


Abstract
Intrathecal administration of nonsteroidal anti-inflammatory drugs in the rat blocks the thermal hyperalgesia induced by tissue injury, which suggests a role for spinal cyclooxygenase (COX) products in this facilitated state. Two isozymes of the COX enzyme have been reported, COX-1 and COX-2, but the agents thus far examined are not isozyme selective. We examined the effects of intrathecally (i.t.) or systemically (i.p.) administered S(+)-ibuprofen (a nonselective COX inhibitor) or 1-[(4-methysulfonyl)phenyl]-3-tri-fluoromethyl-5-(4-fluorophenyl) pyrazole (SC58125; a COX-2 selective inhibitor) on carrageenan-induced thermal hyperalgesia (reduced hindpaw-withdrawal latency). The following observations were made: 1) Thermal hyperalgesia otherwise observed during the first 170 min was blocked in a dose-dependent manner by S(+)-ibuprofen or SC58125 administered i.t. or i.p. before carrageenan treatment. 2) Intraperitoneal, but not i.t., administration of either inhibitor after the establishment of hyperalgesia (170 min after carrageenan injection) reversed thermal hyperalgesia in a dose-dependent manner. Thus, the initial component of thermal hyperalgesia after tissue injury was blocked by systemic or spinal administration of both COX inhibitors, whereas established hyperalgesia was reversed only by systemic inhibitors. This study demonstrates that at least spinal COX-2, if not both COX-1 and COX-2, are necessary for the initiation of thermal hyperalgesia, whereas nonspinal sources of prostanoids (synthesized by COX-2 and perhaps also COX-1) are important for the maintenance of thermal hyperalgesia associated with tissue injury and inflammation.
After tissue injury, an animal will display spontaneous pain behavior and an exaggerated response to moderate stimuli, a state otherwise referred to as hyperalgesia. Consistent with the behavioral effects, injury or inflammation produces a left shift in the relationship between discharge rate and stimulus intensity (Reeh et al., 1986;Kocher et al., 1987) and spontaneous activity in otherwise silent small primary afferent axons (Schaible et al., 1987a,b). This peripheral hypersensitivity of the altered primary afferent can be explained partly by a local release of pro-inflammatory substances, such bradykinin, cytokines or prostaglandins, which activate and sensitize the peripheral nerve ending (Ohuchi et al., 1976; Baccaglini and Hogan, 1983). Early studies of Vane (1971) and Smith and Willis (1971) revealed that agents such as acetylsalicylic acid and indomethacin, which were known to alter the hyperalgesia that occurred secondary to inflammation (e.g., tissue injury), inhibited COX, the enzyme responsible for prostaglandin synthesis. This observation provided a unifying link explaining the ability of these inhibitors, a diverse class of agents called NSAIDs, to normalize the otherwise lowered thresholds (i.e., hyperalgesia). Whereas these agents reversed the lowered threshold observed in the face of local tissue injury, normal nociceptive thresholds in the absence of injury were not affected (Ferreiraet al., 1971; Moncada et al., 1973). Despite this explanation, there were several elements that were inconsistent with the initial hypothesis. Notably, agents such as acetaminophen were poor anti-inflammatory drugs, but were considered active antihyperalgesic agents. Additionally, it was clear that the antipyretic actions of these agents could stem from a central site of thermoregulatory action. Later, it became apparent from studies with supraspinal (Ferreiraet al., 1978) and spinal administration of NSAIDs (Yaksh, 1982) that there were central sites of NSAID action that implicated central prostaglandin synthesis in nociception and hyperalgesia.
In addition to the multiple anatomic sites for potential NSAID action, understanding of the prostanoid synthetic cascade has undergone a revolution in the past decade with the discovery of multiple COX isoforms. Two isozymes of COX have now been cloned and crystallized: COX-1 (Picot et al., 1994) and COX-2 (Kurumbail et al., 1996). Originally, it was believed that COX-1 was expressed constitutively, whereas COX-2 was up-regulated as an immediate-early gene (Kujubu et al., 1991) in response to cellular stimuli, such as hippocampal NMDA receptor activation (Yamagata et al., 1993) or stimulated macrophages after peripheral carrageenan-mediated inflammation (Tomlinson et al., 1994;Katori et al., 1995). However, constitutive expression of COX-2 has been reported in macula densa (Harris et al., 1994) and developing follicles (Sirois and Richards, 1992; Sirois, 1994) as well as brain (Breder et al., 1992, 1995) and spinal cord (Beiche et al., 1996). COX-2 mRNA apparently represents the most common constitutive isoform in the spinal cord, with additional increases in COX-2 levels after adjuvant-induced arthritis. Given these observations, questions arise regarding spinal and peripheral prostanoid synthetic dependence on COX-1, COX-2 or both in association with peripheral tissue injury and inflammation. The individual contribution of these isozymes has not been elucidated because current NSAIDs inhibit both COX-1 and COX-2 (Meade et al., 1993). Newer agents have been developed, however, with observed IC50 values for COX-2 which are 4 to 5 orders of magnitude lower than those for COX-1 (Gierse et al., 1996; Penning et al., 1997). The development of specific COX-2 inhibitors allows investigation of which COX isoform(s) are involved in hyperalgesia. The current study sought to determine the effects of spinal and systemic administration of the NSAID,S(+)-ibuprofen, and a specific COX-2 inhibitor, SC58125, on thermal hyperalgesia evoked in rats by subcutaneous plantar hindpaw injection of λ-carrageenan.
Methods
Animal model.
All experiments were carried out according to protocols approved by the Institutional Animal Care Committee of University of California, San Diego. Male Holtzman-Sprague-Dawley rats (325–400 g; Harlan Industries; Indianapolis, IN) were housed pair-wise in cages and maintained on a 12-hr light/dark cycle with free access to food and water at all times. Studies involving i.p. injections were carried out on naive rats housed under the above conditions. Chronic lumbar i.t. catheters were implanted in rats under halothane anesthesia according to a modification of the procedure described by Yaksh and Rudy (1976). A polyethylene catheter (PE-10) was inserted through an incision in the atlanto-occipital membrane and advanced caudally to the rostral edge of the lumbar enlargement. Studies involving rats with chronic i.t. catheters were carried out 3 to 21 days after implantation, and rats were housed individually after implantation under the same conditions described above. Exclusion criteria were 1) presence of any neurological sequelae, 2) >20% weight loss after implantation or 3) catheter occlusion.
Induction and assessment of hyperalgesia.
To induce a state of local inflammation, 2 mg of λ-carrageenan (Sigma, St. Louis, MO; 100 μl of 20% solution (w/v) in physiological saline) was injected subcutaneously into the plantar surface of the left hind paw of the rat at time zero (T = 0). To assess the thermally evoked paw-withdrawal response, a commercially available device modeled after that described by Hargreaves et al. (1988) was used (George Ozaki, UARDG, Department of Anesthesiology, University of California, San Diego; La Jolla, CA). Specifics of device construction and operation have been published previously (Dirig and Yaksh, 1995; Diriget al., 1997). This device consisted of a glass surface on which the rats were placed individually in Plexiglas cubicles (9 × 22 × 25 cm). The surface was maintained at 25°C by a feedback-controlled, under-glass, forced-air heating system. The thermal nociceptive stimulus originated from a focused projection bulb below the glass surface that was manipulated in a two-dimensional axis on ball bearing slides. This apparatus allowed the stimulus to be delivered separately to either hind paw of each test subject with the aid of an angled mirror mounted on the stimulus source. A timer was actuated with the light source, and latency was defined as the time required for the paw to show a brisk withdrawal as detected by photodiode motion sensors that stopped the timer and terminated the stimulus. A potentiometer was used to control the amperage delivered to the light source and, thereby, the intensity of the stimulus. An amperage of 4.8 Amps was used throughout the study, which resulted in a baseline paw withdrawal latency of 11.48 ± 0.37 sec.
Drugs and injection.
All animals were weighed before testing, and i.t. patency was confirmed in chronically implanted animals by injection of 5 μl physiological saline. Animals were allowed 30 min to acclimate to the device before testing. Baseline latencies were assessed 15 min before carrageenan injection (T = −15). The following drugs were used in this study: S(+)-ibuprofen, R(−)-ibuprofen and SC58125. All drugs were dissolved in 100% dimethyl sulfoxide at such concentrations to deliver the i.t. dose in a total volume of 10 μl by means of a threaded-barrel Hamilton syringe over approximately 1 min, followed by a 10-μl saline catheter flush. Drugs for i.p. delivery were dissolved in the same vehicle at such concentrations that the dose was delivered in a volume of 0.3 ml.
Test paradigm.
Animals with or without lumbar i.t. catheters were assigned randomly to one of several treatment groups. Drugs were injected (i.t. or i.p.) either 10 min before (pretreatment) or 170 min after (post-treatment) carrageenan injection. Paw-withdrawal latencies then were assessed for both the injected (ipsilateral) and uninjected (contralateral) paws every 30 min for 240 min, starting 60 min after carrageenan injection.
For control comparisons, a separate set of animals received subcutaneous paw saline for comparison with carrageenan effects, and another set received subcutaneous carrageenan in the absence of any i.t. or i.p. injections. In the interest of animal conservation, the i.p. vehicle control group received 0.3 ml of vehicle at both 10 min before and 170 min after intraplantar carrageenan. Each animal was used once and immediately sacrificed according to protocol after the experimental time course.
Statistics.
Area under the curve was calculated by use of the trapezoidal rule over the entire time course (baseline, 240 min) for both contralateral and ipsilateral paw-withdrawal latencies. Based on the duration of drug action and the time of post-treatment drug administration, AUC for the ipsilateral hindpaw-withdrawal latencies was split into two bins at the 180-min time point. The ipsilateral AUC from baseline to 180 min postcarrageenan (AUC180) was compared across groups receiving vehicle or drug injection before carrageenan, and ipsilateral AUC from 180 to 240 min after carrageenan (AUC240) was compared for those experimental groups receiving vehicle or drug injection 170 min after carrageenan. The ipsilateral AUC values for various drug and vehicle groups were compared by one-way analysis of variance, and Scheffe’s post hoc correction for multiple comparisons was applied based on its tolerance of different sized experimental groups and conservative pair-wise comparison of all groups.
Results
Carrageenan and vehicle effects.
Within 60 to 90 min after carrageenan injection, the injected paw displayed swelling and erythema. The rats displayed a guarding of the injected paw but would allow it to rest in contact with the 25°C glass surface. If the glass surface was maintained at 30°C, carrageenan-injected animals repeatedly lifted the injected paw and remained agitated, so 25°C glass temperatures were used throughout the study.
Baseline latencies for all experimental groups before carrageenan or drug administration were not different, with a basal withdrawal latency of 11.5 ± 0.4 sec (n = 25; mean ipsilateral hindpaw-withdrawal latency for all vehicle control groups). Contralateral (uninjected) hindpaw-withdrawal latencies remained constant at basal levels for the entire experiment.
In all groups receiving carrageenan injections, withdrawal latencies of the injected paw (ipsilateral) were decreased significantly by 60 min after carrageenan injection to a mean latency of 4.2 ± 0.6 sec (n = 5). This effect was observed in all groups receiving carrageenan injections, and the AUC180for i.p. vehicle and non-drug-injected control groups were not different. There was a modest, but significant (P < .05), increase in AUC180 for the i.t. vehicle group, but latencies still were decreased to 4.5 ± 0.6 sec by 90 min after carrageenan injection (see fig. 1). This thermal hyperalgesia was an effect specific to subcutaneous carrageenan, because subcutaneous physiological saline injection to the paw did not decrease ipsilateral latencies. Besides the mild activation evoked by vehicle, no changes in motor tone or other behavioral effects were observed after i.t. or i.p. administration of vehicle either before or after carrageenan injection. Vehicle-injected animals were agitated and vocalized immediately after injection, but this subsided within 1 min after injection. These effects were transient, and animals displayed a classic carrageenan response (Hargreaves et al., 1988) which was similar to that observed in untreated animals.

Mean latencies of ipsilateral (ipsi; injected) hindpaw-withdrawal latencies are plotted across the 4-hr experimental time course (n = 4–9 ± S.E.M. per experimental group) after paw injection of carrageenan atT = 0. Contralateral PWL (contra; open squares) are plotted to illustrate that latencies of the uninjected hindpaw as well as the saline-injected hindpaw (open circle) did not vary from baseline (T = −15). After paw carrageenan injection, ipsilateral PWLs were decreased significantly from base line by 60 to 90 min in groups receiving i.t. vehicle injection (closed squares), i.p. vehicle injection (closed triangles) or no vehicle administration (closed circles).
Spinal COX inhibitor effects.
As shown in figure2, a dose-dependent blockade of thermal hyperalgesia was observed after i.t. administration of both COX inhibitors. S(+)-ibuprofen, injected 10 min before carrageenan (T = −10), blocked the development of thermal hyperalgesia in a dose-dependent manner for approximately 3 hr after carrageenan injection. R(−)-Ibuprofen, at the highest effective dose of the S(+)-isomer, did not change paw-withdrawal latencies relative to vehicle controls. The AUC180 for S(+)-ibuprofen (80 nmol) was elevated significantly relative to the vehicle controls (P < .01) as well as the 27-nmol S(+)-ibuprofen dose (P < .05). Intrathecal administration of SC58125 (50 nmol) also significantly elevated AUC180 relative to the vehicle control (P < .05; see fig. 6).

As in figure 1, the time course of ipsilateral (ipsi) hindpaw withdrawal latencies are plotted over time. Contralateral (contra) PWLs of animals receiving i.t. vehicle (closed circles) are drawn for comparison and did not vary from baseline. All animals (n = 4–9 ± S.E.M. per experimental group) received i.t. injections at T = −10 (I, arrow) and paw carrageenan injection at T = 0 (C, arrow). The top panel displays the effects ofS(+)-ibuprofen (Ibu) at i.t. doses of 80 (open squares), 27 (closed squares) and 8 nmol (closed triangles), which dose-dependently blocked the decrease in PWL as compared with the ipsilateral PWL after i.t. vehicle administration (open circle). The less active isomer, R(−)-ibuprofen (open triangles), did not change PWLs with respect to vehicle control. The bottom panel presents the similar time course of action of the COX-2 inhibitor, SC58125, at i.t. doses of 50 (open squares), 17 (closed squares) and 5 nmol (closed triangles).

(A) Dose-response curves for i.t. administration ofS(+)-ibuprofen (Ibu) and SC58125 before induction of inflammation, calculated from figure 2 as the area under the curve from 0 to 180 min after paw injection (AUC180). When compared with the mean contralateral paw AUC (dotted lines) and ipsilateral AUC (ipsi; closed squares) calculated from the i.t. vehicle control group, AUC180 was elevated significantly by the maximum doses of both S(+)-ibuprofen (open circles; P < .01, **) and SC58125 (closed circles). The 80- and 27-nmol doses ofS(+)-ibuprofen were significantly different (P < .01, ‡), but the AUC180 forR(−)-ibuprofen (open square) was not different from ipsilateral vehicle control values. (B) AUC180dose-response curves for systemic treatment with the inhibitors before induction of inflammation (recalculated from the data presented in fig.4). Both inhibitors produced significant, dose-dependent increases in AUC180 relative to ipsilateral vehicle control (closed squares; P < .01, **) values, and the middle dose was significantly different from the maximal dose for bothS(+)-ibuprofen (P < .01, ‡) and SC58125 (P < .05, †). (C) AUC values were calculated from 180 to 240 min after carrageenan injection (AUC240), with inhibitors administered systemically 170 min after carrageenan injection. As compared with ipsilateral vehicle values (closed squares), the 10 and 30 mg/kg doses of bothS(+)-ibuprofen and SC58125 significantly increased AUC240 values (P < .01, **), and the 10 and 30 mg/kg doses of S(+)-ibuprofen also were significantly different (P < .01, ‡).
Surprisingly, once thermal hyperalgesia was established, i.t. administration of either inhibitor 170 min after carrageenan injection did not change ipsilateral hindpaw-withdrawal latencies relative to vehicle controls. As shown in figure 3, the maximum effective i.t. doses from figure 2 did not change latencies from the vehicle control group responses when given after thermal hyperalgesia was established.
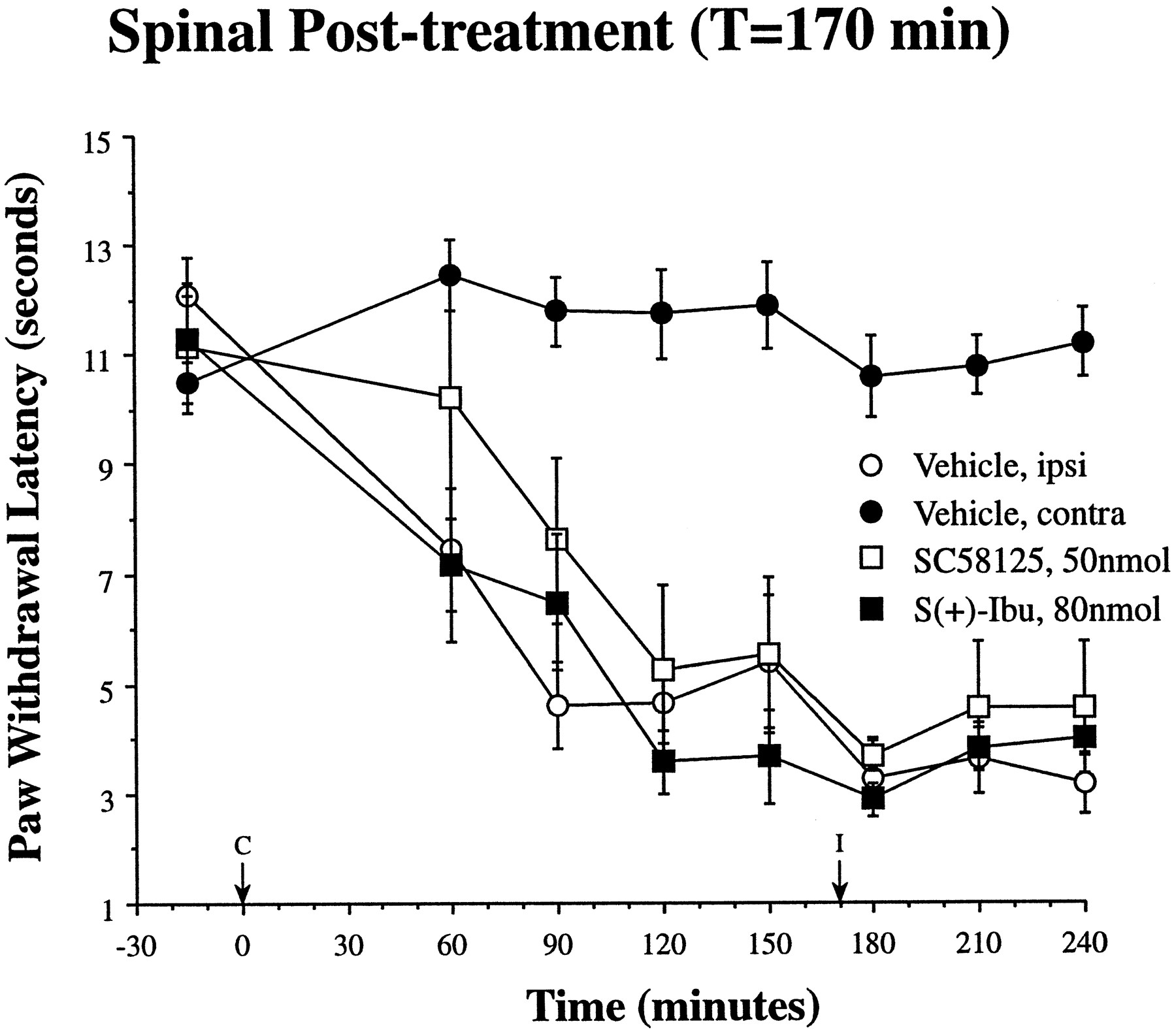
In contrast to the effects of i.t. COX inhibitors administered before carrageenan, the maximum doses of both inhibitors were without effect when administered i.t. once thermal hyperalgesia was established 170 min (I, arrow) after paw carrageenan injection (C, arrow). In comparison with ipsilateral (ipsi; open circles) and contralateral (contra; closed circles) PWL after i.t. vehicle administration (redrawn from fig. 2), the maximum doses ofS(+)-ibuprofen (closed squares) or SC58125 (open squares) did not reverse established thermal hyperalgesia (n = 4–9 ± S.E.M. per experimental group).
Systemic COX inhibitor effects.
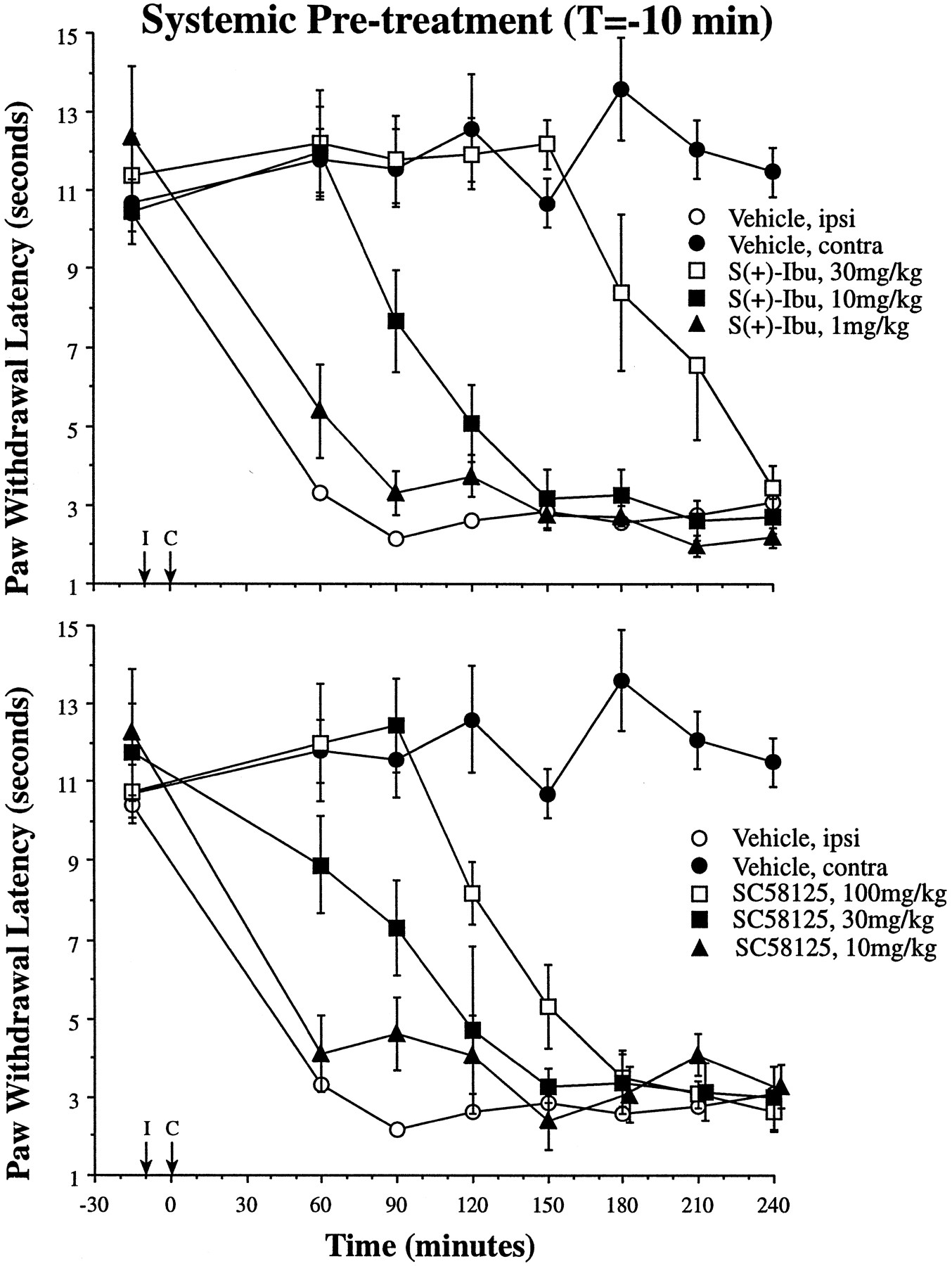
As shown in figure4, i.p. administration ofS(+)-ibuprofen or SC58125 before carrageenan injection (T = −10) blocked the development of thermal hyperalgesia with a duration of action similar to spinal administration. This effect was dose-dependent; AUC180 was increased significantly relative to vehicle control after 10 and 30 mg/kg doses ofS(+)-ibuprofen as well as the 30 and 100 mg/kg doses of SC58125 (P < .01 in each case; see fig. 6). Further demonstrating the dose dependence, the 10 mg/kg S(+)-ibuprofen effect was significantly different from both the 30 mg/kg (P < .01) and the 1.0 mg/kg (P < .05) dosage groups. The same dose dependence was observed with systemic SC58125, where the 30 mg/kg dose was significantly different from the 100 and 10 mg/kg dose (P < .01 in each case). For comparison with the i.t. studies in figures 2 and 3, the maximum effective i.t. doses of both inhibitors were equivalent to a systemic dose of approximately 0.05 mg/kg.

Time courses of hindpaw withdrawal latencies are presented for S(+)-ibuprofen (top) and SC58125 administered i.p. at T = −10 (I, arrow); and paw carrageenan injection was given at T = 0 (C, arrow;n = 4–6 ± S.E.M. per experimental group). Both contralateral (closed circles) and ipsilateral (open circles) PWLs after vehicle administration are drawn for comparison with the 1 (closed triangles), 10 (closed squares) and 30 mg/kg (open squares) doses of S(+)-ibuprofen (top) and the 10 (closed triangles), 30 (closed squares) and 100 mg/kg (open squares) doses of SC58125 (bottom).
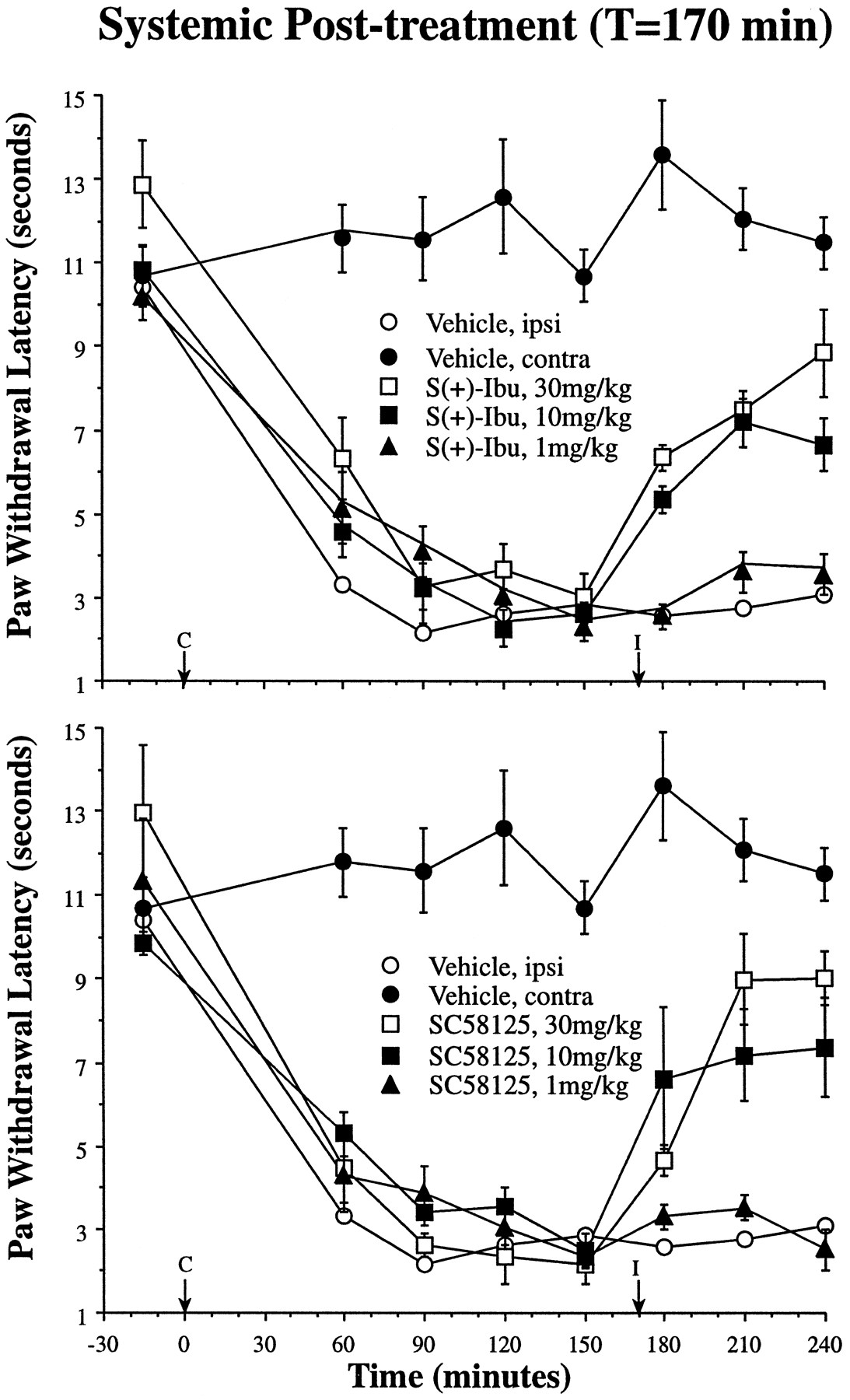
In contrast to the i.t. studies described above, i.p. injection of both agents 170 min after carrageenan injection reversed the carrageenan-mediated thermal hyperalgesia. As shown in figures5 and 6, both S(+)-ibuprofen and SC58125 dose-dependently reversed established thermal hyperalgesia during the last 60 min of the experimental time course (AUC240). This effect was dose-dependent for both S(+)-ibuprofen and SC58125, and the AUC240 was significantly different from vehicle control for both the 10 and 30 mg/kg dose of bothS(+)-ibuprofen and SC58125 (P < .01).

Using the same paradigm as depicted in figure 3, inhibitors were administered i.p. at 170 min (I, arrow) after paw injection of carrageenan (C, arrow). In contrast to the i.t. studies depicted in figure 3, i.p. delivery of inhibitors reversed established thermal hyperalgesia (n = 4–6 ± S.E.M. per experimental group). Intraperitoneal vehicle time courses are redrawn from figure 4, and the dose-dependent reversal of thermal hyperalgesia is shown for 1 (closed triangles), 10 (closed squares) and 30 mg/kg (open squares) doses of both S(+)-ibuprofen (top) and SC58125 (bottom). For comparison with the i.t. dose-response curves depicted in figures 2 and 3, the maximum i.t. doses are equivalent to a systemic dose of 0.05 mg/kg. Ipsi, ipsilateral; contra, contralateral; Ibu, ibuprofen.
Discussion
Central facilitation.
In contrast to the close relationship between stimulus intensity and responses to acute stimuli in the uninjured state, tissue injury and inflammation are associated with hypersensitivity (Raja et al., 1988). This hypersensitivity can be expressed as an increased response and a decreased response latency to a noxious stimulus (hyperalgesia), or a nocifensive response (i.e., pain report or escape attempts) to an innocuous stimulus (allodynia). This altered stimulus-response relationship may be the result of peripheral as well as central mechanisms. It is generally accepted that increased activity in sensory afferent fibers after injury-associated nociceptor sensitization may change the spinal processing of sensory input (for review, see Yaksh, 1993). Thus, peripheral injury or inflammation can generate a state of spinal facilitation in which a moderate stimulus will evoke a profound discharge in dorsal horn nociceptive neurons (Dickenson and Sullivan, 1987; Neugebauer and Schaible, 1988) that depends on spinal NMDA receptor activation. Electrophysiological studies have shown that repetitive C fiber activation can produce activity-dependent alterations and increases in the excitability of spinal cord neurons (Woolf, 1983; Dickenson and Sullivan, 1987). Such facilitated processing of peripheral sensory afferent activity at the spinal cord level would yield an increased ascending neuronal activity (relative to the uninjured state) consistent with what would be a more intense peripheral stimulus (e.g., hyperalgesia) if observed under normal conditions.
λ-Carrageenan and central facilitation.
Plantar injection of λ-carrageenan into the rat hindpaw evokes a local erythema, edema and thermal hyperalgesia, which is assessed as a decrease in paw-withdrawal latencies from a radiant thermal stimulus (Hargreaves et al., 1988) or a decreased threshold to mechanical stimuli (Ferreira et al., 1978). The peripheral component of this hyperalgesic response is apparent from the local edema and erythema and may be caused by the local synthesis and release of inflammatory mediators including bradykinin, cytokines and prostaglandins (Baccaglini and Hogan, 1983; Poole et al., 1995). In fact, this peripheral edema and hyperalgesia can be suppressed by systemic administration of a monoclonal PGE2 antibody, 2B5, in rats (Portanovaet al., 1996) or gene disruption of the prostacyclin receptor in mice (Murata et al., 1997).
Beyond this peripheral role for inflammatory mediators in carrageenan-mediated hyperalgesia, a component of the hyperalgesia is also the result of spinal NMDA receptor activation and increased responsiveness of higher order spinal neurons to peripheral input (i.e., central facilitation). As in other models of central facilitation after peripheral injury, carrageenan-mediated hyperalgesia may be blocked by spinally administered agents that activate opiate receptors (Stanfa et al., 1992), inhibit nitric oxide synthase (Stanfa et al., 1996) or antagonize regulatory sites of the NMDA receptor (Ren et al., 1992a,b). Extending these studies on spinally mediated hyperalgesia and central facilitation, the current study implicates both spinal and nonspinal sites of prostaglandin synthesis (by either COX-2 alone or the combination of COX-1 and COX-2) in the development of thermal hyperalgesia after subcutaneous carrageenan injection. Specifically, different sites and isoforms of COX are involved in the initiation and maintenance of thermal hyperalgesia after tissue injury and inflammation. A clearer conclusion on the isozyme(s) responsible is not possible because S(+)-ibuprofen inhibits both COX-1 and COX-2; definition of the specific isozymes involved awaits future studies with specific COX-1 inhibitors.
Spinal COX inhibition.
Neither systemic nor spinal COX inhibitors had any effect on the thermal escape latency in the uninflamed paw. This agrees with previous studies (Yaksh, 1982;Malmberg and Yaksh, 1992) and emphasizes that COX products do not play a regulatory role in the response to an acute, noxious stimulus, but rather are involved in hyperalgesic responses after tissue injury.
Intrathecal administration of S(+)-ibuprofen or SC58125 before paw injection of carrageenan prevented the development of thermal hyperalgesia for approximately 3 hr. The return of thermal hyperalgesia approximately 3 hr after carrageenan may be proposed to reflect the duration of drug action. We think this unlikely, because the i.t. delivery of even the highest i.t. doses of either inhibitor after thermal hyperalgesia was established (T = 170 min) were ineffective. The data presented in figures 2 and 3 suggest that spinal synthesis of prostaglandins by COX-2 or the combination of COX-1 and COX-2 is necessary for the initiation of thermal hyperalgesia after tissue injury and inflammation, but does not play a role in the maintenance of thermal hyperalgesia induced by carrageenan. The lack of effectiveness of COX inhibitors administered after established inflammation also suggests that there is a limited time in which spinal COX plays a role in the initiation of thermal hyperalgesia after tissue injury. A caveat to this hypothesis is that continuous spinal administration of COX inhibitors would be necessary to fully define the pharmacodynamics of spinal COX activity. Although the results described above do not fully define the duration of action of i.t. COX inhibitors (i.e., only two administration time points), these results are consistent with the hypothesis that there is a limited time frame within which spinal COX inhibition may be effective in blockade of thermal hyperalgesia development.
Systemic COX inhibition.
As with spinal administration of COX inhibitors, systemic (i.p.) administration of either inhibitor before the induction of carrageenan-mediated inflammation blocked the development of thermal hyperalgesia. One interesting observation was that the dose-response curve for SC58125 was shifted to the right, relative to S(+)-ibuprofen, whereas the curves overlap with i.t. or systemic treatment after carrageenan administration. Whether this is a distribution issue is unknown, but the similarity of the time course to the i.t. effects reported above suggests that the data presented in figure 3 may be caused by systemic distribution of the COX inhibitors to a spinal site of action, or that there is a constitutive role for peripheral COX-2 in inflammation. The converse argument, that the effects of i.t. administration were caused by systemic redistribution is unlikely because the maximum effective i.t. dose, if calculated as a systemic dosage, would be approximately 0.05 mg/kg. This dose is 20-fold lower than the lowest, ineffective systemic dose (see fig. 4). Another possible explanation for this right shift in the COX-2 inhibitor dose-response curve after systemic pretreatment is that peripheral COX-1 is playing a primary role in early inflammatory events while low levels of COX-2 are present (i.e., before full induction/expression of peripheral COX-2). Under these conditions, higher doses of the COX-2 inhibitor would be required to produce a behavioral effect (relative to agents that block COX-1). However, after inflammation is fully established and higher COX-2 levels are present, this differential expression and inhibition is no longer present, and the dose-response curves for SC58125 and S(+)-ibuprofen (systemic post-treatment) again overlap.
Systemic treatment with COX inhibitors after establishment of thermal hyperalgesia suggests yet another phenomenon, COX-2 induction. After 3 hr of inflammation, i.t. COX inhibitors were without effect; however, systemic treatment dose-dependently reversed established thermal hyperalgesia. This could have been caused by induction of COX-2 in the periphery or at supraspinal sites, but a spinal site of action was ruled out. Thus, a second conclusion from this study is that a nonspinal (i.e., peripheral or supraspinal) site of prostaglandin synthesis is involved in the maintenance of thermal hyperalgesia. Obviously, a peripheral induction of COX-2 is an attractive hypothesis, and induction of COX-2 expression within 2 to 4 hr after carrageenan-induced pleurisy has been reported previously in mouse macrophages (Tomlinson et al., 1994; Tordjman et al., 1995), but supraspinal sites of action cannot be ruled out by the current study.
Peripheral prostaglandin mechanisms.
The altered stimulus-response relationship observed in the current study as a decreased thermal threshold may have a peripheral and/or central origin. The peripheral hypersensitivity can be explained partly by an injury-induced local release of pro-inflammatory substances, such as bradykinin (Baccaglini and Hogan, 1983) or cytokines (Poole et al., 1995) which can powerfully stimulate the peripheral terminal or prostaglandins, which sensitize pain fibers, producing a left shift and increasing slope of the primary afferent stimulus-response relationship, resulting in a peripherally mediated hyperalgesia (Dray and Perkins, 1993). The effects of systemic COX inhibitors after induction of inflammation (and the lack of spinally administered inhibitors) suggest that a component of the observed thermal hyperalgesia is caused by peripheral synthesis of prostaglandins by either COX-1 and COX-2 or COX-2 alone. The effects of i.t. COX inhibitors suggests additional spinal sources of prostanoids, and whereas the peripheral effects of prostaglandins to sensitize peripheral nerves are well known, spinal mechanisms of prostaglandin action in hyperalgesia associated with tissue injury are less clear.
Spinal prostaglandin mechanisms.
Although i.t. COX inhibitors were ineffective if administered after induction of inflammation, the current study suggests that spinal synthesis of prostaglandins is required for the initiation of thermal hyperalgesia. In vitro studies, with either cell culture or spinal tissue preparations, suggest two possible neuronal effects of prostaglandin receptor activation on central processing and facilitation. PGE2 increases calcium influx in cultured spinal oligodendrocytes (Solivenet al., 1993) and avian sensory neurons (Nicol et al., 1992), and PGE2 also increases tetrodotoxin-resistant sodium influx in rat sensory neurons (Gold et al., 1996). This increased sodium influx is sensitive to the mu opiate receptor agonist, DAMGO ([d-Ala2,(Me)Phe4,Gly(ol)5]enkephalin) (Gold and Levine, 1996). Given the synergy of spinal opiates and NSAIDs in decreasing hyperalgesia after formalin-mediated peripheral tissue injury (Malmberg and Yaksh, 1993), prostaglandins may decrease the threshold for activation of opiate receptor-expressing, primary afferent terminals within the dorsal horn.
Also suggestive of an interplay between sensory system transmitters and prostanoids, PGE2 or PGI2 exposure potentiates the potassium- or capsaicin-evoked release of Substance P from cultured rat sensory neurons (Hingtgen and Vasko, 1994; Hingtgen et al., 1995) and rat spinal tissue slices (Vasko et al., 1993; Vasko, 1995). Substance P can evoke release of PGE2 from rat spinal cord tissue, and both the basal and evoked release from spinal tissue of rats after 24 hr of knee joint inflammation are elevated compared with PGE2 release from the spinal cords of naive rats (Dirig and Yaksh, 1997). A positive feedback loop is suggested from these studies, where increased primary afferent activity after peripheral inflammation sensitizes spinal neurons and increases synaptic glutamate and neuropeptide release from primary afferent terminals. These transmitters act on postsynaptic cells to evoke synthesis and release of prostanoids. Given that prostaglandins can potentiate the evoked release of neuropeptides, prostanoids may feed back on the primary afferent terminal to increase quantal transmitter release from presynaptic terminals. In this fashion, any subsequent action potential that reaches the sensory afferent terminal would result in an increased neuropeptide release, a greater excitation of postsynaptic neurons and increased ascending neuronal activity relative to the peripheral stimulus (i.e., an increased input/output ratio), and thus spinal prostanoid-mediated central facilitation of peripheral sensory input.
In conclusion, the current study suggests that the initiation and maintenance of thermal hyperalgesia after tissue injury may stem partly from prostaglandin synthesis at multiple sites and by multiple COX isoforms. If inhibitors are present before the induction of inflammation, inhibition of prostaglandin synthesis, either systemically or spinally, prevents the development of thermal hyperalgesia. This was evident for both the specific COX-2 inhibitor, SC58125, as well as the mixed COX-1/COX-2 inhibitor,S(+)-ibuprofen. If thermal hyperalgesia was allowed to develop for 3 hr, systemic, but not spinal, treatment with either COX inhibitor reversed thermal hyperalgesia in a dose-dependent manner. These data suggest a tonic role for both COX isozymes in the development of thermal hyperalgesia after tissue injury, with the probable secondary peripheral induction of COX-2 at the site of injury. Further studies are needed to conclusively differentiate COX-1 and COX-2 effects as well as to address the possible supraspinal role of COX-2 in thermal hyperalgesia associated with tissue injury.
Footnotes
-
Send reprint requests to: Tony L. Yaksh, Ph.D., University of California, San Diego, Department of Anesthesiology, 9500 Gilman Drive, Mail Code 0818, La Jolla, CA 92093-0818.
-
↵1 Research supported in part by National Institutes of Health grant NIDA02110 (to T.L.Y.) and Pre-Doctoral National Research Service Award NIDA05726 (to D.M.D.).
-
2 Current address: Searle Research and Development, St. Louis, MO 63198.
- Abbreviations:
- COX
- cyclooxygenase
- COX-1
- cyclooxygenase-1
- COX-2
- cyclooxygenase-2
- NMDA
- N-methyl-d-aspartate
- PG
- prostaglandin
- PGE2
- prostaglandin E2
- PGI2
- prostaglandin I2 (prostacyclin)
- i.t.
- intrathecal
- i.p.
- intraperitoneal
- AUC
- area under the curve
- SC58125
- 1-[(4-methysulfonyl)phenyl]-3-tri-fluoromethyl-5-(4-fluorophenyl)pyrazole
- NSAID
- nonsteroidal anti-inflammatory drug
- PWL
- paw-withdrawal latency
- Received November 10, 1997.
- Accepted February 26, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}