


Abstract
The purpose of our investigation was to characterize the relationships between the pharmacodynamics of synthetic opioids in vivoand the interaction at the mu-opioid receptor. The pharmacokinetics and pharmacodynamics were determined in vivo after a single i.v. infusion of 3.14 mg/kg alfentanil (A), 0.15 mg/kg fentanyl (F) or 0.030 mg/kg sufentanil (S) in rats. Amplitudes in the 0.5 to 4.5 Hz frequency band of the electroencephalogram (EEG) was used as pharmacodynamic endpoint. The EEG effect intensity was related to the (free) concentration in blood (A) or in a hypothetical effect compartment (F, S) on basis of the sigmoidal Emax pharmacodynamic model. The interaction at the mu-opioid receptor was determined in vitro on basis of the displacement of [3H]-naloxone binding in washed rat brain membranes. The value of the sodium shift was used as a measure of in vitro intrinsic efficacy. For the EEG effect the in vivo potencies based on free drug concentrations (EC50,u) were 4.62 ± 0.66 ng/ml (A), 0.69 ± 0.05 ng/ml (F) and 0.29 ± 0.06 ng/ml (S). In the receptor binding studies the affinities at the mu-opioid receptor ( KI) were 47.4 ± 6.6 nM (A), 8.6 ± 4.1 nM (F) and 2.8 ± 0.2 nM (S). For each opioid the ratio between EC50,u and KI was the same with a value of 0.23–0.25, indicating the existence of receptor reserve for the EEG effect. The intrinsic activity (Emax) of the three opioids in vivo was similar with values of 111 ± 10 μV (A), 89 ± 11 μV (F) and 104 ± 4 μV (S). However, the values of the sodium shift varied between 2.8 (S) and 19.1 (A). Further analysis of the in vivo pharmacodynamic data on basis of an operational model of agonism provided evidence for a large receptor reserve, which explains why compounds with different values of the sodium shift all behave as full agonists in vivo.
Synthetic opioids are frequently used in clinical analgesia and anesthesia. Among these opioids important differences in pharmacokinetics and pharmacodynamics exist and this has important implications for the clinical application (Shafer and Varvel, 1991). Currently, a number of new synthetic opioids with more favorable pharmacokinetic and pharmacodynamic properties relative to existing opioids is under development (James, 1994; Rosow, 1993).
In recent years considerable progress has been made in the understanding of opioid receptor pharmacology. Different opioid receptor subtypes have been identified, and it has been demonstrated that opioids may exhibit selectivity with regard to the binding at the various subtypes (Pasternak, 1993). Important differences in the signal transduction pathways between the various receptor subtypes have been observed (Schiller et al., 1992). Furthermore several compounds have been shown to act as partial agonists, indicating that there may be differences in intrinsic efficacy between opioids (Milleret al., 1986; Kajiwara et al., 1986;Berzeteigurske et al., 1995). In several studies the functional role of the different opioid receptor subtypes has been determined (Pasternak, 1993). At present mu-opioid receptors are thought to be responsible for most of the analgesic effects (Mathhes et al., 1996) and some of the other effects such as respiratory depression and sedation (Ling et al., 1983). In clinical investigations the effects of opioids are typically quantified on the basis of rating scales (Shannon et al., 1995). Recently quantitative EEG monitoring has been used successfully to characterize the time course of the pharmacological actions in man after i.v. administration of anesthetic doses. By application of pharmacokinetic-pharmacodynamic modeling it was demonstrated that the value of certain EEG parameters can be directly related to the concentration at a hypothetical effect site on basis of the sigmoidal Emax pharmacodynamic model (Scott et al., 1985;1991; Lemmens et al., 1994; Egan et al., 1996). In this way major differences in potency (i.e., EC50) among the opioids were observed. Furthermore, clear differences in the onset of the effect were observed which could be characterized on the basis of the rate constant keo. The EEG effect was finally shown to occur in a clinically relevant concentration range with regard to the anesthetic effect (Eblinget al., 1990).
Despite the important progress that has been made in both the receptor pharmacology and the clinical pharmacology of opioids, thus far no attempts have been reported to relate the two to each other. In other words the quantitative relationships between receptor binding and pharmacological effect intensity have not been explored in vivo. This is important because the pharmacological actions of a drug are related to the receptor pharmacology in a rather complex fashion. The potency and the intrinsic activity of a drug in vivo are not only dependent on drug-related properties such as affinity to and the efficacy at the mu-opioid receptor but also on tissue related factors such as the efficiency of the receptor-effector coupling and on homeostatic mechanisms that may be operative (Haynes, 1988; Johnson and Fleming, 1989). Recently mechanism based models to characterize the pharmacodynamics of drugs in vivo have been proposed (Tuk et al., 1995; Van der Graaf et al., 1997). In these models the interaction at the receptor has been separated from the multitude of postreceptor events. This separation of drug-specific effect from the tissue specific stimulus-effect propagation is of value to explore and understand changes in pharmacodynamics, caused by factors such as aging, tolerance, disease states and drug interactions and may furthermore be helpful in the design of new compounds.
The purpose of our investigation was to quantitatively explore the relationships between receptor binding and pharmacological effect of synthetic opioids in vivo. Quantitative EEG parameters were used as a pharmacodynamic endpoint, as this provides a continuous and realistic measure of their pharmacologic effect (Cox et al., 1997). The in vivo pharmacodynamic data were analyzed on basis of an operational model of agonism (Black and Leff, 1983) to examine the relationships with the information obtained in the in vitro receptor binding assays.
Methods
Chemicals.
Alfentanil hydrochloride, fentanyl citrate, sufentanil citrate and the internal standard R38527 were donated by Janssen Pharmaceutica BV (Beerse, Belgium). Midazolam was donated by Hoffmann-LaRoche (Basel, Switzerland). Vecuronium bromide was obtained from Organon Technika BV (Boxtel, The Netherlands).
Experimental animals.
Male SPF rats of Wistar descent with a body weight between 250 to 300 g were used in the experiments (Brookman BV, Someren, The Netherlands). The rats were housed individually in plastic cages at constant temperature of 21°C and a controlled light-dark cycle (lights on: 7.00 a.m. to 7.00p.m.). Food (Standard Laboratory Rat Mouse and Hamster Diets, RMH-TM, Hope Farms, Woerden, The Netherlands) and tap water were available ad libitum. One week before the EEG experiment the rats had seven cortical EEG electrodes implanted under fentanyl/fluanisone anaesthesia (Hypnorm, Janssen Pharmaceutica BV, Beerse, Belgium) as described before (Mandema and Danhof, 1990). One day before the experiment, four permanent cannulas were implanted: one in the femoral artery, two in the left jugular vein and one in the femoral vein. The cannulas in the left jugular vein and the femoral vein were used for the administration of alfentanil, midazolam and vecuronium respectively. The cannula in the femoral artery was used for the collection of blood samples. The protocol of the study was approved by the Committee on Animal Experimentation of Leiden University.
Pharmacokinetic-pharmacodynamic experiments.
The pharmacokinetics and pharmacodynamics of the opioids were determined after i.v. administration. Rats were randomly assigned to four different treatment groups of seven to eight rats. Alfentanil (3.14 mg/kg in 40 min), fentanyl (0.15 mg/kg in 20 min) and sufentanil (0.030 mg/kg in 40 min) were administered at a constant rate. A group of control animals received an infusion of 1.5 ml saline for 40 min. To determine the pharmacokinetics of the opioids arterial blood samples (20–1000 μl) were serially collected at fixed time intervals over a period during and after the infusion. Alfentanil, fentanyl and sufentanil blood samples were immediately hemolyzed with 0.5 ml of deionized water and stored at −20°C until analysis.
Two bipolar EEG leads (Cl-Ol and Cr-Or) were continuously recorded using a Nihon-Kohden AB-621G Bioelectric Amplifier (Hoekloos, Amsterdam, The Netherlands) and concurrently digitized at a rate of 210 Hz using a CED 1401plus interface (CED, Cambridge, UK). The digitized signal was fed into a 80486 computer (Inteb BV, Sassenheim, The Netherlands) and stored on hard-disk for off-line analysis. For each 5-sec epoch, quantitative EEG parameters were obtained off-line by fast Fourier analysis using a user-defined program within the data analysis software package Spike 2, version 4.60 (CED, Cambridge, UK). Reduction of EEG data was performed by averaging spectral parameter values over predetermined time intervals.
To prevent opioid-induced seizures which are typically observed upon the administration of these high anesthetic doses, rats received a continuous infusion of midazolam at a rate of 5.5 mg/kg/hr (Coxet al., 1997). To reach steady-state rapidly, midazolam was administered according to a Wagner infusion scheme, with an initial infusion rate at three times the steady-state infusion rate for 16 min (Wagner, 1974). The midazolam infusion was started 30 min before the administration of the opioid. In each of the rats midazolam was able to suppress seizure activity completely.
During and after the infusion of the opioids, severe respiratory depression and muscle rigidity occurred. Rats were artificially ventilated with air using an Amsterdam Infant Ventilator, model MK3 (Hoekloos, Amsterdam, The Netherlands) through a custom made ventilation mask. The ventilation settings were: ventilation frequency 62 beats/min, I-E ratio 1:2 and air supply flow rate 0.7 to 1.0 l/min. When muscle rigidity appeared, the rat received an i.v. bolus injection of 0.15 mg vecuronium bromide and artificial ventilation was started. Administration of vecuronium in a dose of 0.10 mg was repeated each time muscle rigidity reappeared (usually every 5 min) until spontaneous respiratory activity returned and muscle rigidity did not reappear. To determine whether artificial ventilation was adequate arterial pH, -pCO2 and -pO2 values were monitored using a Corning 178 Blood Gas Analyzer (Ciba Corning, Houten, The Netherlands).
During the experiments, body temperature was stabilized between 37.5 and 38.5°C with the aid of Delta Phase Isothermal Heating Pads (Braintree, Braintree, MA) and ventilation with air preheated to 32°C. Body temperature was monitored using a YSI Tele-Thermometer (Yellow Springs Instrument Corporation Inc, Yellow Springs, OH).
Determination of the cerebrospinal fluid over total blood concentration ratio.
The relative free fraction of the opioids in the brain was determined on the basis of the cerebrospinal fluid/total blood concentration ratio in steady-state. For these experiments three groups of seven to eight male Wistar rats were used. The cannulation procedure was as described above. The rats received a steady-state infusion of midazolam as described above. Instantaneous pseudo steady-state concentrations of the opioids were obtained using a computer controlled infusion device. The STANPUMP program (Shafer and Gregg, 1992) was used to clamp blood opioid concentrations at the respective EC50 value, as determined by the pharmacokinetic-pharmacodynamic experiment. Before the start of the opioid infusion, a midazolam infusion was initiated as described above. During the opioid infusion, rats were artificially ventilated and muscle rigidity was managed as described above. Adequateness of artificial ventilation was monitored by measuring arterial pH-, -pCO2- and pO2 levels. Thirty minutes after the start of the opioid infusion, two arterial blood samples of 100 μl were drawn. Simultaneously, a CSF sample was obtained by cisternal puncture. Blood and CSF samples were hemolyzed with 0.5 ml deionized water and stored at −20°C until analysis.
Drug assays.
Blood and CSF concentrations of alfentanil were determined by gas chromatography with nitrogen-phosphorus detection as described previously (Cox et al., 1997). The intra- and interassay variability was generally less than 5% and the lower limit of quantitation was 1 ng/ml for a 0.1-ml sample.
Blood and CSF concentrations of sufentanil were determined using a slightly modified RIA method, as described by Woestenborghs et al. (1994). Hemolyzed blood samples were alkalinized with 1 ml 0.1 N NaOH and subsequently extracted with 5 ml of n-heptane/isoamylalcohol (98.5:1.5, v:v). The organic phase was transferred to a clean tube and subsequently evaporated to dryness under reduced pressure. The residue was then reconstituted in 50 μl methanol and 0.5 ml 2% phosphate buffered bovine serum albumin (BSA) solution. 3H-sufentanil (activity approximately 25,000 dpm) and antiserum were added followed by 2 hr of incubation. Unbound ligand was then precipitated by incubation with 200 μl 2% dextran-charcoal suspension for 1 hr. Supernatant was transferred to scintillation vials containing 4 ml of Packard UltimaGold scintillation fluid (Packard, Meriden, CT). Finally β-activity was measured for 4 min using a Packard Tri-Carb 1500 liquid scintillation analyzer (Packard, Downers Grove, IL). Calibration curves were obtained in duplicate in the range of 0.040 to 10 ng/tube using a blank standard for nonspecific binding and a zero standard for the determination of B0. Activity was expressed as percent binding relative to B0. Relative binding was used to construct a linear logit-log plot. Binding ability (B0) was approximately 33%. Corresponding binding levels were 82 and 10% for 0.040 and 2.0 ng per assay tube, respectively. With the extraction of a blood sample of 1 ml at the end of the experiment sufentanil concentrations of 0.040 ng/ml could be measured. Intraassay variability was 35 and 14% for 0.040 and 2.0 ng per assay tube, respectively.
For the determination of fentanyl concentrations in blood and CSF essentially the same RIA procedure was used as for sufentanil. Binding ability (B0) was approximately 25%. Corresponding binding levels were 89 and 20% for 0.040 and 2.0 ng per assay tube, respectively. With the extraction of a sample of 1 ml at the end of the experiment fentanyl concentrations of 0.040 ng/ml could be measured. Intra assay variability was 37 and 4% for 0.040 and 2.0 ng per assay tube, respectively.
The blood concentrations of midazolam were determined by HPLC with UV detection as previously described (Mandema et al., 1991a). The intra- and interassay variation was less than 6%.
Receptor binding.
Because in our investigation, the emphasis is on in vitro/in vivo correlations in the pharmacodynamics of opiates, the receptor binding characteristics were determined in brain homogenates rather than in cells transfected withmu-opioid receptor. Brain homogenates were prepared according to the method of Lohse et al. (1984). Briefly, rat brain (minus cerebellum) was homogenized in 10 volumes 50 mM Tris-HCl buffer (pH = 7.4) at 25°C. The suspension was centrifuged at 5000 × g for 20 min and the pellet was washed three times with Tris-HCl.
The protein concentration in the homogenate was 2 mg/ml, as determined with the Pierce Micro BCA assay (Pierce, Rockford, IL). Opioid receptor binding was determined by displacement of [3H]-naloxone (New England Nuclear-719, specific activity 57.5 Ci/mmol, New England Nuclear, Boston, MA) at 25°C. Brain homogenate aliquots of 100 μl were incubated with 2.5 nM [3H]-naloxone at various concentrations of the opioids. After 30 min of incubation the samples were filtered through a presoaked glass fiber filter (Whatman GF/B) and eluted six times using 3 ml 50 mM Tris-HCl buffer of 4°C under reduced pressure. The filters were submerged in 3.5 ml Packard UltimaGold scintillation fluid and radioactivity was measured for 4 min by a Hewlett-Packard Tri-Carb 1500 liquid scintillation counter.
In this binding assay the agonistic character of the opioids was evaluated by examining the effect of a high concentration of sodium (100 mM) on the receptor binding affinity. It has been shown that the shift in Ki value is a reflection of the agonist efficacy of the ligand (Pert and Snyder, 1974). The sodium shift was expressed as the ratio of the Ki value in the presence, and in the absence of 100 mM NaCl, respectively. The receptor binding characteristics in the presence of sodium were determined by replacement of 100 μl 50 mM Tris-HCl buffer with 100 μl 400 mM NaCl solution in the incubation mixture.
The receptor binding characteristics of the radioligand [3H]-naloxone ( Kd and Bmax) were determined in a saturation experiment under similar conditions as described above, using various concentrations of the ligand up to 20 nM. Nonspecific binding was determined by calculating the binding of 3H-naloxone in the presence of 10−4 M fentanyl. Free radioligand concentrations were calculated by subtracting the nonspecific binding from the concentrations in the incubation mixture. In the displacement studies the concentration of [3H]-naloxone was equivalent to the Kd value in the saturation experiment. In both displacement and saturation experiments binding was determined in triplicate for each concentration.
Data analysis.
The pharmacokinetics and the pharmacokinetics of the opioids were determined in each individual rat. The blood concentration time profiles were described by a poly-exponential equation:
The sigmoidal Emax pharmacodynamic model was used to describe the relationship between opioid concentration and EEG effect:
The concentration-EEG effect relationship of the three opioids were also simulated according to the operational model of agonism as proposed by Black and Leff (1983):
The pharmacokinetic and pharmacodynamic data were analyzed using the nonlinear least-squares program Siphar version 3.0 (Simed SA, Creteil, France). Pharmacokinetic and pharmacodynamic parameter estimates for the different opioids were statistically evaluated using a one-way ANOVA. In case of non-homogeneity, as determined by Bartlet’s test, the nonparametric Kruskal-Wallis test was used. A significance level of 5% was selected. The receptor binding data were analyzed using the software package GRAPH PRISM, version 2.0 (GraphPad, San Diego, CA).
Results
The changes in the EEG effect (amplitude in the 0.5–4.5 Hz frequency band of the EEG power spectrum) and drug concentrations in the blood as a function of time in four representative rats of the treatment groups are shown in figure 1. During i.v. infusion of the opioids a rapid and pronounced increase in the amplitude of the 0.5 to 4.5 Hz frequency band of the EEG was observed for all drugs. The effect reached a maximum and maintained that level for some time after termination of the infusion. The effect then gradually returned to preinfusion values. The pharmacokinetics of all opioids were most adequately described using a bi-exponential equation. The averaged pharmacokinetic parameters calculated for each opioid in the individual rats are summarized in table1. The clearance of sufentanil was about twice the value obtained for alfentanil. The volumes of distribution at steady-state ranged from 0.75 liter/kg for alfentanil to 5.53 liter/kg for sufentanil. The terminal half-life for alfentanil was considerably smaller than for fentanyl and sufentanil.

Blood concentration (•) and EEG effect (—)vs. time profiles on i.v. infusion of alfentanil (3.14 mg/kg in 40 min), sufentanil (0.030 mg/kg in 40 min), fentanyl (0.15 mg/kg in 20 min) or physiological saline (0.8 ml in 40 min). The solid bars represent the duration of the infusion. The dashed line through the concentration points represents the best fit of data according to the poly-exponential equation.
Pharmacokinetic parameter estimates and CSF/total blood concentration ratio of three opioids in the rat (mean ± S.E.)
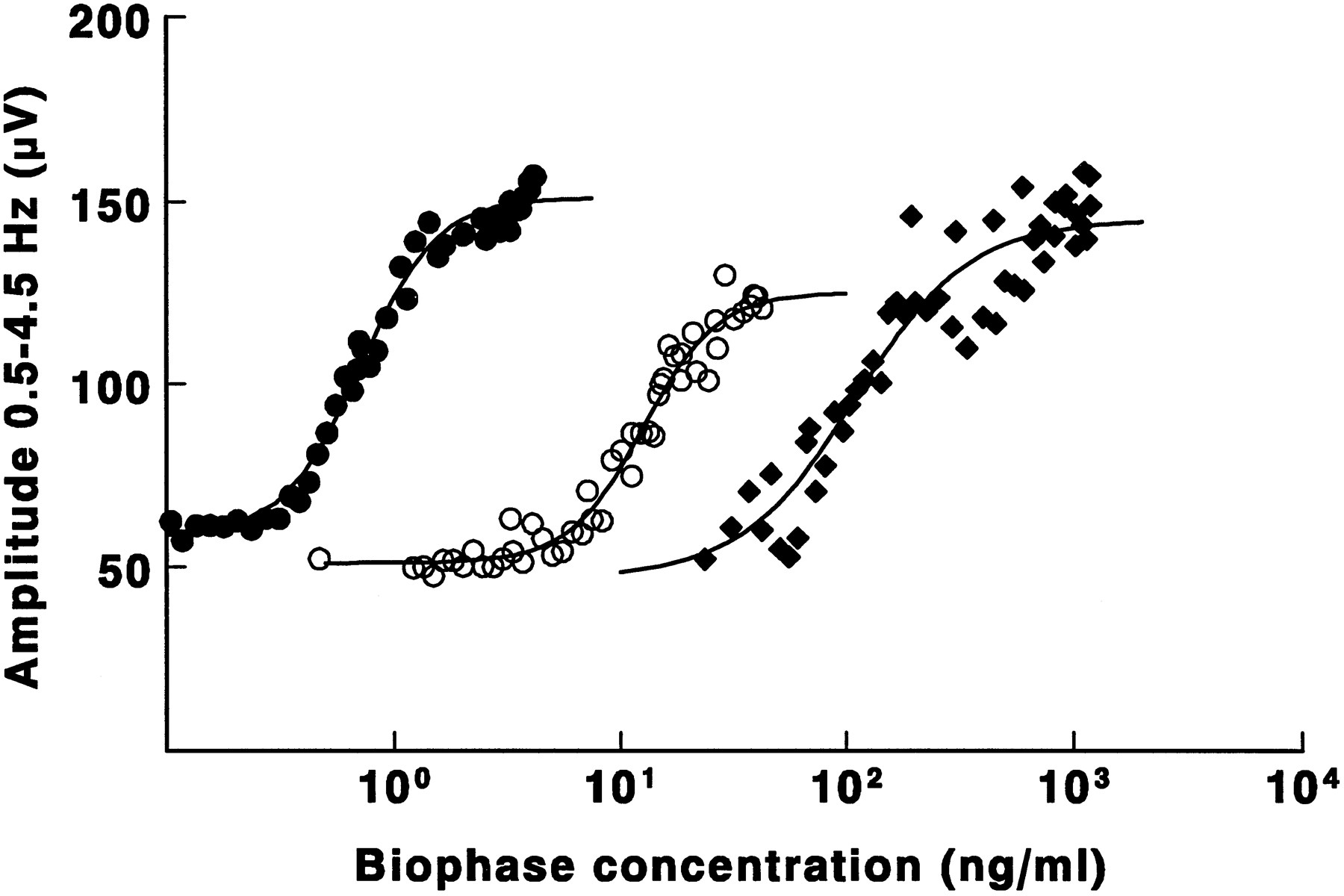
For alfentanil the EEG effect could be related directly to the opioid blood concentrations. For fentanyl and sufentanil profound hysteresis was observed between blood concentrations and EEG effect (fig.2). After minimization of hysteresis, the concentration-EEG effect relationship of the three opioids could be described satisfactorily on basis of the sigmoidal Emaxpharmacodynamic model (equation 2). In figure3 the concentration-EEG effect relationship is shown for the same three individual rats from which the data are shown in figure 1. The averaged pharmacodynamic parameter estimates obtained from the individual rats are represented in table2. Significant differences were observed for the EC50 values of the opioids. Sufentanil (1.43 ng/ml) was the most potent opioid for the EEG effect, and alfentanil (289 ng/ml) was the least potent. No significant differences in Emax, E0 and Hill factor were observed for the three opioids.

EEG effect vs. opioid blood concentration profiles of the opioids. For fentanyl and sufentanil a profound hysteresis loop is observed, whereas for alfentanil hysteresis is absent.

EEG effect vs. effect site concentration plots of alfentanil (⧫), fentanyl (○) and sufentanil (•) in three individual rats. The solid lines represent the best fit through the EEG data according to the sigmoidal Emax pharmacodynamic model.
Pharmacodynamic parameter estimates of three opioids in the rat (mean ± S.E.)
From the start of the opioid infusion midazolam concentrations were constant in all treatment groups with an average concentration of 992 ± 318 ng/ml (mean ± S.D., n = 80).
The determination of the ratio of cerebrospinal fluid/total blood concentrations under steady-state conditions revealed significant differences in the relative free fraction of the opioids in the brain (table 1). The relative free fraction of alfentanil in the brain was considerably lower than those for fentanyl and sufentanil. In general, the measured blood concentrations were in agreement with the concentration levels to which the STANPUMP program was instructed to target, confirming the adequacy of the pharmacokinetic parameter estimates obtained from the pharmacokinetic-pharmacodynamic experiment.
The receptor binding parameters of [3H]-naloxone were Kd = 2.1 ± 0.5 nM and Bmax = 154 ± 28 fmol/mg. The displacement studies with [3H]-naloxone revealed significant differences inin vitro receptor affinity between the different opioids (table 3). Sufentanil showed highest affinity for the mu-opioid receptor ( KI = 2.8 nM), while alfentanil showed the lowest affinity ( Ki = 47.4 nM). In the presence of 100 mM NaCl, Ki shifted to higher values for all opioids. This sodium-shift, expressed in the ratio of the two Ki values, ranged from 2.8 (sufentanil) to 19.1 (alfentanil). In contrast, the receptor binding for the opioid antagonist [3H]-naloxone was not significantly affected: Kd = 1.8 ± 0.3 nM and Bmax = 216 ± 16 pmol/mg protein. A high correlation (r > 0.999) was observed between in vitro mu-opioid receptor affinity (Ki) and in vivo potency (EC50,u) as can be seen from the constant ratio of these two values for each opioid (table 3).
Relationship between in vivo potency (EC50,u) andin vitro receptor affinity (KI) for the three synthetic opioids
The relation between the in vitro and in vivoresults was further investigated by simulations with the operational model of agonism (see “Methods”). Figure4a shows that the model could simulate the concentration-effect curves of alfentanil, fentanyl and sufentanil simultaneously by keeping the agonist-independent parameters (Em and n) constant and assuming that the operational affinity (KA) for each agonist was identical to the Ki value estimated in vitro and that the τ values for each agonist were identical to the sodium shift measured for each agonist multiplied by a constant, agonist-independent factor (4.3). On the basis of the model simulations it can also be understood why all three opioid agonists behaved as full agonists despite the considerable differences in sodium shifts. The simulations in figure 4b show that, due to the high receptor reserve in the system, even compounds with a sodium shift close to unity (i.e., compounds that would behave as almost “silent” agonists in vitro) would still be expected to behave as full agonists in vivo.

a, Operational model of agonism simulations of the self-normalised concentration-EEG effect relationships of the three opioids. The solid lines were simulated using equation 4 with the values for Em (100%) and n (2.3) held constant for all three agonists. The affinity (KA) values were set to theKI values estimated in the presence of NaCl (table 3) and the efficacy parameter (τ) of each agonist was expressed as a constant (4.3) times the sodium shift (table 3). The dashed lines were simulated with the Emax model (equation2) using the parameter values estimated from the experimental data (table 2). b, Operational model of agonist simulations showing the expected concentration-EEG effect relationships on the basis of radioligand binding studies for compounds with constant affinity and different sodium shifts. The lines were simulated using equation 4 with the following parameter values: Em = 100%, n = 2.3, KA = 20 nM and τ = 4.3 × sodium shift.
Discussion
The interaction of 4-anilidopiperidine opioids, such as fentanyl, sufentanil and alfentanil, with the central mu-opioid receptor resulting in analgesic action has been characterized in a number of pharmacological evaluations (Niemeegers et al., 1976; Janssens et al., 1986). Although numerous studies have shown that these opioids interact with the central mu-opioid receptor, the knowledge of the quantitative characteristics of this pharmacological interaction is surprisingly incomplete. Major attention has been focused on receptor affinity whereas the systematic investigation of the agonistic character (potency and intrinsic activity), both in vivo and in vitro, has been rather limited. To our knowledge the comparative intrinsic activitiesin vitro of alfentanil, fentanyl and sufentanil have only recently been reported by James et al. (1991) using the guinea pig ileum assay. Insight in the fundamental pharmacology can provide a suitable basis for the development of new synthetic opioids with both pharmacokinetically and pharmacodynamically superior characteristics for use in clinical anesthesia (James, 1994). Also the systematic investigation of factors that affect the pharmacodynamics of these opioids, such as the development of functional tolerance and the pharmacodynamic interaction with other drugs, will be possible in a mechanistic way. Integrated pharmacokinetic-pharmacodynamic modeling is therefore a useful approach to characterize the in vivopharmacodynamics of synthetic opioids. This approach, in conjunction with in vitro receptor binding characteristics may reveal relationships that provide insight in the underlying fundamental pharmacology of these opioid effects.
Quantitative EEG monitoring has been proposed as an effect measure that is continuous, sensitive, objective and reproducible. As such it possesses the most important characteristics of the ideal pharmacodynamic parameter needed for pharmacokinetic-pharmacodynamic modeling. Also, the EEG effect occurs in a clinically relevant concentration range encountered in anesthesia and can be obtained in both animals (Cox et al., 1997) and man (Scott et al., 1985, 1991).
In our study the concentration-EEG effect relationship of the three opioids could be characterized on the basis of the sigmoidal Emax pharmacodynamic model. This resulted in estimates ofin vivo intrinsic activity (Emax) and potency (EC50) as shown in table 2. An important question is to what extent the values of the pharmacodynamic parameters have been affected by the fact that the rats had also received a single dose of fentanyl during the surgical implantation of the EEG electrodes, 1 wk before the experiment. At present this question cannot be answered. However, an important fact is that the pretreatment has been identical in all rats. This means that a comparison of the pharmacodynamics of the three different opioids is indeed justified. No differences in Emax were observed between the three opioids, indicating a similar intrinsic activity for the compounds. Similar intrinsic activities for the three opioids have also been observed in the guinea pig ileum assay (James et al., 1991). Comparison of the obtained EC50 values, however, resulted in differences inin vivo potency based on total blood concentrations. Sufentanil was approximately 10 and 200 times more potent than fentanyl and alfentanil, respectively.
Differences in the relative free fraction of the opioids in the brain were corrected for by the determination of the CSF over total blood concentration ratio. To obtain equilibrium between the concentration in the blood and in the brain, the steady-state concentration of the opioid in the blood was maintained for 30 min before this ratio was determined. This time appears to be sufficient to reach equilibrium of the drug concentrations in cerebrospinal fluid and blood, since the values of t1/2,keo range between 0 and 4.2 min for the different opioids that have been studied (table 2).
As can be seen from table 1, considerable differences were observed in the relative free fraction of the opioids in the brain, ranging from 1.6% for alfentanil to approximately 20% for sufentanil. These differences indicate the need to correct EC50 values for differences in relative free fraction of the opioids in the brain to reliably compare the in vivo potency of the different drugs. After this correction, differences in in vivo potency between the three opioids were still observed. Sufentanil showed the highest potency of the three opioids and alfentanil the lowest potency. When these in vivo potencies were compared with in vitro binding affinity constants for the centralmu-opioid receptor, as determined by [3H]-naloxone displacement studies, a high correlation between the two parameters was obtained (table 3). Although [3H]-naloxone is not particularly selective formu-opioid receptors, the synthetic opiates that were studied are known to the very selective in the concentration ranges tested. The findings from our investigation justify therefore the conclusion that the EEG effect of synthetic opioids results from their direct, reversible interaction with the central mu-opioid receptor. This is further supported by the fact that the KI values for the three opioids obtained from the displacement studies were in the similar concentration ranges as obtained previously (Leysen et al., 1983; Ilien et al., 1988; Clark et al., 1988; Maguire et al., 1992; Hustveit, 1994). Similar correlations between in vivo potency and in vitro receptor affinity have also been reported for the CNS effects of benzodiazepines (Mandema et al., 1991b) and for the cardiovascular effects of adenosine agonists (Mathot, 1995). Interestingly, the EC50,u values obtained for the three opioids in the EEG model are essentially similar to the respective IC50 values obtained in the guinea pig ileum assay (James et al., 1991). It has been shown that the effect of opioids on the guinea pig ileum is mediated through themu2-opioid receptor subtype (Gintzler and Pasternak, 1983). It is possible that the EEG effects of these opioids are also mediated through mu2-opioid subtype receptors in the CNS. It requires additional investigations to confirm this.
For each ligand the EC50,u value was about 4-fold lower than the KI value in the absence of NaCl. Thus, a maximum pharmacological response of the opioids is already observed when the available opioid receptors are not fully occupied, indicating the existence of a receptor reserve. It cannot be excluded that the opioids have different intrinsic efficacies, but that this is masked by a considerable receptor reserve. It has been shown that the KI value for opioids increases in the presence of sodium ions, whereas the KI values for antagonists are unaffected or even decreased in the presence of sodium (Pert and Snyder, 1974). Sodium shifts among opioids agonists, with values ranging from 2 to 140, have been reported by Kosterlitz and Leslie (1978). The value of the sodium shift may therefore be a measure of intrinsic efficacy of the ligand. From table 3 it can be seen that in the presence of 100 mM NaCl the KI values for the opioids increased significantly. This depressant effect differed widely with values between 2.8 and 19.1 for sufentanil and alfentanil, respectively. The value of 2.2 for the sodium shift of sufentanil in our study is in agreement with the value reported by Leysen et al. (1983). It is of interest to investigate if the sodium shifts of the opioid ligands in combination with the in vivopharmacodynamic estimates of the opioids obtained from the EEG model would be able to predict the efficacy of each ligand. The concentration-EEG effect data were therefore simulated according to the operational model for pharmacological agonism as proposed by Black and Leff (1983). As can be seen from figure 4a, this resulted in concentration-effect relationships for the opioids that adequately described the concentration-effect relationship obtained with the sigmoidal Emax pharmacodynamic model. The simulations showed that the sodium shift measured in vitro provided an accurate prediction of the expression of efficacy in vivo, that is τ could be expressed as the product of sodium shift and an agonist-independent constant. In the operational model of agonism, τ is given by the ratio of the total receptor concentration ([R0]) and the midpoint location of the transducer function (KE) which relates agonist-occupied receptor concentration to pharmacological effect (Black and Leff, 1983). Because [R0] is specific for the tissue and therefore ligand independent, differences in τ are likely to reflect differences in KE values between the opioid ligands. Furthermore, the model predicts that on the basis of the sodium shift it will not be possible to detect a ligand that will behave as a partial agonistin vivo, because a sodium shift close to unity will still result in the expression of maximal possible intrinsic activity (fig.4b).
Another interesting issue is the relationship between our findings in rats and the pharmacological properties in humans. In our study, profound hysteresis was observed for both fentanyl and sufentanil. To derive the effect-site concentration-EEG effect relationship, the hysteresis was minimized successfully using a parametric approach as described by Sheiner et al. (1979).
Concentration-EEG effect relationships of synthetic opioids as obtained in our study have also been obtained in humans (Scott et al., 1985, 1991). Interestingly, the EC50 values obtained for each opioid was in the same concentration range in both rats and humans. Also the three opioids showed equal intrinsic activity (Emax) in both species. Unfortunately, no free drug concentrations were obtained in the study by Scott et al.(1985, 1991) and therefore no direct comparisons could be made between the EC50,u value of the two species. Interestingly, the relative magnitude of hysteresis of the three opioids observed in rats in the present study are essentially similar to those observed in humans. In humans fentanyl and sufentanil show remarkable hysteresis in the plasma concentration-EEG effect relationship (t1/2,keois 6.6 and 6.2 min, respectively), whereas hysteresis for alfentanil is substantially smaller, resulting in a t1/2,keo of 1.1 min (Scott et al., 1985, 1991). The similarities suggest that identical physicochemical and/or physiological processes are involved in the hysteresis phenomenon in both species.
The pharmacokinetic parameter estimates showed significant differences between the three opioids. The systemic clearance was the highest for sufentanil (77 ml/min/kg) approaching total hepatic blood flow (Flaimet al., 1984) and the lowest clearance was observed for alfentanil. However, alfentanil portrayed the lowest volume of distribution at steady-state, and sufentanil showed the largest volume of distribution. Slightly different values for total body clearance and volume of distribution at steady-state for alfentanil and fentanyl were observed, however, by Björkman et al. (1993). These differences may be explained by differences in rat strains used.
From the results of this study it can be concluded that there is a close correlation between the pharmacodynamics of synthetic opioids at the mu-opioid receptor in vitro and the effect on the amplitudes of the 0.5–4.5 Hz frequency band of the EEG in vivo. Mechanism-based modeling of the pharmacodynamics of synthetic opioids may therefore be a useful approach in the design of new compounds, and provides the scientific basis for the extrapolation from preclinical to clinical investigations of the pharmacodynamics of opioids.
Acknowledgments
The authors are grateful for the technical assistance of Erica Tukker and Mariska Langemeijer with the animal surgery and of Jacobien von Frijtag Drabbe Künzel with the receptor binding studies. We also thank Dr. Ad P. IJzerman and Professor Douwe D. Breimer for critically reading the manuscript. The STANPUMP program was kindly provided by Dr. S. L. Shafer, Department of Anesthesia, Stanford University, Palo Alto, CA. The generous donation of the opioids by Janssen Pharmaceutica (Beersse, Belgium) and of midazolam by Hoffman LaRoche (Basel, Switzerland) is highly appreciated.
Footnotes
-
Send reprint requests to: Dr. Meindert Danhof, Leiden/Amsterdam Center for Drug Research, Division of Pharmacology, University of Leiden, Sylvius Laboratory, P.O. Box 9503, 2300 RA Leiden, The Netherlands.
- Abbreviations:
- PK/PD
- modeling-pharmacokinetic-pharmacodynamic modeling
- EEG
- electroencephalogram
- CNS
- central nervous system
- SPF
- specific pathogen free
- I-E ratio
- inspiration expiration ratio
- pCO2
- partial CO2 pressure
- pO2
- partial O2 pressure
- GC
- gas chromatography
- ID
- internal diameter
- HPLC
- high pressure liquid chromatography
- E0
- no drug effect
- Emax
- maximum drug effect
- EC50
- concentration resulting in 50% of the maximum drug effect
- ANOVA
- analysis of variance
- Cl
- total body clearance
- Vd,ss
- volume of distribution at steady-state
- t1/2,αn
- half-life associated with the nth exponential phase
- RIA
- radioimmunoassay, fu, fraction of drug concentration unbound in the body
- Received July 21, 1997.
- Accepted November 13, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}