


Abstract
Activation of protein kinase C (PKC) regulates the activity of a number of neurotransmitter transporters. When Xenopus oocytes expressing the cloned human dopamine transporter (hDAT) were pretreated with bath-applied phorbol 12-myristate 13-acetate (PMA), a PKC activator, [3H]DA uptake decreased irreversibly in a time- and dose-dependent manner (IC50 = 22 nM; maximal inhibition = 63–85%). The inhibition appeared to be PKC-specific because incubation with the inactive form of phorbol ester 4α-phorbol-12,13-didecanoate (400 nM) did not change the uptake activity and PMA (100 nM) inhibition could be partially blocked by the selective PKC inhibitor bisindolylmaleimide I (1 μM). Saturation studies of [3H]DA uptake showed that PMA-induced inhibition was due to a decrease in Vmax with no change in KT. Similar to uptake, PMA pretreatment inhibited both the hDAT transport-associated and substrate-independent leak currents. PMA also decreased membrane capacitance (Cm) by 40%, selectively in hDAT-expressing oocytes. In addition, PMA pretreatment resulted in a 77% decrease in Bmax of [3H]mazindol binding to intact oocytes. In contrast, binding to whole homogenates of PMA-pretreated oocytes was not significantly altered. These results suggest that PMA regulates hDAT expressed in Xenopus oocytes by altering cell surface trafficking of hDAT.
Transporters play a critical role in terminating the synaptic activity of the monoamine neurotransmitters. By transporting released DA, serotonin and norepinephrine back into presynaptic neurons, transporters replenish neurotransmitter stores, limit the temporal and spatial effects of these neurotransmitters at their presynaptic and postsynaptic receptors and thus allow fine-tuning of neurotransmitter actions (see Amara and Kuhar, 1993). Accordingly, regulation, particularly inhibition, of transporter function greatly affects synaptic transmission. Several pyschostimulants including cocaine and amphetamine inhibit the DA, serotonin and norepinephrine transporters, all of which belong to a superfamily of Na+- and Cl−-dependent neurotransmitter transporters. However, DAT has been shown to be the pharmacological target best correlated with the reinforcing effects and abuse potential of psychostimulants (Ritz et al., 1987; Nestler et al., 1993; Pulvirenti and Koob, 1994; Wise, 1996). Recent gene-knockout studies in mice also demonstrated that DAT is an important mediator of the locomotor stimulatory effects of cocaine and amphetamine and that DAT plays a critical role in setting dopaminergic tone in the central nervous system (Giros et al., 1996). In addition, the neurotoxins 6-hydroxydopamine and 1-methyl-4-phenylpyridinium have long been postulated to enter DA neurons via DAT (Melamed et al., 1985; Javitchet al., 1985). Therefore, studies of DAT function and its regulation may have physiological, pathological and therapeutic importance.
A single gene for DAT has been cloned from rat, cow and human (Kiltyet al., 1991; Shimada et al., 1991; Usdinet al., 1991; Giros et al, 1992; Vandenberghet al., 1992; Eshleman et al., 1995). The deduced primary amino acid sequence contains multiple putative consensus sequences for phosphorylation by protein kinases, suggesting that DAT may be regulated by posttranslational modification. There are sequence variations among the species, including the number of potential phosphorylation sites, e.g., hDAT, has two PKC sites whereas rDAT has three. rDAT, either endogenously expressed in striatal synaptosomes and primary mesencephalic cultures or heterologously expressed in cells, exhibits varied sensitivity to PKC activators (Kitayama et al., 1994; Copeland et al., 1996;Huff et al., 1997). Until recently (Zhang et al., 1997), it was uncertain whether hDAT could be regulated by PKC as well.
Biochemical studies of the stoichiometry have shown that uptake by DAT of each DA+ molecule (positively charged at physiological pH) is coupled to cotransport of 2 Na+ and 1 Cl− (Krueger, 1990; McElvain and Schenk, 1992; Gu et al., 1994). This results in a minimum net flux of two positive ions entering the cell per transport cycle and indicates that the DA uptake process should be electrogenic. Indeed, when hDAT is expressed in Xenopusoocytes, substrates such as DA and amphetamine induce an inward, transport-associated current (Sonders et al., 1997). Both the transport-associated current and substrate uptake are Na+-dependent and voltage-dependent. In addition, a substrate-independent constitutive “leak” current, which is also voltage-dependent, is associated with hDAT expression (Sonders et al., 1997). This leak current is blocked by both DAT substrates and antagonists and is more pronounced in a buffer in which Li+ has been substituted for Na+. Whether hDAT transport-associated and leak currents are sensitive to PKC regulation has not been studied.
We have performed experiments to investigate the possible role of PKC in regulating hDAT expressed in Xenopus oocytes. We found that the PKC activator PMA (bath-applied) inhibited [3H]DA uptake, the hDAT transport-associated current, the substrate-independent leak current, and radioligand binding to hDAT on intact oocytes. PMA also selectively decreased Cm of hDAT-expressing oocytes. However, binding to hDAT in whole homogenates from PMA-pretreated oocytes was unchanged. These results suggest that activation of PKC regulates surface trafficking, rather than activity per se, of hDAT.
Materials and Methods
hDAT cRNA preparation and oocyte expression.
Capped cRNA was transcribed from linearized oocyte expression vector pOTV containing the 1.9-kb hDAT cDNA insert as described (Sonders et al., 1997) using mMessage mMachine (Ambion, Austin, TX). Stage V or VIXenopus laevis oocytes were manually defolliculated, injected with water-diluted cRNA (∼10 ng/oocyte) and maintained at room temperature in FRB (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2 and 5 mM HEPES, pH 7.5) supplemented with 2.5 mM NaPyruvate, 0.5 mM theophylline, 100 U/ml penicillin, 100 μg/ml streptomycin and 50 μg/ml gentamycin. Oocytes were used 3 to 5 days after injection.
[3H]DA and [3H]alanine uptake.
For saturation uptake studies, three to four oocytes/group were incubated in 0.5 ml [3H]DA (10 or 100 nM, 3,4-[7-3H]-dihydroxyphenylethylamine, specific activity 20.3 Ci/mmol, Du Pont New England Nuclear, Boston, MA) and unlabeled DA (final concentrations of 10 nM - 1 mM) for 10 min at room temperature and quickly washed three times in 2 ml FRB. [3H]DA accumulation was determined by dissolving each oocyte in 0.2 ml of 1% SDS, and the radioactivity was quantitated by liquid scintillation spectroscopy. Vmax and K m values were calculated from nonlinear curve fitting by Inplot4 (GraphPad, San Diego, CA). To compare the effects of various reagents, uptake assays were performed with a single concentration of 100 nM [3H]DA or 50 nM [3H]alanine (l-[2,3-3H]alanine, specific activity 58 Ci/mmol, Amersham, Arlington Heights, IL) for 10 min at room temperature. In all experiments with [3H]DA, nonspecific uptake was determined in uninjected oocytes and was less than 1% of that taken up by DAT-expressing oocytes. In some experiments water-injected oocytes were used, and similar results were observed.
Two-electrode voltage clamp electrophysiology.
Two-electrode voltage clamp recordings were performed in oocytes at room temperature using glass microelectrodes filled with 3 M KCl (Sonders et al., 1997). FRB was superfused at a rate of 2 to 3 ml/min (bath volume 0.5 ml). A Warner OC-725B amplifier (Warner Instruments, Hamden, CT) was used with a DigiData 1200 interface. pClamp6 software (Axon Instruments, Foster City, CA) was used to control stimulation parameters, for data acquisition and for analysis. MacLab data acquisition software (AD Instruments, Castle Hill, Australia) and a MacLab/2e interface were simultaneously used to monitor and record experiments. Currents were low-pass filtered at 100 Hz and digitized at 2048 Hz. The voltage dependence of DAT-mediated currents was studied using the following protocol. A sequence of jumps in membrane potential in 10 mV increments was used to measure steady-state currents at potentials between −120 and +40 mV. Oocytes were held at −60 mV before jumps to each test potential (400 msec). Current values were measured and averaged during the last 100 msec of the test interval when they had reached steady-state. Currents were recorded before and again 2 min after DA or cocaine had been superfused. Currents attributable to the actions of drugs were determined by performing off-line subtraction of currents recorded during buffer perfusion from those recorded during drug perfusion (IDrug - IBuffer). In leak current studies, NaCl in FRB was substituted by LiCl; and the pH was adjusted to pH 7.5 with KOH.
Capacitive transients elicited by the voltage jumps from the holding potential of −60 mV to −70 and −50 mV were measured at 5-min intervals over the course of each experiment. The area under the transients was integrated using Clampfit 6.0, and Cm was determined from the slope of the linear regression of the area vs. voltage relationships.
[3H]Mazindol binding to intact oocytes and homogenates.
Saturation radioligand binding to intact oocytes was performed in 1 ml FRB containing 5 nM [3H]mazindol ([4′-3H]mazindol, specific activity 17 Ci/mmol, Du Pont New England Nuclear) and unlabeled mazindol (final concentrations of 5 nM - 1 μM) on ice for 60 min. The binding was terminated by a 15-sec wash in 2 ml of ice-cold FRB, and the radioactivity was determined by liquid scintillation spectroscopy. Nonspecific binding was defined by binding in the presence of 1 μM GBR 12909. Bmax andK d values were calculated from nonlinear curve fitting by Inplot4. Oocyte homogenates were made by sonicating 6 oocytes in 0.5 ml ice-cold FRB. Binding to oocyte homogenates was performed in 0.25 ml FRB containing 5 nM [3H]mazindol in the absence (total binding) and presence (non-specific binding) of 1 μM GBR 12909 on ice for 60 min. The binding was terminated by rapid filtration (Schleicher & Schuell, Keene, NH, no. 30 glass fiber filters) and washing using a cell harvester (Brandel, Gaithersburg, MD). The radioactivity associated with the filters was determined by scintillation spectroscopy.
Phorbol ester treatment.
PMA, 4αPDD and BIM were made as 1000x stocks in DMSO and diluted with FRB to final concentrations before use. In uptake and binding studies, oocytes were pretreated in the bath with PMA or 4αPDD for 3 min followed by a 27-min incubation in FRB or for 30 min (no wash) before the 10-min [3H]DA uptake assay or the 60-min [3H]mazindol binding assay in the absence of the drugs. BIM was added together with PMA when applicable. 0.1% DMSO was used as the control in all experiments. For electrophysiological experiments, oocytes were perfused with PMA for 3 to 5 min; and currents were then recorded in the absence of PMA 15 and 30 min later.
Materials.
BIM and 4αPDD were purchased from Calbiochem (San Diego, CA). Mazindol and GBR 12909 were purchased from Research Biochemicals International (St. Louis, MO). Cocaine was a gift of NIDA (Bethesda, MD). All other drugs were purchased from Sigma Chemical Co. (St. Louis, MO).
Results
[3H]DA and [3H] alanine uptake into oocytes.
To investigate the possible regulation of DAT by PKC, hDAT-expressing oocytes were preincubated with the PKC activator PMA. The effect of bath-applied PMA on [3H]DA uptake into Xenopus oocytes was both concentration- and time-dependent. Figure1 shows that preincubation with various concentrations of PMA for either 3 min (followed by a 27-min incubation in FRB) or 30 min resulted in a marked decrease by as much as 63 to 85%, respectively, in subsequent uptake of 100 nM [3H]DA into oocytes. The calculated IC50 values for PMA were 22.9 ± 2.5 and 21.9 ± 18.3 nM, respectively. Saturation studies of the effect of PMA (30 min) on [3H]DA uptake revealed that the decrease in [3H]DA uptake induced by a maximally effective concentration of PMA (100 nM) resulted from a 69% decrease in the Vmax value, without a significant change in the apparent KT value for the substrate (table 1). Incubation with 400 nM 4αPDD, an inactive analog of PMA, did not change DA uptake (fig.2). Addition of the selective PKC inhibitor BIM (1 μM) during the 30-min incubation with PMA partially prevented the PMA-induced inhibition of DA uptake (fig. 2). Neither vehicle (0.1% DMSO) nor BIM alone changed DA uptake (fig. 2). These results suggest that the PMA effect is likely, at least partially, mediated via PKC activation. Furthermore, 100 nM PMA did not change the endogenous Na+-dependent alanine transporter activity. [3H]alanine uptake measured in oocytes pretreated for 30 min with 100 nM PMA was 104.8 ± 3.8% (n = 3 oocytes from 1 batch) as compared to the vehicle control groups. This suggests that the PMA effect is transporter specific.

Concentration-response curves for the effect of PMA on [3H]DA uptake. The 10-min [3H]DA (100 nM) uptake assays were performed as described in “Materials and Methods” in oocytes pretreated with bath-applied 0.1 nM to 1 μM PMA for 30 min (•; three batches of oocytes, three oocytes from each of batch) or for 3 min followed by a 27-min incubation in FRB (○; two batches of oocytes, three oocytes from each batch). Control values for [3H]DA uptake (in FRB only) were 1.72 ± 0.14 fmol/sec. Data shown are mean values ± S.D.
PMA effect on KT and Vmax values for [3H]DA uptake

Specificity of PMA effect on [3H]DA uptake. hDAT-expressing oocytes were pretreated in the bath with the indicated agents for 30 min, and then 10-min [3H]DA (100 nM) uptake assays were performed. The concentrations tested were 0.1% DMSO (control 4 batches of oocytes), 400 nM 4αPDD (7 batches of oocytes), 100 nM PMA (12 batches of oocytes), 1 μM BIM (5 batches of oocytes) and 100 nM PMA + 1 μM BIM (9 batches of oocytes). Data shown are mean values ± SEM. ***P < .001, PMA vs. all other four groups, **P < .01, PMA+BIM vs. PDD and BIM, and *P < .05, PMA+BIM vs. DMSO using one-way analysis of variance followed by Newman-Keuls post hoc comparisons.
hDAT-associated currents.
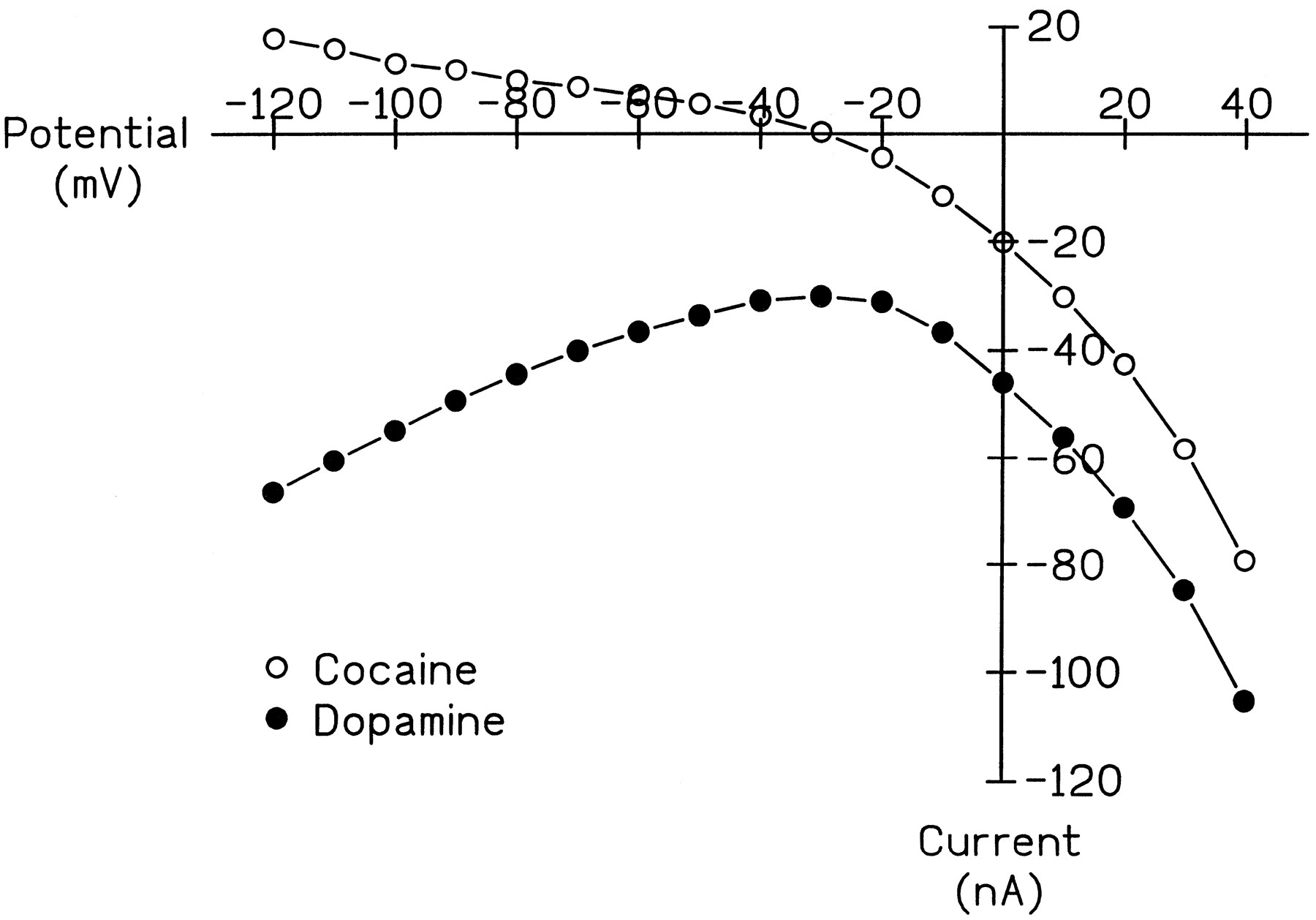
Two-electrode voltage clamp experiments were performed to investigate possible changes induced by PMA in the electrophysiological properties of hDAT. The change in the I-V relationships recorded in hDAT-expressing oocytes induced by either DA or cocaine using the voltage-jump protocol (fig.3) were similar to those reported from voltage ramp protocols (Sonders et al., 1997). At least two currents, an electrogenic transport-associated current and a substrate-independent leak current, are associated with hDAT expression in oocytes (Sonders et al., 1997). The inward transport current induced by DA was predominant over the range of hyperpolarized voltages from -20 to -120 mV (fig. 3). In contrast, cocaine blocked a constitutively active, inward leak that resulted in an outward current at all voltages tested (fig. 3). DA also blocked this leak; however, this was evident only at the more depolarized potentials (-10 to +40 mV; fig. 3). We investigated the effect of PMA on these two currents separately.

Representative I-V relationships for DA- and cocaine-induced currents. Voltage jump protocols were carried out as described in “Materials and Methods” in the same hDAT-expressing oocyte in the following order: in FRB, DA (20 μM), FRB, cocaine (3 μM). Subtractive currents for IDrug - IBufferare shown. Data are representative of results from recordings made in five oocytes from three batches.
Individual oocytes were pretreated in the bath with 100 nM PMA for 3 to 5 min; and after either a 15- or 30-min wash, the substrate-induced currents were recorded. PMA pretreatment inhibited the transport-associated current at each of the potentials tested (fig.4). The inhibition was greater after 30 min of washing than after 15 min (fig. 4). Over the same time period, the resting membrane potentials of the oocytes pretreated with PMA, as compared with vehicle (0.1% DMSO) pretreatment, were not altered; and DMSO-pretreated oocytes exhibited no decrement in DA-induced currents (data not shown). The extent of PMA inhibition (50–81%) was similar with that observed for [3H]DA uptake. These observations help to confirm the close relationship postulated to exist between DA uptake and the transport-associated current (Sonderset al., 1997).

Effect of PMA pretreatment on hDAT transport-associated currents. The voltage jump protocol was executed in the same oocytes in the presence of 100 μM DA first in FRB before PMA treatment (•) and then 15 (○) and 30 (⋄) min after PMA treatment. Oocytes were treated with bath-applied 100 nM PMA for 3 to 5 min and washed with FRB. Subtractive currents for IDA - IBuffer are shown. Data shown are mean values ± S.E.M. for three oocytes from two batches.
The other major DAT-associated current is the constitutively active, substrate-independent leak current. This conductance is reduced by both substrates and antagonists and is 2.4-fold more permeable to Li+ than Na+ (Sonderset al., 1997). Although Li+ permeates the leak pathway, it does not support transport. Therefore, in experiments where Li+ was substituted for Na+, only the leak current was observed (compare figs. 3 & 5). Incubation with 100 nM PMA for 3 min, followed by a 15-min wash, resulted in a 58% inhibition of the DA-induced leak current when compared with vehicle pretreatment (fig.5). Thus, the magnitude of the decrease induced by PMA treatment in the leak current correlated well with that for the transport current shown in fig. 4. These results demonstrate that exposure to PMA inhibited both transport and leak currents in hDAT-expressing oocytes.

Effect of PMA pretreatment on hDAT leak currents. Oocytes were pretreated in the bath with 0.1% DMSO (control; •) or 100 nM PMA (○) for 3 min and washed with FRB. The voltage jump protocol was executed 15 min after PMA treatment in LiCl-substituted FRB, first in the absence and then in the presence of 100 μM DA. Subtractive currents for IDA - IBuffer are shown. Data shown are mean values ± S.E.M. for three oocytes from one batch.
Membrane capacitance.
Changes in cell surface area induced by PMA were determined from capacitive transients. Baseline Cm did not differ among the three groups tested: control oocytes (either uninjected or hDAT-expressing; 194 ± 32 nF, n = 3 oocytes from two batches), uninjected oocytes subsequently pretreated with PMA (168 ± 29 nF, n= 3 oocytes from one batch) and hDAT-expressing oocytes subsequently pretreated with PMA (224 ± 16 nF, n = 4 oocytes from one batch). Thus, any transporter-associated capacitance was insignificant relative to Cm. In control oocytes, Cm was not altered during a 45-min experiment in which voltage jumps were elicited at least once every five min (fig.6). Similarly, pretreatment with 100 nM PMA for 3 to 5 min induced no significant change in Cm of uninjected oocytes in the 45 min after treatment (fig. 6). However, similar PMA pretreatment in hDAT-expressing oocytes induced a significant, time-dependent decrease in Cm of ∼40% at 30 min posttreatment (fig. 6; P < .05, hDAT/PMA compared with control or uninjected/PMA using analysis of variance followed by Newman-Keuls post hoctests). To determine whether the PMA-induced change in Cm was related to the level of hDAT expression, we used the currents induced by 20 μM DA while the oocytes were clamped at −120 mV as an indication of the level of hDAT expression and included all of the hDAT-expressing oocytes tested at 30 min after exposure for 3 to 5 min to 100 nM PMA (n = 7 oocytes from four batches). However, the PMA-induced change in Cm (range 38–86% of baseline) and the level of hDAT expression (range −23.9 to −105 nA) were not significantly correlated (r = −0.11).

Effect of PMA pretreatment on membrane capacitance (Cm) in uninjected or hDAT-expressing oocytes. Cm was measured from the capacitive transients elicited by the voltage jumps as described in “Materials and Methods” and is expressed as a percentage of the mean of two baseline measurements in each oocyte. The voltage jump protocol was executed in uninjected (○) or hDAT-expressing (•) oocytes before PMA treatment (baseline), the oocytes were treated with bath-applied 100 nM PMA for 3 to 5 min and washed with FRB, and then voltage jumps were elicited at the times indicated. Controls (▵) were either uninjected or hDAT-expressing oocytes that were not treated with PMA but that were subjected to the same voltage jump protocol. Baseline Cm did not differ among the three groups: 198 ± 15 nF (n = 10). Data are mean values ± S.E.M. for three to four oocytes from one to two batches. Cm in hDAT-expressing/PMA-treated oocytes, collapsed across time, was significantly different that in uninjected oocytes (P < .05; one-way between and one-way within analysis of variance followed by Newman-Keuls post hoc comparisons).
[3H]Mazindol binding.
The decrease in hDAT Vmax and currents may result from a change in cell surface DAT trafficking, changes from an active to inactive state of DAT, or both. The capacitance results suggest a loss of cell surface membrane and, thus, altered trafficking. To investigate further the mechanism(s) involved, saturation binding curves for [3H]mazindol were constructed using intact oocytes (fig. 7). We have previously observed that [3H]mazindol labels a single site with low nanomolar affinity on intact hDAT-expressing oocytes (Sonderset al., 1997). Pretreatment with bath-applied PMA (100 nM) for 30 min, as compared with vehicle, markedly decreased total measurable [3H]mazindol binding sites by 78% whereas it nonsignificantly increased the apparentK i value (fig. 7; table2). The diminution in Bmax correlated well with the extent of inhibition observed in the [3H]DA uptake Vmax (table 1) and the hDAT currents (figs. 4 and5). However, the same PMA pretreatment did not significantly alter specific [3H]mazindol binding (5 nM) to oocyte homogenates (control 16.1 ± 1.3 fmol/oocyte vs. PMA: 15.0 ± 1.4 fmol/oocyte, mean ± S.E.M.; n = 3 oocytes from each of two batches). These results suggest that PMA alters cell surface trafficking of hDAT, rather than the conversion of DAT to an inactive state.

[3H]Mazindol saturation binding to intact oocytes. [3H]Mazindol saturation binding was carried out as described in “Materials and Methods” on intact oocytes pretreated with bath-applied 0.1% DMSO (•) or 100 nM PMA (○) for 30 min and washed with FRB. GBR 12909 (1 μM; G) was used to define nonspecific binding. Data shown are mean values ± S.E.M.; three batches of oocytes, three oocytes from each batch.
PMA effect on [3H]mazindol binding parameters in intact oocytes
Discussion
The Xenopus oocyte expression system provides a useful system to investigate electrogenic proteins, such as hDAT, because both biochemical and electrical properties can be studied. It is also useful for signal transduction studies. Therefore, it was the system of choice for the current studies in which the effects of short-term PKC regulation were characterized on uptake, electrophysiological and binding properties of hDAT. Our studies suggest that activation of PKC by bath-applied PMA decreases all of these activities associated with hDAT expressed on the cell surface of Xenopus oocytes, most likely by altering cell surface trafficking of hDAT.
PMA, a compound widely used to activate PKC, markedly decreased the uptake of [3H]DA by hDAT expressed inXenopus oocytes. At a maximally effective concentration (100 nM), bath-applied PMA decreased the apparent Vmaxwithout changing the KT value for DA. In contrast, 4αPDD, the inactive analog of PMA, at a concentration of 400 nM did not alter DAT activity, suggesting that PKC activation is required. The partial reversal of PMA-induced inhibition of uptake by the highly selective PKC inhibitor BIM (Toullec et al., 1991; Corey et al., 1994) provided additional evidence for PKC involvement. PMA regulation of rDAT activity has been investigated using several different preparations. In rat striatal synaptosomes and primary mesencephalic cultures, 10 μM PMA produced only a 22% inhibition of [3H]DA uptake (Copeland et al., 1996); and in COS and LLC-PK1 cells 100 to 200 nM PMA produced a 35% inhibition of DAT activity (Kitayamaet al., 1994; Huff et al., 1997). However, Tianet al. (1994) observed no effect of PMA on [3H]DA uptake into rat striatal synaptosomes. Recently, it was reported that PMA decreased by 72% the Vmax of hDAT stably expressed in C6 glioma cells but that the affinity of PMA was relatively low (IC50 = 30 μM; Zhang et al., 1997). In contrast, the PMA-induced inhibition of hDAT activity observed here was both potent (IC50 = 22 nM) and efficacious (75% decrease), suggesting that hDAT in the oocyte expression system is highly sensitive to regulation by PKC.
When modulation of the activity of other Na+- and Cl−-dependent neurotransmitter transporters by PKC has been investigated, inhibition of activity has generally been observed. For example, rat GABA transporters in primary astrocyte cultures, synaptosomes, transfected cells and Xenopusoocytes (Gomeza et al., 1991; Osawa et al., 1994;Tian et al., 1994; Sato et al., 1995b); SERT in human platelets (Anderson and Horne, 1992) and in HEK-293 cells (Qianet al., 1997); a glycine transporter in HEK-293 cells (Satoet al., 1995a) and a taurine transporter inXenopus oocytes (Loo et al., 1996) all exhibited diminished activity. However, Corey et al. (1994), who injected the PKC activators intracellularly rather than bath applying them, described a PKC up-regulation of a GABA transporter expressed inXenopus oocytes. In all cases PMA altered the Vmax of the transporter of interest.
Neurotransmitter transporters are driven by the electrochemical gradients of co/counter-transport of ions. The PMA-mediated inhibition of hDAT uptake appears to be DAT-specific because the same PMA treatment did not inhibit [3H]alanine uptake by the endogenous alanine transporter, which is also Na+-dependent (Jung and Richter, 1983). The resting membrane potentials of the oocytes were also unaltered by PMA. Taken together, these results suggest that it is unlikely that the changes in hDAT function and binding that we observed were due to altered membrane viability and/or electrochemical gradients.
Electrophysiological recording has recently been used for real-time measurement of the voltage dependence, ionic coupling and channel-like properties of a number of different electrogenic transporters (reviewed by Lester et al., 1994; DeFelice and Blakely, 1996; Sonders and Amara, 1996). Use of the two-electrode voltage clamp technique in hDAT-expressing oocytes allowed us to investigate for the first time the effect of PMA on hDAT currents, and we observed that the effect of bath-applied PMA on both of these currents paralleled its effects on [3H]DA uptake. The fact that inhibition was sustained upon washing after exposure to PMA further points to its effect being mediated through PKC activation rather than by a direct drug effect. Whereas the resting membrane potentials of oocytes were unchanged by PMA pretreatment, the transport-associated current was inhibited to a similar extent as uptake. Similar results have recently been reported for hSERT-mediated uptake and currents in a stably-expressing cell line (Qian et al., 1997). PMA pretreatment also diminished the hDAT substrate-independent leak current to the same extent as the transport-associated current. These results suggest that the transport pore and the leak pore may reside at the same site on DAT and that this site can be regulated by PKC. However, an alternative explanation is that the PMA-induced inhibition is solely caused by changes in cell surface trafficking (see below),i.e., PKC may regulate DAT trafficking rather than directly altering the transport/leak pore(s). The similarity among the effects of PMA on uptake, currents and binding provides further evidence that the substrate and leak currents are indeed properties of DAT itself. Nonetheless, the possibility still cannot be completely excluded that an extrinsic protein may be involved in the electrophysiological properties of DAT, and that this protein is regulated by PKC at the same time and to the same extent.
PMA pretreatment decreased the number of [3H]mazindol binding sites measured on the intact hDAT-expressing oocytes but not in oocyte homogenates. In control oocytes most hDATs appear to be localized to the cell surface; our preliminary studies using sucrose density gradient centrifugation and radioligand binding assays showed that ∼86% of the hDAT are associated with the plasma membrane (S.-J. Zhu, unpublished observations). Unfortunately, there is no hydrophilic radiolabeled DAT ligand available for detection of DAT exclusively at the cell surface; [3H]mazindol will permeate the intact oocyte plasma membrane. Nonetheless, it is likely that the Bmax measured on intact cells with [3H]mazindol primarily reflects the number of hDATs present on the cell surface and not those in the intracellular pool (H. Bönisch, personal communication). This is because [3H]mazindol binding to DAT is negligible at low Na+ concentrations (Javitch et al., 1984), such as those present intracellularly in the oocyte (6 mM; Barish, 1983). The marked difference between PMA-induced changes in [3H]mazindol binding to the intact and homogenate oocyte preparations suggests that a significant proportion of hDAT is no longer present at the cell surface following PMA pretreatment. However, future studies using approaches such as biotinylation/immunoblot assays, as has been used with SERT (Qianet al., 1997), are required to confirm the mechanism(s) underlying these observations. Nonetheless, based on the fact that the reduction in the amount of measurable binding sites on the cell surface is similar to the decrease in [3H]DA uptake velocity and hDAT currents, we postulate that PKC regulation of DA uptake is largely via membrane trafficking of DAT. This has been suggested for three other Na+- and Cl−-dependent neurotransmitter transporters (Corey et al., 1994; Loo et al., 1996; Qianet al., 1997). Since there were no significant changes in KT or K d, it is unlikely that PMA alters the ratio of active to inactive transporters as suggested by Kitayama et al. (1994). Our results, and those of Zhang and colleagues (1997) showing a 32% PMA-induced decrease in the number of [3H]WIN 35,428 binding sites on intact hDAT-expressing C6 cells, also differ from those reported by Kitayama et al. (1994) in which PMA did not change [3H]WIN 35,428 binding measured on intact rDAT-expressing COS cells. However, our results agree with [3H]mazindol binding studies in which PMA did not alter the Bmax in rat synaptosomal and mesencephalic membrane preparations (Copeland et al., 1996). The discrepancy in PKC-mediated changes in uptake and binding between groups may result from the differences between rDAT and hDAT (i.e., the number and position of the PKC phosphorylation sites) and/or differences between the expression systems including membrane trafficking mechanisms.
Additional support for the suggestion that PMA induces a change in cell surface trafficking comes from our Cmmeasurements. In Xenopus oocytes, measurements from freeze fracture replicas indicate that the area of the cell surface is 9-fold more than the area predicted for a smooth sphere (Zampighi et al., 1995); and this infolding provides the potential for relatively large PMA-induced reductions (70–80%; Vasilets et al., 1990; Bourinet et al., 1992) in cell surface membrane area. Our results indicate that 30 min after a 3- to 5-min exposure to 100 nM PMA, Cm was reduced by ∼40% in hDAT-expressing oocytes. Surprisingly, there was no significant reduction in Cm in uninjected oocytes also pretreated with PMA. Whether this difference is related to the high level of hDAT expression (∼5 × 1010copies/oocyte, table 2) is unclear. Similar to the results presented here, comparisons carried out using uninjected and hDAT-expressing oocytes from the same six oocyte batches reveal no significant differences in Cm between the two groups (R. D. Mayfield and N. R. Zahniser, unpublished data). Nonetheless, cRNA injections may enhance the observation of PMA-induced Cm changes by introducing a molecule, in this case hDAT, that is subject to PKC-dependent redistribution. This redistribution may trigger the change in surface area as a natural consequence of using membrane to achieve redistribution or may prevent steady-state fusion if it is the exocytotic limb that is modified. In any case, treatments for longer times and/or with higher concentrations of PMA can result in large reductions in Cm in both uninjected and mRNA-injected Xenopus oocytes (Vasiletset al., 1990; Bourinet et al., 1992). Additional evidence that this effect is common to PKC activation comes from the reports of Hirsch et al. (1996) and Loo et al.(1996) that treatment with sn-1,2- dioctanoylglycerol decreases Cm, as well as maximal currents, associated with expression of Na+/glucose cotransporters and a mouse retinal taurine transporter in oocytes.
Whether PKC regulation of DAT and other neurotransmitter transporters is due to direct phosphorylation of the transporter remains unclear. PMA treatment increases in vivo phosphorylation of rDAT stably expressed in LLC-PK1 cells (Huff et al., 1997). However, the PMA-induced regulation and subcellular redistribution of the GABA and glycine transporters have been suggested to occur through a novel regulated secretory pathway and/or another indirect mechanism since removal of the predicted PKC phosphorylation sites did not alter the response of the transporter to PMA (Coreyet al., 1994; Sato et al., 1995a). An alternative approach lead Hirsch et al. (1996) to the same conclusion for the Na+/glucose cotransporter. If PKC regulation does not involve phosphorylation of the consensus sites on DAT, it is possible either that phosphorylation of DAT occurs at noncanonical site(s) or that another PKC-sensitive protein may be mediating hDAT trafficking in the oocytes.
There are also consensus sites for PKA phosphorylation on both hDAT and rDAT. Kadowaki et al. (1990) reported that dibutyryl-cAMP and forskolin enhanced [3H]DA accumulation in rat hypothalamic cell cultures containing dopaminergic neurons by as much as 2-fold. However, Tian et al. (1994) and Copelandet al. (1996) failed to demonstrate a change in [3H]DA uptake when synaptosome preparations were incubated with the cAMP analog 8-Br-cAMP under the same experimental conditions as PMA, while [3H]GABA uptake was inhibited by 30%. We have also observed that incubation with dibutyryl-cAMP, 8-Br-cAMP or forskolin did not significantly change [3H]DA uptake by the hDAT in oocytes (data not shown). Taken together, these results suggest that neither hDAT nor rDAT are sensitive to PKA regulation.
PKC-mediated inhibition of DA uptake would be expected to enhance and prolong synaptic DA neurotransmission after DA release in vivo. Its impact on postsynaptic DA receptors could be similar to DAT antagonists such as cocaine. Although no studies have directly demonstrated that DAT activity can be regulated by PKC-coupled presynaptic receptors and the specific PKC isoforms expressed in brain, our results suggest that such PKC-mediated effects are possible and should be the focus of future investigations.
Acknowledgments
The authors thank Dr. R. Dayne Mayfield for help with the statistical analyses and Mr. Gaynor Larson for assistance with the figures.
Footnotes
-
Send reprint requests to: Dr. Nancy R. Zahniser, Department of Pharmacology C236, University of Colorado Health Sciences Center, 4200 East Ninth Ave., Denver, CO 80262.
-
↵1 This work was supported by National Institutes of Health grant DA04216, postdoctoral fellowship DA50706 to S.J.Z. and career development award DA05706 to N.R.Z.
- Abbreviations:
- BIM
- bisindolylmaleimide I
- Cm,membrane capacitance
- DA, dopamine
- DAT
- dopamine transporter
- DMSO
- dimethyl sulfoxide
- FRB
- frog Ringer’s buffer
- GABA
- γ-aminobutyric acid
- hDAT
- human dopamine transporter
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- I-V
- current-voltage
- 4αPDD
- 4α-phorbol-12,13-didecanoate
- PKA
- cAMP-dependent protein kinase
- PKC
- protein kinase C
- PMA
- phorbol 12-myristate 13-acetate
- rDAT
- rat dopamine transporter
- SDS
- sodium dodecyl sulfate
- SERT
- serotonin transporter
- Received January 29, 1997.
- Accepted May 27, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}