A). Complete inhibition was seen at 3 μM, and this effect could be completely blocked by 10 μM atropine (data not shown). In contrast, SB 202026, up to a concentration of 10 μM, inhibited ACh release by a maximum of around 40%. Further, SB 202026 (0.1–10 μM) antagonized the inhibitory effect of 1 μM OXO-M on ACh release by over 50% (fig.3B), a result that confirms its partial agonist profile in this tissue.
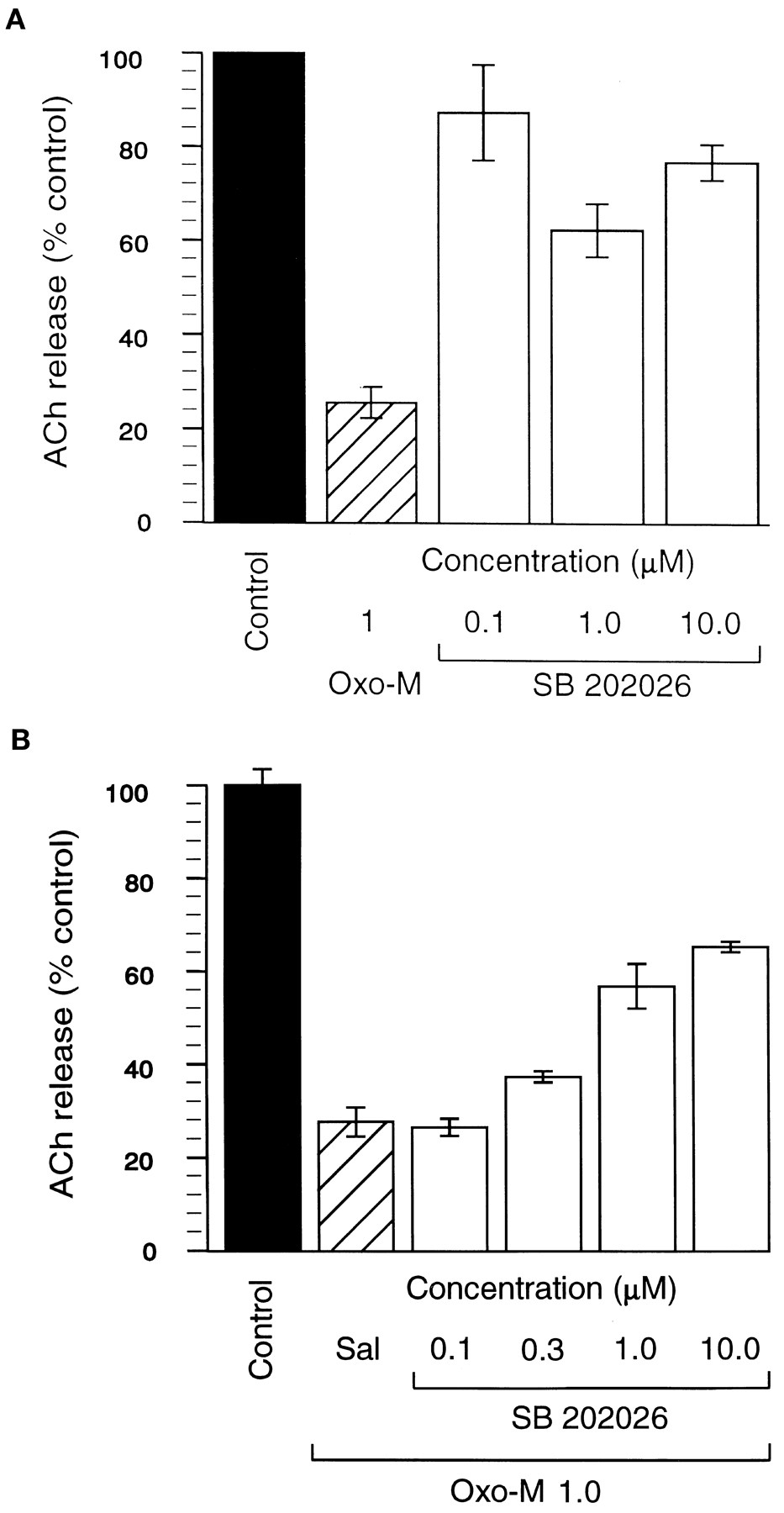
A: Inhibition of ACh release by OXO-M and SB 202026. Each bar represents the mean of six experiments. Results are expressed as percentage inhibition compared with vehicle ± S.E.M. Filled bar, control; hatched bar, OXO-M 1 μM; clear bars, SB 202026 0.1 to 10 μM. B: Inhibition of the effects of OXO-M on ACh release by SB 202026. Results are expressed as percentage inhibition compared with vehicle ± S.E.M. Filled bar, control; hatched bar, OXO-M 1 μM alone; clear bars, OXO-M (1 μM) and increasing concentrations (0.1–10 μM) of SB 202026.
Contraction of guinea pig ileum.
Dose-response relationships for carbachol and SB 202026 are shown in figure4. The data obtained were analyzed by the method of Furchgott and Bursztyn (1967) to determine the relative intrinsic efficacies of the test compounds to carbachol. In the absence of PBZ, an alkylating agent that effectively reduces the tissue receptor reserve, both compounds produced similar maximal responses; this suggests high efficacy on the ileum preparation. After incubation of the tissue with PBZ, carbachol was still able to induce a maximal contraction, whereas the maximal effect of SB 202026 was reduced to approximately 23% of the maximal possible contraction.

Contraction of the isolated longitudinal muscle of the guinea pig ileum in response to carbachol and SB 202026, in the absence and presence of PBZ. •-• SB 202026 alone; •....• SB 202026 in the presence of PBZ; □-□ carbachol alone; □....□ carbachol in the presence of PBZ. Each point represents the mean of three or four experiments. Results are expressed as a percentage of the maximal response to carbachol in each experiment ± S.E.
Functional Studies In Vivo
Body temperature, tremor and salivation in mice.
Results are summarized in table 3. Both oxotremorine (0.03–1.0 mg/kg) and SB 202026 (0.03–10.0 mg/kg) induced hypothermia, oxotremorine being approximately twice as potent as SB 202026 in this model. Thus the doses that caused a 3°C fall in core body temperature were 0.052 and 0.1 mg/kg s.c., respectively. Oxotremorine induced severe tremor at doses similar to those that induced hypothermia, whereas SB 202026 induced mild or no tremor at doses up to 10 mg/kg s.c. Both compounds induced salivation at all doses tested, an effect that is mediated by M3 receptors located peripherally (Schiavone and Brambilla, 1991) and for which there is a high receptor reserve in the mouse (Ringdahl et al., 1987).
Summary of the effects of SB 202026 and oxotremorine in mice
Induction of hippocampal RSA.
Results are illustrated in figure 5. SB 202026 potently (0.018 mg/kg i.v.) induced RSA in the anesthetized rat, with an effect equivalent to that produced by the 15-fold higher dose (0.32 mg/kg i.v.) of arecoline (typical response, 90 μV). There was no significant difference between the maximal changes in power induced by the two compounds (P > .05). These effects could be inhibited by scopolamine hydrobromide, 0.5 mg/kg i.v. (data not shown).

Induction of RSA in the anesthetized rat. •-• SB 202026; □-□ arecoline. Each point represents the mean of six experiments. Results are expressed as the peak amplitude of RSA achieved at each dose of test compound ± S.E. There was no significant difference between the maximal effects attained by the two compounds.
Cardiovascular effects in the anesthetized rat.
The effects of i.v. administration of arecoline and SB 202026 on mean BP and HR are shown in figures 6A and B, respectively. Arecoline (0.1 mg/kg i.v.) induced marked (80–90%) transient, dose-related hypotension (60 mm Hg) and bradycardia (265 beats/min) at doses similar to those that induced RSA. The maximal hypotension (23 mm Hg) or bradycardia (49 beats/min) induced by SB 202026 at the RSA dose (0.018 mg/kg i.v.) in these models was significantly lower than the arecoline response, and effects due to SB 202026 did not increase with increasing dose, a result that indicates a partial agonist effect. The differences between the effects of the two compounds were statistically significant on both parameters (P < .001).
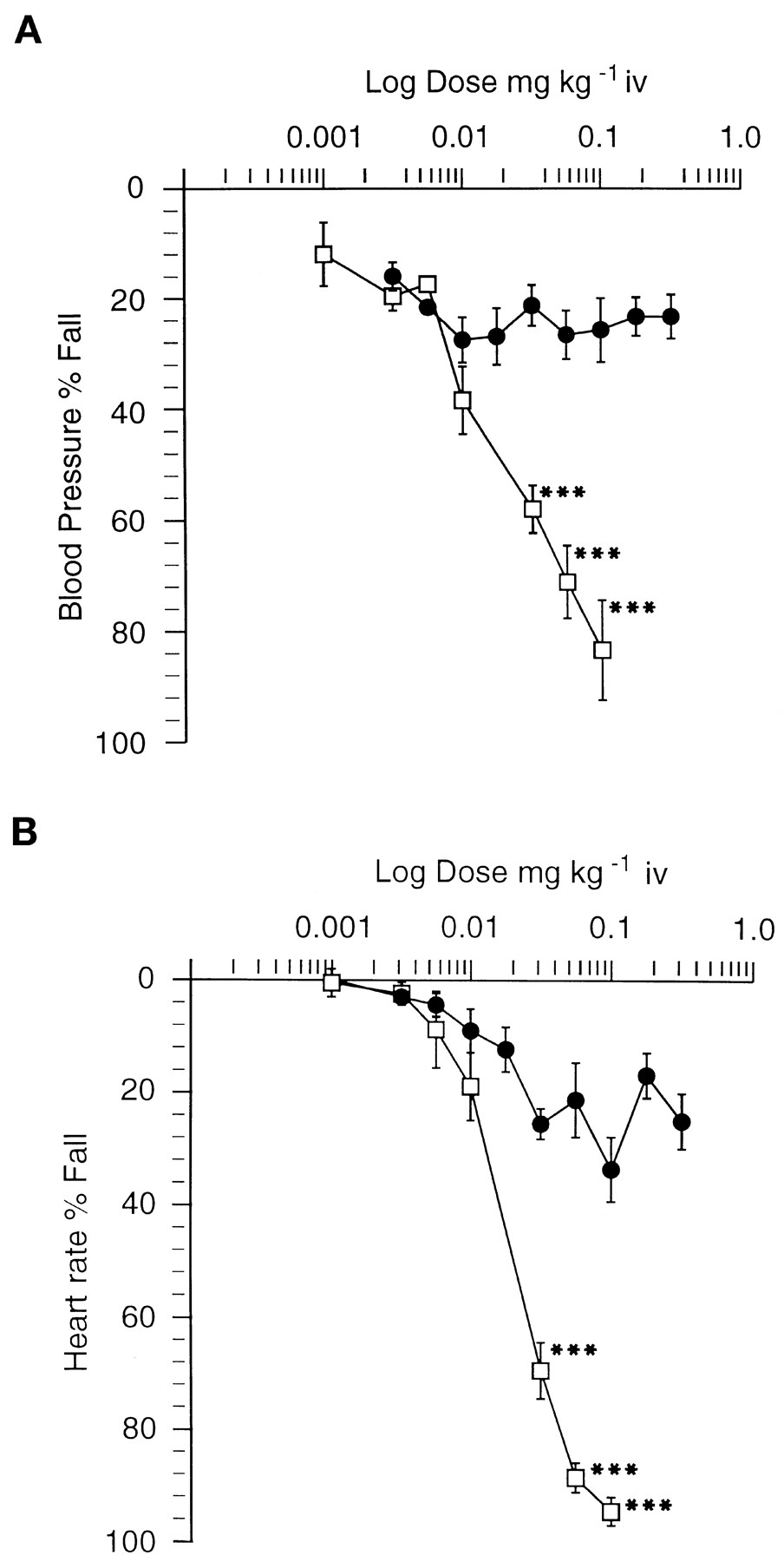
A: The effects on mean BP after acute i.v. administration in the anesthetized rat. •-• SB 202026; □-□ arecoline. Each point represents the mean of four experiments. Points represent the mean transient fall in BP ± S.E. as a percentage of the predose level. The magnitude of the effect of arecoline was significantly greater than that of SB 202026. *** P < .001. B: The effects on mean HR after acute i.v. administration in the anesthetized rat. •-• SB 202026; □-□ arecoline. Each point represents the mean of four experiments. Points represent the mean transient fall in HR ± S.E.M. as a percentage of the predose level. The magnitude of the effect of arecoline was significantly greater than that of SB 202026. *** P < .001.
Discussion
SB 202026 has been shown to be a highly potent muscarinic partial agonist and, unlike the high-efficacy agonists tested, to exhibit functional selectivity for effects mediated by M1receptors. In ligand binding studies, SB 202026 was found to have high affinity for muscarinic receptors in rat cortex (IC50 = 14 nM), whereas it lacked affinity for nicotinic and a range of other neurotransmitter receptors. The ratio of binding affinities of SB 202026 against muscarinic agonist and antagonist ligands (QNB/OXO-M) was consistent with that of an agonist with low intrinsic efficacy (Brown et al., 1988a), which makes it a suitable compound with which to assess potential for functional selectivity (Ringdahlet al., 1987).
In functional studies conducted in vitro, the ability of SB 202026 to induce a response was found to differ between tissues, a result that supports the hypothesis that functional selectivity can be achieved. The results reported here indicate that, for SB 202026, this functional selectivity is in favor of actions mediated by M1 receptors. SB 202026 demonstrated high efficacy in depolarizing the SCG, an effect previously shown to be mediated by M1 receptors (Newberry et al., 1985; Newberry and Priestley, 1987; Boddeke, 1991). This effect was observed in the presence of the selective M2 antagonist AFDX-116 to block the hyperpolarization that has been shown to be mediated by M2 receptors (Newberry and Priestley, 1987). In contrast, SB 202026 had low efficacy in inhibiting ACh release, which is modulated by M2 receptors located presynaptically in the rat cortex (Hoss et al., 1990; Richards, 1990; Quirionet al., 1989). In the presence of SB 202026 (1–10 μM) the response to OXO-M was dose-dependently inhibited, which indicates an antagonist action of SB 202026 and provides further confirmation of its low intrinsic efficacy. SB 202026 also exhibited a partial agonist profile in studies using guinea pig ileum. Though not the most abundant receptor in the gut (M2 receptors represent over 60%) (Levey, 1993), M3 receptors mediate contraction in this tissue (Caulfield, 1993; Flier and Underhill, 1989). SB 202026 was able to induce contraction of the guinea pig ileum to a maximum similar to that of carbachol. However, in the presence of PBZ, this response was significantly reduced. PBZ is one of a number of an alkylating agents that can be used to investigate the receptor reserve in a tissue and the relative efficacies of agonists on that tissue (Furchgott and Bursztyn, 1967). The fraction of receptors that can be blocked irreversibly without a reduction of the maximal effect of an agonist is a measure of receptor reserve, and the relative responses of agonists under these conditions provide an indication of their relative intrinsic efficacies (Ariens, 1983). Thus, after inactivation of a proportion of the receptor reserve in the gut, the maximal response to SB 202026 was reduced to approximately 23% of its original response, whereas carbachol was still able to induce a maximal contraction under the same conditions. These findings illustrate the differences in intrinsic efficacy between carbachol and SB 202026 and emphasize the low intrinsic efficacy of SB 202026.
The functional selectivity seen in vitro was reflectedin vivo. SB 202026 potently induced RSA in the hippocampus of the anesthetized rat, an effect that is characteristic of muscarinic agonists (Bevan, 1984b; Olpe et al., 1987; Enz et al., 1992). SB 202026 and arecoline administered i.v. induced similar increases in power, though arecoline was approximately 20-fold less potent. This is a quantitative model of cholinergic activation in a region of the brain that is considered critical to cognitive processing (Stewart and Fox, 1990) and undergoes severe degeneration in AD (Bowen, 1983). Antagonism of this response by pirenzepine (Barnes and Roberts, 1991) suggests that RSA is mediated by muscarinic receptors of the M1 subtype. Findings that postsynaptic M1 receptors are preserved in AD (Pearce and Potter, 1991) suggest that compounds with demonstrated efficacy at these sites could have therapeutic utility in AD. Studies investigating the effects of pirenzepine on spatial learning in an animal model of cognition (Haganet al., 1987) provide further support for an important role for M1 receptors in learning and memory processes. Induction of RSA can therefore be used as a model to identify agents that may be useful in treating the cognitive deficit inherent in Alzheimer’s disease.
Compared with oxotremorine and arecoline, SB 202026 had low efficacy in systems where M2 or M3 receptors mediate physiological effects. Thus, in the mouse, oxotremorine induced severe tremor, which is thought to be mediated by M3 receptors (Sanchez and Lembol, 1994), whereas SB 202026 did not, which suggests that it would have a low propensity to induce extrapyramidal effects in the clinical setting. This finding is consistent with a low receptor reserve for the induction of tremor in the mouse (Ringdahl et al., 1987). Further, only minimal effects on HR and BP were seen after i.v. administration of SB 202026 to the anesthetized rat. After i.v. administration of muscarinic agonists, cardiovascular effects are mediated peripherally by activation of M2 and M3 receptors located on the heart and vasculature, respectively (Wilffert et al., 1983; Clague et al., 1985). Stimulation of M2 receptors on the heart causes bradycardia through vagal stimulation (Caulfield, 1993), whereas stimulation of M3 receptors in the vasculature causes vasodilation (and subsequent hypotension) indirectly through induction of NO release (Caulfield, 1993). M2 actions may also contribute indirectly to the hypotension observed through stimulation of sympathetic mechanisms (Eglen and Whiting, 1990). The results with SB 202026 suggest that the compound is behaving as a partial agonist in these tissues; the maximal fall in both BP and HR caused by SB 202026 was 70% less than that observed after administration of arecoline. The receptor reserve for these responses is known to be low (Eglen and Whiting, 1990; Freedman et al., 1993), which explains the reduced maximal effect with SB 202026 when compared with arecoline, which has high efficacy at M2 and M3 sites. Together, these findings suggest that in vivo, SB 202026 possesses lower efficacy in the cardiovascular system and is more CNS selective than the agonist arecoline.
Central selectivity of muscarinic agonists is important in the development of treatments for AD. SB 202026 meets this criterion, because its potent actions in the brain can be separated by dose from effects mediated by peripheral muscarinic receptors. In addition to the findings reported here, SB 202026 has been shown to have potent cognition-enhancing activity in both rat and marmoset models at doses 3 to 10-fold lower than those that induce limiting side effects (Clarket al., 1996; Loudon et al., 1996). Receptors in the cortex and hippocampus are thought to be closely involved in learning and memory processes (Cortes et al., 1987), and indeed the M1 subtype predominates in these areas (Corteset al., 1987; Waelbroeck et al., 1990; Leveyet al., 1991; Flynn and Mash, 1993). The partial agonist profile demonstrated in the in vitro and in vivofunctional models used, when taken together with the known distribution of muscarinic receptor subtypes (Levey, 1993), leads us to conclude that the separation between doses that induce desirable central effects and those that cause undesirable side effects is due to functional selectivity for central systems bearing M1 receptors.
Until recently the clinical value of achieving functional selectivity by controlling efficacy has been difficult to predict, because it is dependent on the relative agonist potencies in the target tissue and in those responsible for causing undesirable side effects (Freedmanet al., 1993). Furthermore, this relationship is likely to differ between different animal species and the human. SB 202026 has now been tested in the clinic, both in healthy volunteers (Cooperet al., 1996) and in patients (Kumar and Orgogozo, 1996). Cognitive improvement has been demonstrated in AD patients (Kumar and Orgogozo, 1996) at doses that were well tolerated.
In summary, SB 202026 is a muscarinic partial agonist that has been found both in vitro and in vivo to possess a degree of functional selectivity for central receptor systems over those peripheral systems that are responsible for many of the side effects associated with muscarinic receptor stimulation. It has proved superior to higher-efficacy agonists (e.g., arecoline) in that cognition-related effects (e.g., induction of hippocampal RSA) can be achieved in the absence of significant side effects, a result that supports the contention that SB 202026 possesses a centrally selective profile. This has since been confirmed in animal cognitive models (Clark et al., 1996; Loudon et al., 1996) and in clinical studies (Kumar and Orgogozo, 1996). SB 202026, then, may represent a significant advance for the potential use of muscarinic agonists in the treatment of AD. SB 202026 is currently being evaluated in Phase III clinical trials.
Footnotes
-
Send reprint requests to: J. M. Loudon, SmithKline Beecham Pharmaceuticals, New Frontiers Science Park, Third Avenue, Harlow CM19 5AW, UK.
- Abbreviations:
- AD
- Alzheimer’s disease
- SB 202026
- R-(Z)-(+)-α-(methoxyimino)-1-azabicyclo[2.2.2] octane-3-acetonitrile
- QNB
- quinuclidinyl benzilate
- OXO-M
- oxotremorine-M
- GABA
- γ-amino butyric acid
- fHHSiDF
- fluoro-hexahydro-siladifenidol
- CHO
- Chinese hamster ovary
- HM
- human muscarinic receptor subtype
- SCG
- superior cervical ganglion
- PBZ
- phenoxybenzamine
- RSA
- rhythmical slow wave activity
- BP
- blood pressure
- BPM
- beats per minute
- NO
- nitric oxide
- ChEIs
- cholinesterase inhibitors
- Received April 18, 1997.
- Accepted August 11, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}