


Abstract
Oxidative stress is considered a cause or propagator of acute and chronic disorders of the central nervous system. Novel 2,4-diamino-pyrrolo[2,3-d]pyrimidines are potent inhibitors of iron-dependent lipid peroxidation, are cytoprotective in cell culture models of oxidative injury, and are neuroprotective in brain injury and ischemia models. The selection of lead candidates from this series required that they reach target cells deep within brain tissue in efficacious amounts after oral dosing. A homologous series of 26 highly lipophilic pyrrolopyrimidines was examined using cultured cell monolayers to understand the structure-permeability relationship and to use this information to predict brain penetration and residence time. Pyrrolopyrimidines were shown to be a more permeable structural class of membrane-interactive antioxidants where transepithelial permeability was inversely related to lipophilicity or to cell partitioning. Pyrrole substitutions influence cell partitioning where bulky hydrophobic groups increased partitioning and decreased permeability and smaller hydrophobic groups and more hydrophilic groups, especially those capable of weak hydrogen bonding, decreased partitioning, and increased permeability. Transmonolayer diffusion for these membrane-interactive antioxidants was limited mostly by desorption from the receiver-side membrane into the buffer. Thus, in this case, these in vitro cell monolayer models do not adequately mimic the in vivo situation by underestimating in vivo bioavailability of highly lipophilic compounds unless acceptors, such as serum proteins, are added to the receiving buffer.
A series of novel 2,4-diamino-pyrrolo[2,3-d]pyrimidines were described as potent inhibitors of iron-dependent lipid peroxidation, and proved to be cytoprotective in cell culture models of oxidative injury and neuroprotective in brain injury and ischemia models (Bundy et al., 1995; Andrus et al., 1997; Hall et al., 1997). First-generation compounds like tirilazad mesylate (PNU-74006F), one member of a series of 21-aminosteroids that inhibit iron-dependent lipid peroxidation and are used for the treatment of subarachnoid hemorrhage (Hall et al., 1994), have poor oral bioavailability and limited brain access and consequently are administered parenterally (Laizure et al., 1993). Therefore, major considerations in the discovery of the pyrrolopyrimidines were oral activity and superior brain penetration given that the objective was to treat chronic neurodegenerative diseases associated with oxidative mechanisms of pathology (Lees, 1993). One a priori criterion for the neuroprotective activity of pyrrolopyrimidines was the ability to reach target cells deep within brain tissue in efficacious amounts. To aid the selection of lead compounds with this property, we measured the transcellular permeability and cellular partitioning of pyrrolopyrimidine analogs in a cell culture model designed for highly lipophilic molecules.
Structural determinants of permeability and partitioning are discussed for a series of structurally similar homologs. In addition, detailed studies were conducted concurrently with two radiolabled compounds from the pyrrolopyrimidine series representing different physicochemical, permeability, and cell partitioning attributes to discern the roles of protein binding and cell partitioning on permeation and to complement ongoing pharmacological and pharmacokinetic studies. The data proved useful in predicting which compounds were most likely to leave the blood and penetrate underlying tissue. In a companion paper, brain uptake dynamics and cellular penetration of these compounds are confirmed in vivo (Sawada et al., 1999).
Materials and Methods
Reagents.
Madin-Darby canine kidney (MDCK) epithelial cells at passage 52 were from the American Type Culture Collection (Rockville, MD). Tissue culture-grade dimethyl sulfoxide (DMSO) and BSA (fraction V, lot 129F0136) were from Sigma Chemical Co. (St. Louis, MO). 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (free base was from Boehringer-Mannheim Corp. (Indianapolis, IN). PBS was purchased from GIBCO BRL (Grand Island, NY) and used in 1 mg/mld-glucose and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid adjusted to pH 7.4. Polycarbonate filter inserts (Transwell) with a diameter of 24 mm and a 0.4-μm pore size were purchased from Costar (Cambridge, MA). Reagents for HPLC were of the highest purity available. [14C]Mannitol (56 mCi/mmol) and [14C]testosterone (50 mCi/mmol) were purchased from Amersham Corp. (Arlington Heights, IL). The 2,4-diamino-pyrrolo[2,3-d]pyrimidine analogs were supplied as solids by Medicinal Chemistry, Pharmacia & Upjohn (Kalamazoo, MI). [14C]PNU-87663 monomethanesulfonate salt (22.2 μCi/mg; 443.9 g/mol) and [14C]PNU-89843·HCl (30.8 μCi/mg; 322.2 g/mol) were obtained as >98% pure by HPLC from the Pharmacia & Upjohn Radiosynthesis Group (Kalamazoo, MI). These were reconstituted in DMSO to give 0.5 mCi/ml, and the solutions were stored at −20°C under argon without significant oxidation/degradation.
Unlabeled Compound Permeability Assay.
Continuous monolayers of MDCK cells were cultured on Transwell filter inserts as described previously (Sawada et al., 1994). The apical (donor) and basolateral (receiver) chambers were rinsed for 10 min with PBS and equilibrated for 20 min with PBS containing 3.0% (w/v) and 0.5% (w/v) BSA, respectively. All incubations were done at room atmosphere at 37°C with continuous mixing by nutation (Nutator; Clay Adams, Parsippany, NJ). Donor solutions were prepared by slowly injecting a small volume of a 4 mg/ml stock solution of compound in DMSO into PBS-3% BSA while mixing vigorously to give 50 μM compound and <0.5% (v/v) DMSO. After mixing at 37°C for 15 min, the donor solution was passed through a 0.2-μm-pore filter, and 1.5 ml was added to the cells in the apical chamber. An aliquot was taken from the donor solution before addition to the cells to determine the initial concentration by reversed-phase HPLC. Thereafter, samples were taken every hour from both the donor (25 μl) and receiver (100 μl) solutions to follow the kinetics of disappearance and appearance during apical-to-basolateral flux of compound across the cell monolayer. The insert with cells was moved to 2.5 ml of fresh receiver solution at each time interval to maintain sink conditions. In cases where flux was fast, sampling times were adjusted so that <10% of the initial drug mass permeated per time interval. In all cases, the donor solutions were depleted by <10% of their initial volume over the course of the assay. After the last time point, each insert with cells was rinsed quickly three times with cold PBS, and the cells were collected by scraping. After extraction (Folch et al., 1957), the amount of compound in the organic phase was measured by reversed-phase HPLC to determine the amount of compound that had become cell associated and to obtain mass balance.
Uptake, Efflux, and Transmonolayer Flux of 14C Compound.
The details of the procedures for these experiments are reported by Sawada et al. (1994). For measuring uptake and efflux kinetics at the apical membrane, MDCK cell monolayers were grown in plastic 6-well dishes. In uptake experiments, cells were incubated with 1 ml of PBS-BSA containing 0.1 μCi/ml (10.1 μM) [14C]PNU-87663 or 0.5 μCi/ml (50.7 μM) [14C]PNU-89843. For efflux experiments, cell monolayers were preloaded with either 7.8 μM [14C]PNU-87663 or 21.3 μM [14C]PNU-89843 in 1 ml of PBS-3% BSA for 45 min. After a brief rinse with PBS, 1 ml of PBS-BSA was added to each well, and the entire volume was replaced with fresh PBS-BSA at each time interval to maintain sink conditions.
Transcellular flux of 14C compound was measured using filter-grown cell monolayers and 18 μM [14C]PNU-89843 or 16 μM [14C]PNU-87663. All incubations were done at 37°C at room atmosphere with constant mixing by nutation. At the end of each experiment, the cells were rinsed with cold PBS and solubilized (Sawada et al., 1994), and the amount of cell-associated radioactivity was measured.
Solubility.
The solubility of the radiolabeled compounds was measured in PBS containing 0.05, 0.1, 0.5, or 3% (w/v) BSA and 9.1 μM [14C]PNU-87663 or 8.7 μM [14C]PNU-89843. Solutions were prepared as described above and assayed for total radioactivity. Aliquots (1 ml) were centrifuged for 10 min at 150,000g at 20°C, and the amount of radioactivity remaining in solution was determined. Donor solutions for all subsequent experiments were prepared in this way to ensure that the compound was soluble and not present as a microprecipitate.
Compound Stability.
The stability of the14C compounds was examined under experimental conditions to avoid measurements in which significant degradation or metabolism occurred. Donor solution and the aqueous and organic solvent extracts (Folch et al., 1957) of plastic-grown MDCK cell monolayers collected after 30, 90, and 180 min were subjected to reversed-phase HPLC. Control samples without cells and using [14C]PNU-89843 also were analyzed.
Reversed-Phase HPLC.
Donor and receiver solutions containing unlabeled pyrrolopyrimidines were diluted 4-fold with methanol or acetonitrile to yield the appropriate mobile phase. The organic phase of cell extracts was dried under nitrogen and reconstituted in mobile phase. After centrifugation for 5 min at 14,000g to remove precipitated protein, samples were injected in mobile phase. The mobile phase varied with compound and consisted of either 25% to 75% (v/v) methanol diluted with aqueous phase containing 0.5% (v/v) triethyamine and adjusted to pH 5 with glacial acetic acid or 20% to 40% (v/v) acetonitrile containing 0.1% (v/v) trifluoroacetic acid. Conditions were optimized to obtain a capacity factor (k′) of 2 to 3. Columns used were a 250 × 4.6 (i.d.)-mm Zorbax RX-C18 or SB-CN (5-μm particle size) column (DuPont, Wilmington, DE) or a 150 × 4.6 (i.d.)-mm Inertsil-C8 (5-μm particle size) column (Metachem, Torrance, CA), with a 1-cm guard column. The samples were chromatographed with an isocratic method at a flow rate of 1.0 to 1.5 ml/min. Detection was at 242 nm or using fluorescence with excitation at 320 nm and an emission cutoff filter of 389 nm. Five-point standard curves were generated with 0.05 to 10 μg/ml in mobile phase without protein present. The 14C compounds were chromatographed with an isocratic method using a 250 × 4.6 (i.d.)-mm Zorbax SB-CN (5-μm particle size and 80-Å pore size; Mac-Mod Analytical, Chadds Ford, PA) column, at ambient temperature with a constant injection volume of 45 μl and a Flo-One/Beta model CR radioactive flow detector equipped with a 2500-μl flow cell (Radiomatic Instruments, Tampa, FL). The mobile phases consisting of 0.5% (v/v) triethylamine in distilled, deionized water at pH 5.0 (acetic acid) and methanol were mixed on line at organic-to-aqueous ratios of 80:20 (v/v) (PNU-87663) and 75:25 (v/v) (PNU-89843). Detection was at 242 nm with retention times of 6.3 min (PNU-87663) and 5.7 min (PNU-89843) at a flow rate of 1.0 ml/min. The liquid scintillation fluid (Flo-Scint II, Radiomatic Instruments, Meriden, CT) was added to the column effluent at a rate of 4 ml/min.
Data Analyses.
Apparent permeability coefficients (Pe) were obtained from the slope of the rate of appearance of unlabeled parent compound into the receiver solution, after donor disappearance reached equilibrium, using:
Uptake, efflux, and transcellular permeability coefficients for the14C compounds were calculated as described bySawada et al. (1994). Binding constants and partition coefficients were calculated using previously derived equations (Raub et al., 1993) and 6.58 × 10−3 cm3 for the volume of the cell monolayer (Vcell) in an area of 9.4 cm2 (1 well of a 6-well plate).
Log partition coefficients for n-octanol/water (logPoct) were estimated with CLOGP version 3.53 software (Daylight Chemical Information Systems, Inc., Irvine, CA) according to incremental contributions of fragments. Apparent CLOGP values were calculated, when strongly ionized aliphatic tertiary amines were present, using pKa values of 10.5 for piperazine and 10.3 for N-methylated piperazine (Perrin et al., 1981) according to Yalkowsky and Morozowich (1980). Although the pyrrolopyrimidines are weakly basic with a diffuse charge and pKa ≈ 6.5, this pKa value was ignored in calculation of apparent CLOGP because it accounts for only a decrease of 0.05 log unit.
Cellular accumulation of a compound was calculated as the percentage of the initial mass in the donor solution extracted from the cell monolayer at the end of the flux experiment. Under the conditions of the experiment and depending on the diffusivity of the compound, this accumulation was not always at equilibrium. The efficiency of the extraction protocol, and especially the relative partitioning of compound between the organic and aqueous phases, was monitored indirectly by mass balance. Therefore, the cell-associated fraction is an estimate of the partition coefficient, which is underestimated in cases in which appreciable water solubility exists, or with the more hydrophilic, ionizable analogs, and where mass balance was poor or <70%.
Results
Permeability and Lipophilicity.
A congener series of 26 pyrrolopyrimidines with discrete additions, deletions, and substitutions (Tables 1 and2) were chosen for this study from hundreds of compounds to derive a qualitative structure-permeability relationship to understand and predict tissue partitioning and permeation. Flux of compound from donor to receiver through a continuous cell monolayer occurred with a variety of different kinetics from relatively unrestricted permeability, where appearance nearly equals disappearance (Fig. 1A), to membrane-restricted permeability, where the rate of disappearance is nonlinear and exceeds the rate of appearance to different degrees (Fig.1, B–D). In all cases, permeability of these lipophilic compounds was controlled by the aqueous boundary layer (Raub et al., 1993).
Chemical structures, calculated log Poct, transepithelial permeability coefficient, cell partitioning, and mass balance of pyrrolopyrimidine analogs assayed using MDCK cell monolayers
More chemical structures, calculated log Poct, transepithelial permeability coefficient, cell partitioning, and mass balance of pyrrolopyrimidine analogs assayed using MDCK cell monolayers

Transcellular flux kinetics of different pyrrolopyrimidine compounds. Disappearance of compound from the donor solution (▪) and appearance in the receiver solution (•) bathing the basolateral surface of the cell monolayer were measured by HPLC with time. The results were plotted as mass fractions relative to the mass in the donor at time zero or MD(0). A, strongly ionized compounds or those that are highly protein bound are poorly permeable, and the cell associated fraction is low. B, as cell accumulation increases, both the rates of disappearance and appearance increase and disappearance kinetics become hyperbolic. C and D, marked and rapid cell accumulation results in distinctly different kinetics of loss and appearance with relatively slow (C) or fast (D) appearance kinetics.
The permeability coefficients for diffusion of these compounds across the cell monolayer (Pm) were calculated from the Pe values (Tables1 and 2) by correcting for the aqueous boundary layer. Because of the addition of a strongly basic piperazine, the compound set was divided into weakly basic (•) and piperazinyl (▪) subsets. Permeance of the weakly basic analogs was inversely related to lipophilicity (Figure2). Linear regression analysis of triplicate values for 14 weakly basic compounds gave a slope of −3.0 ± 0.5 (n = 42) with a correlation coefficient (r2) of 0.89 (F = 32) over 4 log units. Given data variability, this simpler interpretation was favored statistically over an exponential or a sigmoidal relationship. We used calculatedPoct values due to the difficulty in measuring this value accurately and reliably for such lipophilic compounds. Measured Poct values for the radiolabeled PNU-89843 and PNU-87663 were ∼25% less than the calculated values. It has been reported that there is good agreement (±0.5 log unit) between observed and calculated values, especially in the range of CLOGP 4 to 5 (Hansch and Leo, 1995). Given that the congener series used in our study has distinct substituent changes that are well predicted by fragment analysis, we assume that the relative hydrophobicities are consistent across this series. In addition, because all of the weak bases have a similar pKa near neutrality, the CLOGP is approximately equal to the CLOGDpH 7.4 across this series. The original template is compound 7. Placement of hydrophobic groups in the pyrrole C-6 or N-7 positions decreased permeability, whereas smaller hydrophobic groups, more hydrophilic groups, and groups capable of hydrogen bonding increased permeability.

Correlation between the transmonolayer permeability coefficient (Pm) and lipophilicity of the weakly basic pyrrolopyrimidines. Pm values are the mean of triplicate determinations obtained from the slope of the kinetics of appearance of compound in the receiver solution measured by reversed-phase HPLC, defined by eq. 1, and corrected for the unstirred water layer described in Materials and Methods. Lipophilicity is an estimate using the calculated logPoct/water or CLOGP based on a computational fragmental method. Apparent CLOGP values for the piperazinyl analogs (▪) are adjusted for ionization at pH 7.4. The data points are identified by number according to the list of compounds in Table 1. The two unidentified compounds (*) are examples that are extensively metabolized.
To mimic the protein gradient from blood to brain (Raub et al., 1993;Sawada et al., 1994), donor and receiver solutions contained 3% and 0.5% BSA, respectively. The addition of protein also improved the solubility and the mass recovery of the most lipophilic compounds. For example, >95% of a 50 μM compound 7 in 3% BSA remains in solution after centrifugation at 150,000g, indicating that precipitation in the donor is minimal and that low permeability is not limited by poor solubility in the donor. The solubility of compound in the receiver solution was not as significant a problem because the concentrations of compounds obtained per time interval did not exceed 0.5 μM and solubility of compound 7, for example, in 0.5% BSA is at least 8 μM. However, increased loss to adsorption in the presence of only 0.5% BSA might contribute to lower mass recoveries, yet there was no apparent relationship between lipophilicity or charge and decrease in mass balance (Tables 1 and 2). Furthermore, thePe values measured using HPLC correlate well with values measured using trace concentrations (∼1–2 μM) of radiolabeled compounds 3 (PNU-89843) and 7 (PNU-87663), suggesting that lower recoveries did not significantly affectPe values. With an assay precision error of 12%, Pe values are overestimated as mass balance decreases where aPe with 70% mass balance is ∼40% faster than expected if mass balance were 100%. Compounds that appear to deviate significantly from the inverse relationship betweenPm and lipophilicity were metabolized. For example, the two unidentified analogs (*) in Fig. 2 had mass recoveries of <15%. and the HPLC chromatograms show multiple peaks at 254 nm. Mass recoveries average 80% with a pooled standard deviation of 4% (n = 26) and most likely were limited by assay accuracy and compound adsorption rather than by metabolism and/or degradation.
The rates of diffusion of the piperazinyl substituted analogs were significantly slower than the weak base analogs of similar lipophilicity (Fig. 2), but they also demonstrate a linear correlation (r2 = 0.90; F = 36) with an identical slope of −2.6 ± 0.4 (n = 24) over a 2-log unit range. The addition of the ionizable piperazine at position N-7 generally increases permeability of the weak base homolog (compare compounds 7 and 8 and compounds 3 and 4), and this was either negated or decreased by N-methylation (compare compounds 7–9 and compounds 3–5) or by dimethylation (compare compounds 13–15). Substitution of the two pyrrolidines with piperazines (compounds 22 and 24; Table 2) conferred significant hydrophilicity with a net charge of −1 to −2, causing these compounds to behave differently with regard to permeability (Fig. 2).
Permeability and Cell Partitioning.
At the end of each flux experiment, the cells are collected and extracted with organic solvents, and the amount of compound recovered in the organic phase or that became cell associated is quantified and expressed as the percentage of the initial concentration of compound that was in the donor solution. In most, but not all cases (see Fig. 1, A and B), a pseudoequilibrium was reached (see Fig. 1, C and D), and this is especially true for the more lipophilic compounds. A plot of lipophilicity versus percent of compound that became cell associated suggests that separate optima exist for the weak base and N-7-piperazinyl compounds where an increase in cell partitioning is independent of the apparent CLOGP value (Fig.3).

Relationship between lipophilicity and the fraction of compound that is cell associated at the end of the flux experiment. Lipophilicity is an estimate using the calculated logPoct/water or CLOGP based on a computational fragmental method. The cell-associated fraction is an estimate of partitioning of weakly basic (•) and piperazinyl analogs (▪) from buffer-albumin to the cell monolayer at pseudoequilibrium. The data points are the mean of triplicate samples and are identified by number according to the list of compounds in Table 1.
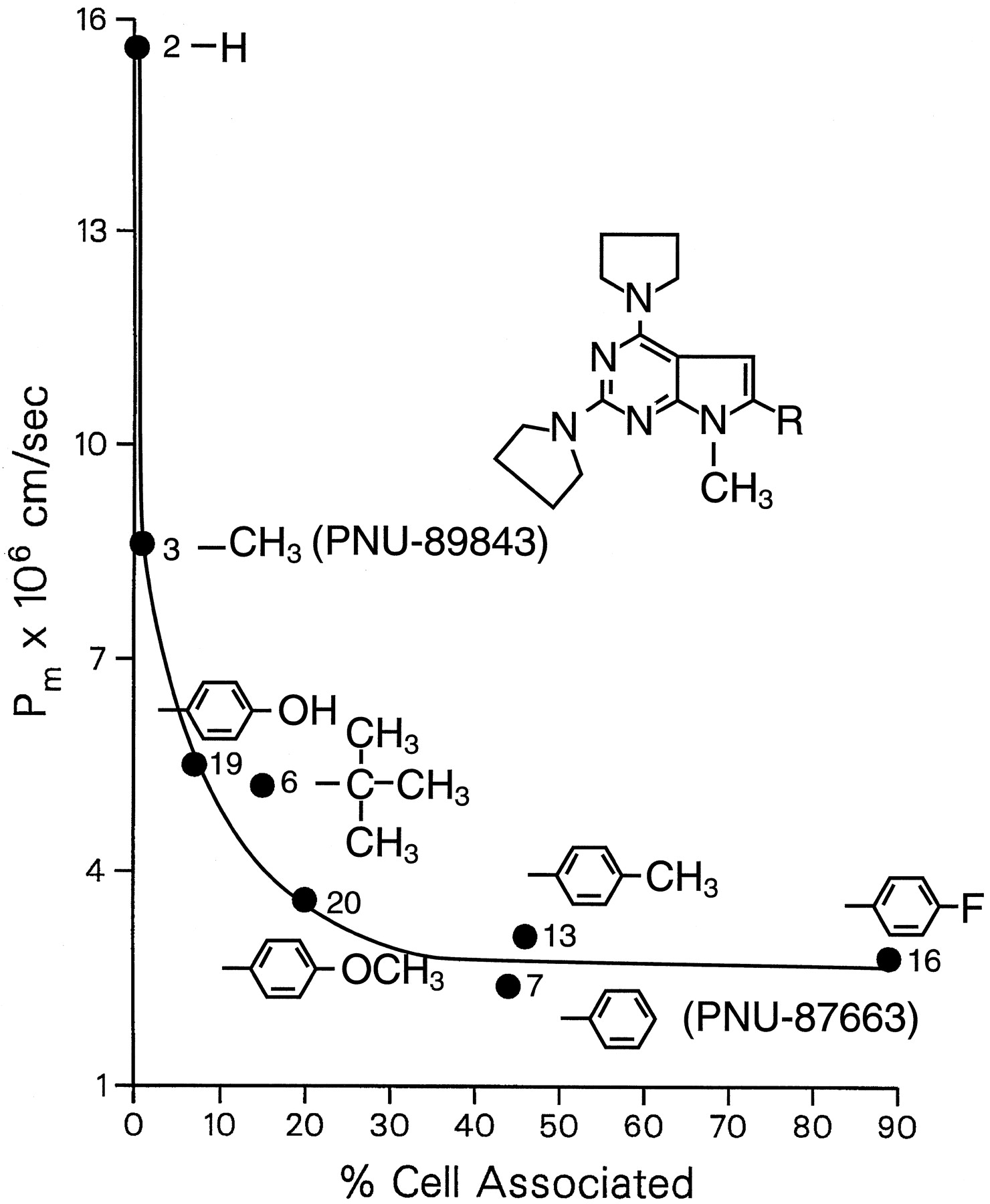
The increase in cell partitioning was accompanied by a decrease in permeability. For example, compounds 4, 5, and 8 were the least cell associated (Fig. 3) and the most permeating compounds (Fig. 2) of theN-piperazinyl pyrrolopyrimidines. Within the series of structurally related weak base homologs, Fig.4 clearly showed the inverse relationship between Pm and cell partitioning. When the percentage of compound that became cell associated was <1% to 2%, permeability was relatively fast. Increasing the percentage of cell-associated compound to >2% resulted in decreased permeability. and this occurred with the addition of a large hydrophobic group, like a phenyl (compound 7) or a tert-butyl (compound 6), in the pyrrole C-6 position. When a group capable of hydrogen bonding, such as a hydroxyl was placed in the para position of the C-6 phenyl, cell accumulation decreased and permeability increased (compare compounds 19 and 7). Methylation of this hydroxyl (compound 20) resulted in increased cell association and decreased permeation.
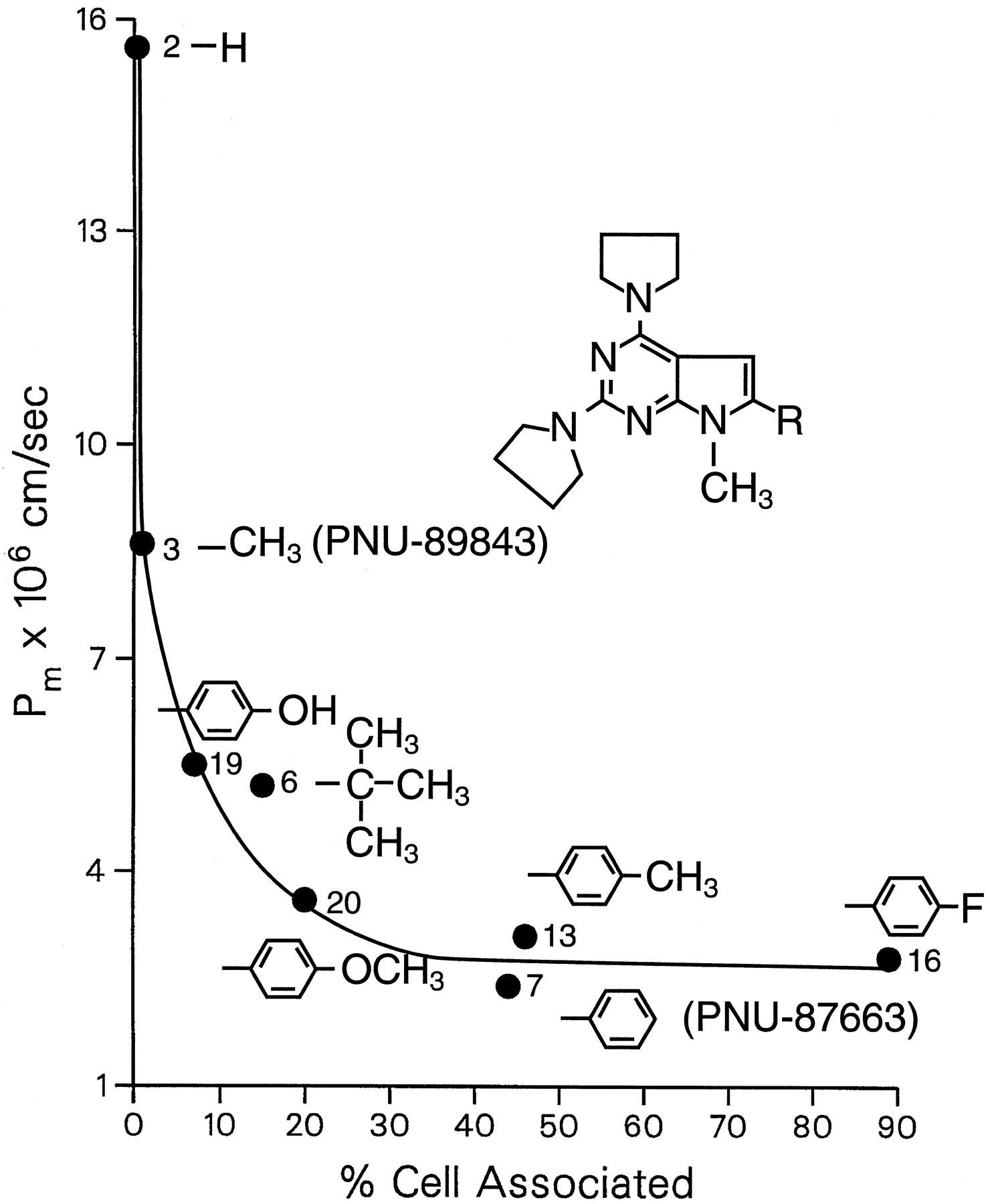
Influence of structural determinants of a homologous series of weakly basic pyrrolopyrimidine analogs on permeability and cell partitioning. Pm and the fraction cell associated were obtained as described in the legends to Figs. 2 and 3. The data points are the mean of triplicate samples and are identified by number according to the list of compounds in Table 1.
To better understand the partitioning and permeation of the pyrrolopyrimidines, two compounds, representing the opposing characteristics of the weak base series, were radiolabeled and contrasted using a more defined kinetic model of this in vitro cell culture system (Fig. 5) (Raub et al., 1993; Sawada et al., 1994). Compound 3 (PNU-89843), with a methyl group in position C-6, was minimally accumulated and very permeable. Compound 7 (PNU-87663), with a phenyl in position C-6, was markedly accumulated and slowly permeating.

Kinetic model of passive diffusion of a protein (P)-interacting drug (D) across a cell within a confluent monolayer. Lateral diffusion into neighboring cells and from the apical to basolateral membranes is limited by the tight junction. Free and bound species diffuse across the aqueous boundary layer, and cell uptake is the free drug partitioning into the apical membrane in a thermodynamic step across the water-lipid interface. The free drug concentration is determined by the operational dissociation constant (Kd), and its membrane insertion is dictated by the intrinsic partition coefficient (Kintr). Once in the hydrocarbon core or inner leaflet of the lipid membrane bilayer, drug can partition into and diffuse through the cytoplasm (pathway 1) or diffuse laterally throughout the cell plasma membrane (pathway 2), reaching equilibrium. Efflux from the cell into the aqueous receiver solution is the mirror image of the uptake kinetic steps. The effective permeability coefficient is a summation of all of these steps (Raub et al., 1993;Sawada et al., 1994).
Transmonolayer Diffusion of PNU-87663 and PNU-89843.
The amount of radioactivity associated with the cells and both solutions was measured to obtain mass balance and confirmed that metabolism and/or degradation was negligible. Reversed-phase HPLC analysis showed that [14C]PNU-87663 chromatographed as a single peak after 4 h of continuous incubation with confluent MDCK cell monolayers. In contrast, metabolism of [14C]PNU-89843 begins within 1 h and continued slowly with time. Both compounds were stable for 4 h in buffer alone, confirming that [14C]PNU-89843 was metabolized by the cells. Except for transmonolayer flux where the rate of diffusion superseded metabolism, the kinetic data for [14C]PNU-89843 were collected within 1 h.
Flux across the cell monolayer was examined in the apical-to-basolateral and basolateral-to-apical directions to determine whether these compounds were preferentially transported. Cultured epithelial cell monolayers express polarized, energy-dependent transporters, such as the multidrug resistance P-glycoprotein efflux pump (Burton et al., 1997), that result in underestimated apical-to-basolateral flux. Bidirectional flux of PNU-89843 and PNU-87663 was not asymmetric, indicating that these compounds were not preferentially transported and permeated via passive diffusion (data not shown).
Comparison of PNU-87663 and PNU-89843 transmonolayer kinetics showed a marked difference in permeability (Fig.6). Disappearance of PNU-87663 was exponentially rapid with time with aPe of 1.15 ± 0.38 × 10−4 cm/s and slowed down after 30 min, approaching steady state. Increasing the concentration of BSA in the receiver from 0.5% to 3% had minimal effect on the exponential loss of compound but did alter the steady-state phase (Fig. 6A). This was attributed to the more rapid appearance in the receiver where maintenance of the cell-donor equilibrium was altered. The disappearance of PNU-89843 occurs with a rate (Pe = 0.85 ± 0.17 × 10−4 cm/s) identical (P > .05, Student’s t test) to that of PNU-87663 because both were aqueous boundary layer controlled; however, the kinetics was not biphasic (Fig. 6B).

Transmonolayer flux of (A) [14C]PNU-87663 and (B) [14C]PNU-89843. Cumulative mass fractions (mean and S.D. of triplicate samples) relative to the total mass in the donor att0 are shown as a function of time for disappearance from the donor (○) and appearance in the receiver (■). In all cases, the donor solution at pH 7.4 contained 3% BSA. The BSA concentration in the receiver solution at pH 7.4 was either 0.5% (dashed line) or 3% (solid line). At the end of the experiment, the amount of radioactivity recovered in the cells was 39 ± 2% (PNU-87663) and 5.1 ± 1.1% (PNU-89843), giving mass balances of 97 ± 5% and 111 ± 10%, respectively. The curves are the best fit of nonlinear least-squares regression analysis according to the model of Sawada et al. (1994).
The concomitant appearance of these compounds in the receiver solution was contrastingly different. PNU-87663 appearance was relatively slow and linear over time, with a Pbl of 2.82 ± 0.28 × 10−7 cm/s with 3% BSA in the receiver (Fig. 6A). This was 400-fold slower than uptake and was decreased 62% when the concentration of BSA in the receiver was decreased to 0.5%. Significant accumulation in the receiver solution occurred after the initially rapid loss of compound from the donor solution slowed down, and Pe values were determined after steady state was reached. PNU-89843 was 17-fold more permeable than PNU-87663 with an appearancePbl of 48.3 ± 1.67 × 10−7 cm/s with 3% BSA in the receiver (Fig.6B). Unlike PNU-87663, the disappearance and appearance kinetics of PNU-89843 were mirror images. Like PNU-87663, the rate of appearance was dependent on the BSA concentration because the appearancePe decreases 43% in the presence of 0.5% BSA in the receiver solution (data not shown).
Other additives were added to the receiving compartment to better mimic the in vivo setting: liposomes, lipid emulsions, rat brain homogenates, and cell monolayers plated on the underside of the filter and/or in the plastic receiving well. In all cases, the rate of uptake from the donor was unaffected and the rate of appearance was increased minimally, but proportionately as if the receiver volume was expanded (data not shown).
Cell Uptake Kinetics of PNU-87663 and PNU-89843.
The uptake kinetics involve the diffusion of free and bound compound across the aqueous boundary layer, followed by partitioning of free compound into the apical membrane, diffusion throughout the cell, and attainment of equilibrium (Fig. 5). The initial rates of uptake for both compounds were minimally affected by BSA concentration because uptake of both the free and the BSA-bound compound was aqueous boundary layer controlled, and a linear correlation between the uptakePe values and the free drug concentration supported this (data not shown). A dependence on BSA concentration was realized with differences in the equilibrium reached after 10 min (Fig. 7). Cell uptake increases as the concentration of BSA decreases due to the increase in free or unbound compound. These data were analyzed using nonlinear least-squares regression analysis per the biophysicokinetic model described to calculate an apparent distribution coefficient (Ke) between total compound (free and protein bound) in the aqueous solution and the cell. By using eq. 4 from Raub et al. (1993), an intrinsic distribution coefficient (Kintr) and an apparent dissociation binding constant (Kd) were calculated. The Kd value was operational because the stoichiometry of the interaction between compound and BSA was not known. PNU-87663 had a significantly greaterKintr than PNU-89843, or 1750 ± 100 versus 750 ± 50. Accordingly, PNU-87663 was less bound to BSA with a Kd of 130 μM compared with aKd of 20 μM for PNU-89843.

Accumulation of (A) [14C]PNU-87663 and (B) [14C]PNU-89843 at the apical membrane domain of plastic-grown cell monolayers is measured as disappearance from the donor solution as a function of BSA concentration. The data are the mean and S.D. of triplicate samples expressed as the fraction of mass remaining in the donor solution relative to the total mass in the donor at t0. The curves are the best fit of nonlinear least-squares regression analysis according to the model of Sawada et al. (1994).
Efflux Kinetics of PNU-87663 and PNU-89843 from Cell.
The appearance of compound in the receiver during transmonolayer flux was limited by desorption or efflux of compound from the basolateral cell membrane into the receiver bathing solution. Therefore, this event was governed by the membrane partition coefficient of the compound and its affinity for BSA (Fig. 5). Efflux of PNU-87663 and PNU-89843 was dependent on the concentration of BSA (Fig.8), but PNU-89843 was 40- to 70-fold faster than PNU-87663. In all cases, the cell-associated compound desorbed with first order kinetics, suggesting that there was a single kinetic pool.

Efflux kinetics of cell-associated (A) [14C]PNU-87663 and (B) [14C]PNU-89843 from the apical membrane of plastic-grown cell monolayers into buffer containing different BSA concentrations. The cell monolayers were allowed to accumulate 14C compound to equilibrium in 1% BSA and quickly rinsed, and the cumulative mass fraction that appeared in the bathing solution (receiver) with time is measured relative to the total mass that was cell associated att0. The mean and S.D. of triplicate samples are shown and fit by nonlinear least-squares regression analysis according to the model of Sawada et al. (1994).
Discussion
Passive transcellular or transmembrane permeation of hydrophobic molecules is governed by a number of physicochemical and thermodynamic properties, including solubility, lipophilicity, polarity, and serum/protein interactions. Biophysicokinetic models are one approach to separate and quantify these components to recognize and understand their relative importance in the design of compounds for optimal delivery and, thus, efficacy. In this report, we surveyed a homologous series of novel, lipophilic antioxidants by using cultured cell monolayers and a previously developed kinetic model (Raub et al., 1993;Sawada et al., 1994). The objective was to understand the structural dependence of permeance of pyrrolopyrimidines and to use this information to predict tissue penetration, especially brain, and residence time to aid in the selection of lead candidate compounds.
Contrary to what is often assumed generally, permeability of pyrrolopyrimidines in vitro is inversely related to lipophilicity or to increased cell partitioning. An inverse relationship between permeability and lipophilicity is not uncommon and has been described in many studies as a parabolic relationship (Hansch and Clayton, 1973;Nook et al., 1988; Grieg, 1989; Bernards and Hill, 1992; Ridout et al., 1992; Saji et al., 1992, Nau et al., 1994). Such data have generated controversy because it is widely accepted that permeability increases as lipophilicity increases, reaching a maximum. The way in which permeability is measured contributes to the discrepancy. Some studies rely solely on a rate of disappearance from the donor solution. This rate of disappearance increases with increased lipophilicity, reaching a plateau value limited only by the hydrodynamics of the system. A decrease in permeability under such circumstances has been attributed to a decrease in free solute concentration caused by lowered aqueous solubility or micelle formation. Here, we measured both disappearance and appearance and found that the disappearance kinetics were rapid and of the same order of magnitude for all compounds consistent with unstirred water layer-limited diffusion. The free solute concentration was accounted for by removing precipitates from the donor solution through ultracentrifugation before use. Although independent physical studies indicate that the pyrrolopyrimidines are surface active, they were relatively weak surfactants and do not form micelles at the concentrations used here in the presence of albumin (Epps and McCall, 1997).
The addition of albumin, while decreasing adsorption to noncellular surfaces and increasing solubility, complicated data interpretation such that the observed Pm value included a binding equilibrium effect to account for the change in free solute concentration. However, the effect of protein binding on decreased permeability in the present dataset for pyrrolopyrimidines was minimal and inconsistent with the observed weak binding constants for PNU-89843 and PNU-87663. The Kdvalue for PNU-89843 was 6.5-fold lower than that for PNU-87663, yet PNU-89843 was, according to the linear relationship between permeability and lipophilicity, significantly more permeable. These data imply that although protein interactions contribute to the observed Pm values, it was not the cause of the inverse relationship between permeability and lipophilicity.
Other factors that have been attributed to the parabolic effect and that have been accounted for here include measuring permeability under nonequilibrated conditions, the principle of bulk (molecular size) tolerance, metabolism, and active transport. All permeability coefficients are calculated from appearance kinetics after disappearance kinetics reach steady state or when the cell concentration is at steady state and mass balance is achieved. For this homologous series, molecular size was not correlated with increased lipophilicity; however, it was clearly shown that partitioning and, hence, permeability were consistent with Overton’s rules of steric influence on diffusion of a molecule through a lipid membrane (Wright and Diamond, 1969). Metabolism was negligible under the conditions of the assay. We also ruled out that decreased flux can be attributed to active efflux, which can markedly decrease net apical-to-basolateral flux.
Our results were similar to those reported by Ridout et al. (1992) that percutaneous absorption of phenolic analogs was negatively controlled by increased partitioning. Using intestinal epithelial cell monolayers,Wils et al. (1994a) also showed that highly lipophilic (logPoct-buffer, pH 7.4 > 3.5) molecules have lowered transcellular permeability. They proposed that this observed slow permeability in vitro predicts low oral absorption. Others have claimed that a thresholdPe value of 1 × 10−6 cm/s is obligatory for maximal oral absorption (Artursson and Karlsson, 1991; Wils et al., 1994b). This may occur only when buffer-to-membrane partitioning in the donor is the rate-controlling mechanism for diffusion, which was not valid in the present situation. The pyrrolopyrimidines have greater than 50% oral bioavailability in rats (Bundy et al., 1995). We contend that the slow permeability of highly lipophilic molecules was a consequence of these in vitro assays, which have an aqueous receiving solution of limited volume. This study and previous data showed that desorption from the receiver-side membrane into the buffer is the limiting step and that the in vitro models did not adequately mimic the in vivo situation (Raub et al., 1993; Sawada et al., 1994). In illustrating the caveats of relying solely on disappearance kinetics for predicting oral absorption, Nook et al. (1988) noted the importance of blood extraction, or the “receiver” equivalent. This explanation can be nicely demonstrated in vitro by adding to the receiver solution more protein or protein that binds compound with high affinity (Sawada et al., 1994). Attempts to improve this transfer were unsuccessful, and none of the alternate approaches outperformed serum proteins. We suggest this is due to their inability to readily diffuse through the 0.4-μm-pore filter to access the basolateral membrane and act as efficient acceptors. Lipophiles still have to partition from the membrane into the underlying aqueous solution. Additional cell monolayers underneath the filter simply enlarge the receiver compartment and may mimic the in vivo situation better if situated more closely to the cell monolayer barrier separated only by a basement membrane.
The interplay of hydrophilic and hydrophobic characteristics and their position on the molecule illustrate what determines the permeability of pyrrolopyrimidines. The importance of the pyrrole C-6 position for cell partitioning was shown. These data are consistent with the proposed role for hydrogen-bonding groups in membrane permeability of nonelectrolytes (Wright and Diamond, 1969) and peptides (Burton et al., 1992) and in brain penetration (van de Waterbeemd and Kansy, 1992), but the net effect was positive rather than negative. Hydrophilic substitutions that increased aqueous solubility had a lesser affect on cell accumulation if made on positions other than the hydrophobic moiety at position C-6. This assignment was consistent with film balance data indicating that amphiphilic PNU-87663 partitions to the aqueous-phospholipid interface oriented with the protonated pyrrolidine groups in the aqueous phase and the hydrophobic pyrrole buried in the fatty acyl chains of the membrane (Epps and McCall, 1997).
Delineation of transcellular flux into uptake by the apical membrane, cell partitioning, and efflux from the basolateral membrane showed that efflux of PNU-89843 from the basolateral membrane (effluxPbl) (Table3) was 17-fold faster than that for PNU-87663. These results with PNU-89843 and PNU-87663 could be compared with data for earlier-generation antioxidants PNU-78517F (Table 3) and PNU-74006F (data not shown) because identical studies were performed using the same biophysicokinetic analyses and hydrodynamic conditions (Raub et al., 1993). The marked difference between these compounds was the rate of efflux from the basolateral membrane. However, effluxPbl was not simply a cell-to-buffer partitioning event because it was derived by including the kinetics of uptake on the donor side and therefore is influenced by the concentration of compound in the cell. Free PNU-89843 immediately partitioned into the apical membrane and, given its lowerKintr, repartitions into the receiver solution down the concentration gradient created by sink conditions. Consequently, marked cell concentrations were never accumulated. The same result could be achieved with those compounds that have a largeKintr by shifting the equilibrium in favor of the receiver solution and inducing a gradient as likely occurs in vivo. Evidence for this was obtained in vivo where efflux of PNU-89843 from brain was greater than 5-fold faster than PNU-87663 (Sawada et al., 1999).
Comparing different highly membrane interactive antioxidants under equivalent conditions
We predict from these results that the pyrrolopyrimidines permeate the brain (and other organs) beyond the blood-brain barrier faster than PNU-74006F or PNU-78517F. Laser scanning fluorescence microscopy showed that PNU-87663 penetrates deep into the brain soon after i.v. injection (Sawada et al., 1999). Evidence against a similar distribution for PNU-74006F and PNU-78517F was indirect and less convincing, yet the results suggest that these compounds reside mostly within the endothelium before they are rapidly cleared (Epps and McCall, 1997). The equilibrium amount of compound and its residence time were dependent on the rate of clearance from the blood. As blood levels fall, tissue levels of PNU-89843 will also decline immediately, whereas tissue levels of PNU-87663E decline more slowly. That this occurs in vivo was confirmed by measuring brain and plasma drug levels in mice 5 and 60 min after i.v. administration. Although plasma levels for PNU-87663 and PNU-89843 decreased (84–86%) similarly within ∼1 h, the brain/plasma ratio for PNU-87663 was ∼5-fold greater than that for PNU-89843 (Sawada et al., 1999).
In conclusion, in vitro assays showed that pyrrolopyrimidines were a more permeable structural class of antioxidants, in support of oral activity and superior brain penetration. A qualitative structure-permeability assessment indicated that substitutions at the pyrrole C-6 position influence membrane partitioning and that permeability was inversely related to lipophilicity or cell partitioning. These results indicated that the assessment of tissue penetration of highly lipophilic compounds using these in vitro assays involved more than a series of connected aqueous partitioning steps and that addition of serum proteins, or as yet unidentified endogenous carriers, to the receiving compartment can improve this assessment.
Acknowledgments
We thank Steve E. Buxser, Doug E. Decker, Dennis E. Epps, and Ferenc J. Kézdy for valuable discussions and suggestions during the course of this work. We also give special recognition to our coauthor Norman Ho, without whom this and other works would not have been possible and who, during the writing of this article, retired from Pharmacia & Upjohn.
Footnotes
-
Send reprint requests to: Thomas J. Raub, Ph.D., Drug Absorption & Transport, Mailstop 7271–209-623, Pharmacia & Upjohn, Inc., 301 Henrietta St., Kalamazoo, MI 49007. E-mail:thomas.j.raub{at}am.pnu.com
-
↵1 Present address: 3M Pharmaceuticals, St. Paul, MN 55133.
-
↵2 Present address: 59-303 Pupukea Road, Haleiwa, HI 96712.
- Abbreviations:
- CLOGP
- calculated log octanol-water partition coefficient
- Ke
- apparent distribution coefficient
- Kintr
- intrinsic distribution coefficient
- MDCK
- Madin-Darby canine kidney
- Pbl
- permeability coefficient for basolateral membrane efflux
- Pe
- apparent permeability coefficient
- Pm
- permeability coefficient for monolayer
- Poct
- octanol-water partition coefficient
- DMSO
- dimethyl sulfoxide
- Received May 6, 1998.
- Accepted September 8, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}