


Abstract
The antitumor anthracycline, doxorubicin (DOX), can cause heart failure (HF) upon cumulative administration. Lowering the cumulative dose of DOX proved useful to minimize HF risk, and, yet, there is a growing concern that HF might occur after doses that were thought to be safe. Clinical trials that prospectively address such concerns are lacking. Because HF risk correlates with cardiac exposure to DOX, cumulative doses associated with HF risk were re-explored by modeling the accumulation of anthracycline pools in human myocardium. Ex vivo myocardial samples were used in vitro to simulate DOX rapid infusions. The accumulation of anthracycline pools was measured and incorporated into equations from which a risk versus dose curve was obtained. The experimental curve identified a 5% risk dose that was congruent with a previously reported clinical value (380 versus 400 mg/m2, respectively); however, 1–2% risk occurred after lower doses than reported. Simulations of gain-of-function polymorphism of carbonyl reductase 3, which converts DOX to its poorly diffusible alcohol metabolite, doxorubicinol (DOXOL), expanded anthracycline pools and caused 5% or 1–2% risk doses to decrease to 330 or 180–230 mg DOX/m2, respectively. These data show there is no safe dose of DOX. Diminishing cardiac exposure to circulating DOX may represent a cardioprotective strategy. We show that DOX slow infusions or liposomal DOX, which reduce cardiac exposure to DOX, caused formation of smaller anthracycline pools, did not generate DOXOL, increased the 5% risk dose to 750–800 mg/m2, and prevented HF risk aggravation by carbonyl reductase polymorphism.
Introduction
The anthracycline doxorubicin (DOX) is active in many tumors, but its clinical use is limited by the possible development of heart failure (HF). DOX-related HF risk increases slowly over a range of cumulative doses but increases sharply once a threshold dose is exceeded (Ewer, 2013). Such an exponential relation between cumulative dose and HF risk suggests that low-dose DOX inflicts a damage that equilibrates with defense mechanisms such as enzymatic or nonenzymatic antioxidants, whereas high-dose DOX overwhelms the detoxifying capacity of the heart (Minotti et al., 2004; Menna et al., 2008).
Cumulative doses associated with 5% risk of HF have been reported to provide oncologists and cardiologists with limits for a safe administration of DOX. A mean cumulative dose of 400 mg DOX/m2 was reported to cause 5% risk of HF in adults (Swain et al., 2003). However, much lower doses were shown to cause HF (Limat et al., 2003) or to induce subclinical echocardiographic abnormalities that in the general population are associated with the future occurrence of HF (Drafts et al., 2013). Further concerns are caused by the enzymatic reduction of a carbonyl group in the side chain of DOX, leading to the formation of a secondary alcohol metabolite, doxorubicinol (DOXOL, Fig. 1). DOXOL is many times more toxic than DOX (Minotti et al., 2004). Patients with gain-of-function polymorphism of carbonyl reductase (CBR)3, which converts DOX to DOXOL, may develop HF after low cumulative doses (Blanco et al., 2012). In sum, and for many reasons, there seems to be no safe dose of DOX (Menna et al., 2012). Oncologists or hematologists are raising concerns also about cumulative doses that cause HF in 1–2% of patients.

DOX reduction to DOXOL. DOX is composed of a tetracyclic ring, an aminosugar moiety, and a side chain with a carbonyl group. Two-electron reduction of the side chain carbonyl group converts DOX to its secondary alcohol metabolite, DOXOL.
Molecular mechanisms of cardiotoxicity are multifactorial and only in part understood; they include, among many others, iron-catalyzed oxidative stress, calcium dysregulation, and DNA damage by topoisomerase 2β (Minotti et al., 2004; Zhang et al., 2012). Pharmacokinetic determinants have been elucidated in greater detail. Cardiotoxicity correlates with anthracycline plasma peak concentration (Cmax) and diffusion in the heart (Minotti et al., 2004). Because DOX is incompletely cleared from cardiac tissue (Stewart et al., 1993; Salvatorelli et al., 2012), each infusion generates an anthracycline pool that accumulates in the heart (Minotti et al., 2010). HF probably occurs as the accumulation of anthracycline pools exceeds the defense mechanisms of cardiac tissue. An unbalance between anthracycline accumulation and defense mechanisms may occur any time in the life of cancer patients, sometime years after the last anthracycline dose (Minotti et al., 2010).
Clinical management of patients treated with DOX would greatly benefit from clinical trials that prospectively re-explored dose versus risk relations. Unfortunately, there is a lack of such trials at this point in time. DOX and other anthracyclines are considered as old drugs that do not justify the financial efforts that this kind of study may require. Studies of laboratory animals would be of uncertain exploratory value as they are limited by species- and strain-related differences in the levels of formation of secondary alcohol metabolites (Mordente et al., 2003). Because cardiotoxicity correlates with cardiac accumulation of anthracyclines, we used a translational model of human heart that was tailored to simulate DOX infusions and to characterize the size of infusion-related anthracycline pools. Mathematical models were used to convert the size of anthracycline pools into risk versus dose curves from which 5% risk doses of DOX were extrapolated. This experimental approach was used to answer questions about risk of HF from reportedly safe cumulative doses of DOX.
Materials and Methods
Myocardial Samples.
We used myocardial samples from 27 male patients (median age 73 years, range 44–82) and 15 female patients (median age 68 years, range 52–85) undergoing aorto–coronary bypass grafting or valve replacement. All samples were taken during cannulation of beating nonischemic right atrium for preparation of cardiopulmonary bypass. This was a standard procedure, and all samples were to be routinely discarded by the surgeons; therefore, the patients were not subjected to any unjustified loss of tissue. This protocol was approved by the Ethics Committee of University Campus Bio-Medico of Rome. Written informed consent was obtained from patients.
Myocardial Exposure to DOX.
Myocardial samples were processed to obtain thin myocardial strips (∼0.2 g, ∼10 mm long, ∼2 mm wide). Each strip was placed in 2 ml human plasma and was subjected to simulations of rapid infusions of 60 mg DOX/m2 for treatment of breast cancer (Gianni et al., 1997). Because HF risk depends on plasma anthracycline Cmax, DOX was used at its Cmax value and myocardial exposure was limited to the time interval over which Cmax was reported to decrease by no greater than 50% (time at Cmax). The strips were then placed in anthracycline-free plasma to reproduce systemic anthracycline elimination and to stimulate myocardial efflux of DOX or of DOXOL that had been formed inside the strips (referred to as endogenous DOXOL). Where indicated, Cmax was decreased and time at Cmax was increased to simulate myocardial exposure to DOX slow infusion (El-Kareh and Secomb, 2000) or myocardial exposure to free DOX that circulated in the bloodstream after infusions of uncoated liposomal DOX (Swenson et al., 2003). In all cases the incubation time was limited to 4 hours to ensure that myocardial strips retained their viability (Salvatorelli et al., 2006). Because many tissues express CBR, generate DOXOL, and release it in bloodstream, the strips were also exposed to purified DOXOL to simulate the action of circulating DOXOL (referred to as exogenous DOXOL). Inasmuch as exogenous DOXOL from liposomal DOX or DOX slow infusion remained at Cmax for ≥24 hours (Leca et al., 1991; Swenson et al., 2003), DOXOL time at Cmax was simulated by normalizing 24 hours of exposure in vivo to 4 hours of incubation in vitro. Time at Cmax of DOXOL after DOX rapid infusion was simulated in accordance to such normalization.
Pharmacokinetic details of myocardial exposure to DOX and exogenous DOXOL are reported in Table 1.
Clinically-modeled exposure of human myocardium to DOX and exogenous DOXOL
Adapted from Leca et al., 1991; Gianni et al., 1997; El-Kareh and Secomb, 2000; Swenson et al., 2003.
Uptake, Metabolization, and Retention of DOX.
Myocardial retention of DOX was determined by the sum of the levels of DOX recovery from the soluble and membrane fractions of the strips, whereas anthracycline efflux was determined by the levels of DOX recovery from plasma. Because plasma per se was preliminarily shown to not convert DOX to DOXOL, the recovery of DOXOL from plasma reflected the efflux of endogenous DOXOL from strips in plasma. DOXOL formation was therefore calculated by the sum of DOXOL recovery from strips with DOXOL recovery from plasma, and net values of myocardial DOX uptake were calculated as (DOX retention + DOX efflux in plasma + DOXOL formation). Clearance was determined by the equation [100 × (efflux:uptake)]. In the experiments of strips exposed to exogenous DOXOL, the uptake of DOXOL was determined as (DOXOL retention + DOXOL efflux). Separate determinations of anthracycline uptake and efflux could not be performed when myocardial strips had to be exposed to DOX or DOXOL for 4 hours without plasma change (simulations of DOX slow infusions or exposure to exogenous DOXOL from liposomal DOX or DOX slow infusion).
Assays.
At the end of incubations, the strips were washed with ice-cold 0.3 M NaCl, homogenized in 1 ml same medium, and centrifuged for 90 minutes at 105,000g to separate soluble and whole-membrane fractions. These were extracted with an equal volume of 90:10 methanol:acetonitrile and acidified with 0.2% trifluoroacetic acid. A total of 100 μl supernatant was loaded onto a (100 × 4.6 mm, 2.7 μm) Poroshell 120 EC-C18 column (Agilent Technologies, Palo Alto, CA), which was operated at 25°C and was eluted at the flow rate of 0.8 ml/min for a total 25-minute run time [20-minute linear gradient from (95–5%) to (50–50%) 50 mM sodium phosphate-acetonitrile]. DOX and DOXOL were detected fluorimetrically (excitation at 480 nm, emission at 560 nm), identified by co-chromatography with authentic standards, and quantified against appropriate standard curves. Retention times were 15.3 minutes for DOX and 13.9 minutes for DOXOL. Plasma aliquots were extracted and analyzed by similar procedures. Within-day and between-day coefficients of variation were 2% and 7%, respectively. DOX and DOXOL were obtained through the courtesy of Nerviano Medical Sciences (Milan, Italy).
Quantification of Cardiac Anthracycline Pools.
Cardiac anthracycline pools were quantified by the sum of the net levels of myocardial retention of DOX and DOXOL. The latter were multiplied by 40 to account for the stronger negative effects of DOXOL on systolic contraction and diastolic relaxation as compared with DOX (Boucek et al., 1987; Mushlin et al., 1993). Cardiac pools of DOX were therefore determined by the following formula:

Following quantification of cardiac anthracycline pools, simulations of gain-of-function CBR3 polymorphism were conducted by considering that CBR genetic variants formed ∼twofold higher levels of DOXOL (Blanco et al., 2008, 2012). The levels of DOX were considered to decrease by the amount required to generate excess DOXOL. Myocardial exposure to exogenous DOXOL was also assumed to increase twofold.
Five Percent Risk Pools and Risk versus Dose Curves.
Experimental conditions that mimicked rapid infusion of 60 mg DOX/m2 caused formation of an anthracycline pool of a mean size of 4.7 µM. We assumed that the same anthracycline pool occurred after each subsequent anthracycline infusion. Therefore, the anthracycline pool associated with the 5% risk dose of DOX, 400 mg/m2, was calculated as:
This was taken as the cardiac pool of DOX that caused 5% risk of HF (5% risk pool) and was incorporated into equations that produced risk versus dose curves (see Results).
Data Analysis.
Cardiac anthracycline pools (nanomoles/gram tissue) were normalized to micromolar equivalents on considering that cardiac tissue has a density very similar to that of water (1 g/ml) (Mushlin et al., 1993). Data were means ± S.D. of seven experiments (simulations of liposomal DOX infusion) or nine experiments (DOX rapid or slow infusion). Differences between data sets and curves were analyzed by analysis of variance with Bonferroni’s test for multiple comparisons, Student’s t test, or receiver operator characteristics, as appropriate. All analyses were performed by Prism 5 (GraphPad Software, La Jolla, CA). Other details are reported in the figure legends.
Results
Pharmacokinetics of Different DOX Schedules in Human Myocardial Strips.
Under experimental conditions that mimicked DOX rapid infusion, human myocardial strips incorporated DOX and converted it in DOXOL. DOX underwent partial efflux and clearance, whereas DOXOL did not. Simulations of CBR polymorphisms did not cause significant changes of DOX uptake and retention but caused a twofold increased formation of endogenous DOXOL (Table 2).
Myocardial pharmacokinetics of DOX and endogenous or exogenous DOXOL
Myocardial exposure to anthracyclines and exogenous DOXOL was performed as per Cmax and time at Cmax values reported in Table 1 and Materials and Methods. For exogenous DOXOL, CBR polymorphism was assumed to cause a twofold increased Cmax. Values were means ± S.D. of seven experiments for liposomal DOX infusion or nine experiments for DOX rapid infusion and DOX slow infusion. DOX clearance was significantly higher for liposomal DOX than DOX rapid infusion, and DOX retention was significantly higher for DOX rapid infusion than liposomal DOX (P < 0.001 by two-tailed unpaired Student’s t test).
In other experiments, DOX was used at one-fourth of the concentration adopted for simulating rapid infusions, and myocardial exposure to DOX was prolonged over 2 hours. These conditions were intended to simulate cardiac exposure to DOX in patients infused with uncoated liposomal DOX, a formulation that delivers high amounts of anthracycline in tumors but causes lower plasma Cmax and longer time at Cmax of free anthracycline (Swenson et al., 2003; Minotti et al., 2004). Under such defined conditions, DOX uptake was similar to that of DOX rapid infusion, but clearance was higher and net retention was significantly lower; furthermore, the strips did not generate measurable amounts of endogenous DOXOL.
In a third set of experiments, DOX was used at one-tenth of the concentration adopted for simulating rapid infusions, and myocardial exposure to DOX was prolonged over 4 hours.This was done to simulate DOX slow infusion, which is characterized by very low plasma Cmax and prolonged circulation of DOX (Leca et al., 1991; El-Kareh and Secomb, 2000). Net DOX retention was significantly lower compared with DOX rapid infusion, and again, the strips did not generate measurable levels of endogenous DOXOL (see also Table 2).
We considered that for liposomal DOX or DOX slow infusion, endogenous DOXOL formation was limited by the low levels of DOX retention in the strips. We therefore assumed that CBR polymorphism was unlikely to increase DOXOL formation in these conditions (see Table 2). This assumption was also based on the notion that the CBR3 variant associated with HF risk aggravation exhibited an increased affinity for its cofactor, NADPH, but not for DOX (Lakhman et al., 2005).
Determinants of Endogenous DOXOL Formation.
Because DOXOL is important for DOX pharmacokinetics and toxicity, we characterized reasons it was formed only by rapid infusions. We considered that the reductive metabolization of anthracyclines to secondary alcohol metabolites occurs primarily in cytosol (Salvatorelli et al., 2012); therefore, we characterized whether switching from DOX rapid infusion to liposomal DOX or DOX slow infusion changed anthracycline distribution in the strips and diminished DOX levels in the soluble (cytoplasmic) fraction. DOX levels in the membrane fraction gradually decreased as DOX rapid infusion was replaced by liposomal DOX or DOX slow infusion (Fig. 2A). DOX levels in the cytoplasmic fraction also decreased as DOX rapid infusion was replaced by liposomal DOX; however, DOX levels after DOX slow infusion were apparently identical with those induced by DOX rapid infusion (Fig. 2B).

DOX distribution in human myocardial strips. DOX was measured in membrane and cytoplasmic fractions of human myocardial strips exposed to simulation of DOX rapid infusion and liposomal DOX or DOX slow infusion. (A) *P < 0.05 or **P < 0.001 for DOX rapid infusion versus liposomal DOX or DOX slow infusion, respectively, whereas ***P < 0.01 for liposomal DOX versus DOX slow infusion. (B) *P < 0.001 for DOX rapid or slow infusion versus liposomal DOX. Data were means ± S.D. of seven to nine experiments, as specified in Materials and Methods, and were analyzed by analysis of variance with Bonferroni’s post-test. DOX, DOX rapid infusion; DOX s.i., DOX slow infusion; lipo DOX, uncoated liposomal DOX.
The experiments in Fig. 2 characterized the combined effects of Cmax and time at Cmax on anthracycline partitioning and distribution in the strips. In previous studies, we characterized that for any given length of myocardial exposure to anthracyclines, minor changes of Cmax were independent determinants of major changes of DOX retention or clearance (Salvatorelli et al., 2012). In this work, DOX slow infusion was simulated over 4-hour incubations without plasma change, which precluded determinations of DOX efflux. We modified the experimental conditions and characterized how DOX retention or clearance was influenced by the very low Cmax of DOX slow infusion. Briefly, myocardial strips were exposed to 10, 2.5, or 1 μM, which was similar to experiments of DOX rapid infusion, liposomal DOX, or DOX slow infusion, respectively. After 30 minutes, the strips were placed in anthracycline-free plasma to stimulate total clearance and to calculate the relative contribution of anthracycline elimination from membrane or cytoplasmic fractions. DOX clearance increased as DOX concentration was decreased from 10 to 2.5 or 1 μM. Moreover, anthracycline elimination from the cytoplasmic fraction gradually increased as DOX concentration was decreased, such that the clearance of 1 μM DOX was determined primarily by DOX elimination from the cytoplasmic fraction (Fig. 3).
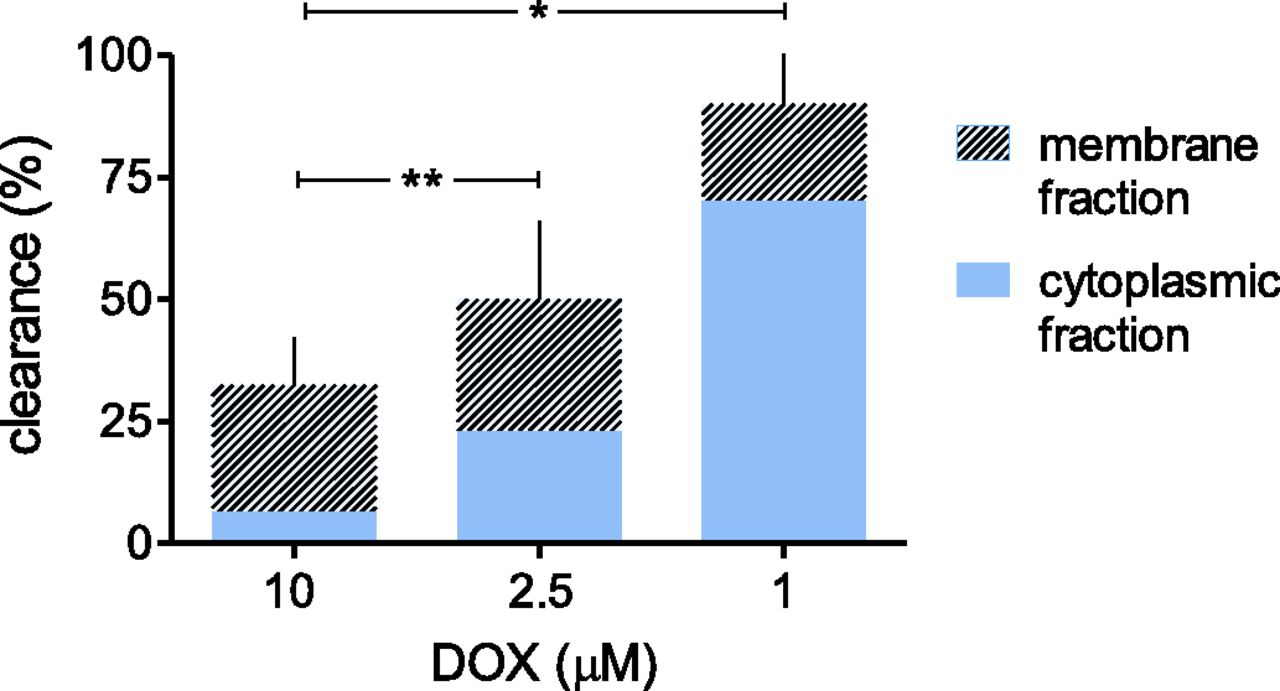
DOX clearance from human myocardial strips and role of DOX elimination from membrane and cytoplasmic fractions. Human myocardial strips were exposed to 10, 2.5, or 1 μM DOX, similar to DOX concentration in simulations of DOX rapid infusion, liposomal DOX, or DOX slow infusion. After 30 minutes, the strips were placed in anthracycline-free plasma to promote anthracycline elimination. DOX clearance was determined as described in Materials and Methods. DOX levels in cytoplasmic or membrane fractions were compared with samples exposed to 1–10 μM DOX for 4 hours to permit a continuous equilibration of DOX across strips and plasma. Net differences for DOX levels in cytoplasmic or membrane fractions from the two sets of strips were expressed as percentages of clearance. Values were means of three to six experiments. *P < 0.001 for clearance of 1 μM versus 2.5 or 10 μM DOX, and **P < 0.05 for clearance of 2.5 versus 10 μM DOX (analysis of variance with Bonferroni’s post-test).
Collectively, data in Figs. 2 and 3 suggested that the cytoplasmic fraction served as an exchange compartment from which DOX moved bidirectionally toward the membrane fraction or back in plasma. In simulations of DOX rapid infusions, an equilibrium was attained and cytoplasmic levels of DOX were high enough to permit DOXOL formation by CBRs. In simulations of liposomal DOX infusions, DOX movements toward the membrane fraction were decreased, whereas DOX movements toward plasma were increased, such that cytoplasmic DOX retention was too low for DOXOL to be formed and for CBR variants to increase its formation. In simulations of DOX slow infusion, DOX movements toward the membrane fractions were nearly abolished, whereas DOX movements toward plasma were remarkably increased. It follows that, in simulations of slow infusions, the cytoplasmic levels of DOX looked similar to those induced by rapid infusions but actually reflected a labile anthracycline pool that was directed primarily toward plasma and did not permit adequate interactions of DOX with cytoplasmic CBRs (Fig. 4).

DOX equilibration across plasma and cytoplasmic or membrane fractions of human myocardial strips. Schematic representation of bidirectional movements of DOX from the cytoplasmic fraction of myocardial strips toward the membrane fraction or plasma. Only rapid infusions allowed DOX to equilibrate across the three compartments and to attain levels permissive to metabolization to DOXOL in the cytoplasmic fraction (indicated by addition of -HOH). See text also for explanations. DOX, DOX rapid infusion; DOX s.i., DOX slow infusion; lipo DOX, uncoated liposomal DOX.
Defective Diffusion of Exogenous DOXOL in Human Myocardial Strips.
Myocardial strips were used to simulate cardiac exposure to exogenous DOXOL that formed after DOX rapid infusion, liposomal DOX infusion, or DOX slow infusion. Exogenous DOXOL never diffused from plasma in strips, not even when DOXOL concentrations were increased twofold to simulate gain-of-function polymorphism of systemic CBRs (see also Table 2). The values of partition coefficient (−0.03 for DOXOL versus 0.53 for DOX), and especially of distribution coefficient (−0.69 for DOXOL versus 0.1 for DOX), demonstrate that DOXOL is significantly more polar than DOX (ChemAxon, 1998–2017, https://chemicalize.com). An increased polarity may limit diffusion of exogenous DOXOL from plasma in strips, and probably explains also the complete myocardial retention of endogenous DOXOL as opposed to the partial efflux of DOX.
Cardiac Anthracycline Pools.
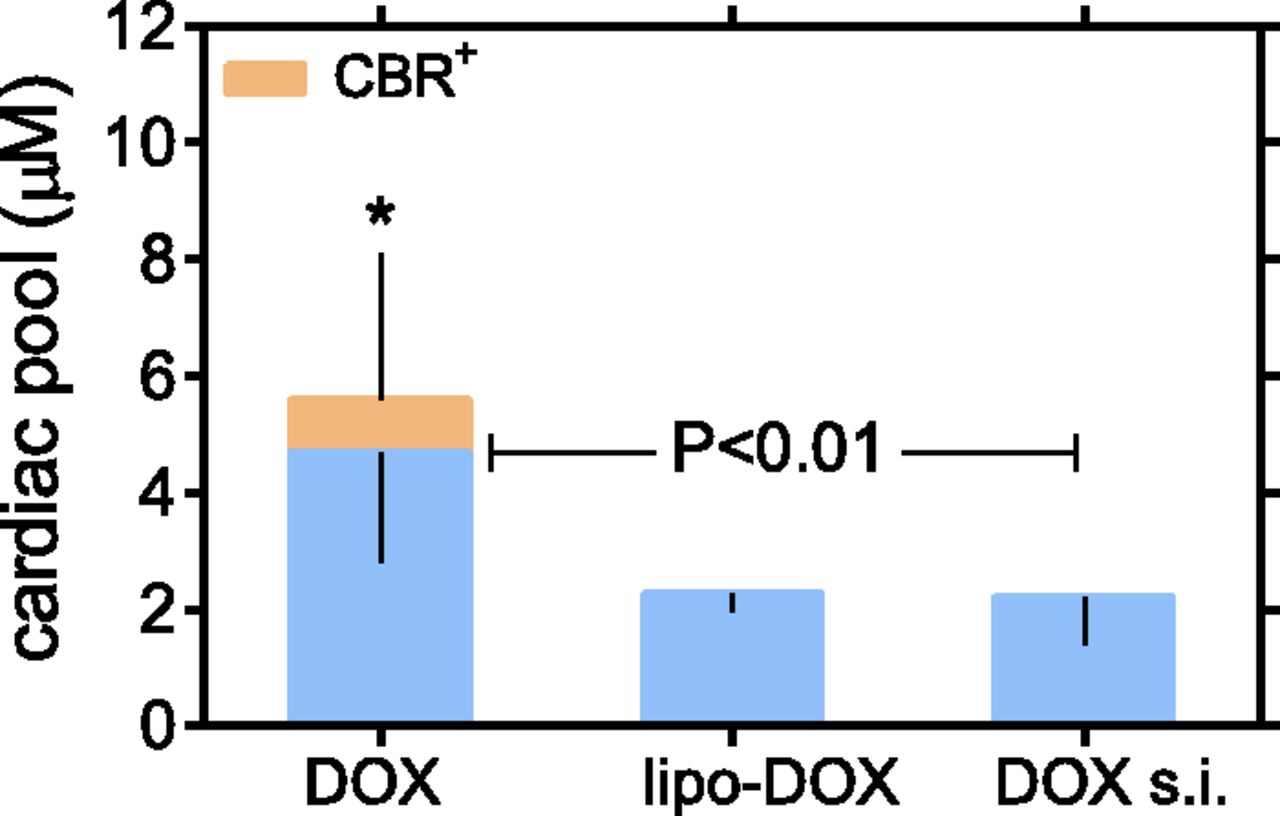
Having characterized that exogenous DOXOL did not partition from plasma to strips, infusion-related cardiac anthracycline pools were determined by the sum of the net levels of myocardial retention of DOX and endogenous DOXOL. The size of anthracycline pools was calculated by correcting the levels of endogenous DOXOL for its 40-fold higher toxicity as compared with DOX. The anthracycline pool induced by DOX rapid infusion was significantly larger than those induced by liposomal DOX or DOX slow infusion, and was the only pool that increased under conditions of CBR polymorphism (Fig. 5).

Cardiac anthracycline pools. Cardiac anthracycline pools were quantified as described in Materials and Methods. Regardless of CBR polymorphism, the pool induced by DOX rapid infusion was significantly larger than those induced by liposomal DOX infusion or DOX slow infusion (P < 0.01 by analysis of variance with Bonferroni’s post-test). *Denotes that CBR polymorphism significantly augmented the cardiac pool from DOX rapid infusion (P = 0.037, paired Student’s t test). CBR+, CBR polymorphism; DOX, DOX rapid infusion; DOX s.i., DOX slow infusion; lipo-DOX, uncoated liposomal DOX.
Developing a Risk versus Dose Curve for DOX Rapid Infusion.
The exponential relation between HF risk and anthracycline doses meets the criteria for the broad group of mathematical functions of the type (y = x2), where y can be equated with HF risk and x2 can be equated with the number (n) of DOX infusions. This type of function must incorporate a correction constant (c) that is specific to DOX (Ewer, 2013), according to the following eq. 1:
 (1)
(1)To adapt eq. 1 to the relation between HF risk and cardiac anthracycline pools, we considered that for an HF risk of 5% n2 was given by the second power of the ratio of 5% risk pool to the size of the anthracycline pool induced by a single infusion:

Both the 5% risk pool and the post-infusion pool were known, which allowed to extrapolate c from eq. 1 and to calculate a value of 0.113. We next considered that the destructive force of anthracycline pools increases exponentially as the cumulative dose of DOX approaches and exceeds the 5% risk dose of 400 mg/m2 (Ewer, 2013). Equation 1 was therefore corrected by a factor that normalized the destructive force of any given dose of DOX to the destructive force of the 5% risk dose. This factor was expressed by the second power of the ratio of pooln to 5% risk pool, where pooln was the anthracycline pool induced by n infusions. Risk of HF was therefore calculated by the following eq. 2:
 (2)
(2)Inasmuch as the infusion dosage was known (see Table 1), n was converted into mg DOX/m2 and eq. 2 was used to build a risk versus dose curve.
Equation 2 produced an exponential curve that was congruent with the curve derived from three clinical trials of DOX (Swain et al., 2003); however, receiver operator characteristics analyses showed that slopes were slightly, but significantly different for doses of ≤400 mg DOX/m2. In fact, the experimental curve identified an excess risk of HF from doses between 200 and 400 mg/m2 (Fig. 6).

Equation-built risk versus dose curve of DOX. (A) Shows the risk versus dose curves derived from clinical trials (Swain et al., 2003) or obtained experimentally from eq. 2 in the text. The two curves were different for doses of <400 mg DOX/m2 (receiver operator characteristics analysis). (B) Shows that the experimental curve identified HF excess risk over a dose range between 200 and 400 mg DOX/m2 (receiver operator characteristics analysis over 50 mg increments).
Risk versus Dose Curve for Liposomal DOX and DOX Slow Infusion.
Equation 2 was used to build risk versus dose curves also for liposomal DOX and DOX slow infusions, and for all DOX conditions in simulations of CBR polymorphism. This required that correction constants be determined for each condition (Table 3). Both liposomal DOX and DOX slow infusion showed exponential risk versus dose curves; however, these were shifted rightward as compared with DOX rapid infusion, thus demonstrating that liposomal DOX and DOX slow infusion caused HF risk at higher cumulative doses. Simulations of CBR polymorphism caused a leftward shift of the curve of DOX rapid infusion, which denoted an increased HF risk at low cumulative doses. In contrast, simulations of CBR polymorphisms caused no effect on risk versus dose curves of liposomal DOX or DOX slow infusion (Fig. 7).
Correction constants for equation-based risk versus dose curves
Correction constants were obtained by setting eq. 1 at 5% risk of HF. In the same equation, the number of infusions associated with 5% risk of HF was expressed by the ratio of 5% risk pool to infusion-related pool, as determined for each DOX schedule in absence or presence of CBR polymorphism.

Anthracycline risk versus dose curves and effects of CBR polymorphism. Risk versus dose curves for DOX rapid infusion (A), liposomal DOX (B), or DOX slow infusion (C) were obtained by eq. 2. Dashed lines identify 5% HF risk and 5% risk doses for each experimental condition. CBR+, CBR polymorphism.
Five Percent Risk Doses.
Five percent risk doses were extrapolated from risk versus dose curves (see also Fig. 7). The 5% risk dose of DOX rapid infusion was refined from 400 to 380 mg/m2 and was decreased to 330 mg/m2 by simulations of CBR polymorphism. The 5% risk dose of liposomal DOX or DOX slow infusion was 800 or 750 mg/m2, respectively, and was not decreased by simulations of CBR polymorphism.
Discussion
From Cardiac Anthracycline Pools to HF Risk.
We characterized whether HF risk from DOX could be recalculated on the basis of anthracycline pools that accumulated after pharmacokinetic simulations of DOX infusions. The 5% risk pool, which served to build eq. 2 and risk versus dose curves, was tailored to the dose of DOX that caused 5% risk in patients ≤65 years of age and without pre-existing cardiomyopathy (Swain et al., 2003). Our experiments therefore identify 5% risk doses that apply primarily to low-risk adults. This having been acknowledged, we refined the 5% risk dose of DOX from 400 to 380 mg/m2. Special populations (children, the elderly, patients with cardiovascular disease) would develop 5% risk after lower cumulative doses than 380 mg/m2 (Salvatorelli et al., 2015).
Simulations of DOX slow infusions diminished cardiac accumulation of DOX by 50% compared with DOX rapid infusion and shifted the risk versus dose curve toward the right, such that 5% risk of HF was approximated to 750 mg DOX/m2. These findings denote the advantages of reducing Cmax, which is the main pharmacokinetic characteristic of slow infusions. The risk versus dose curve was shifted to the right also when myocardial samples were exposed to simulations of liposomal DOX infusions. It is worth noting that liposomal formulations cause higher anthracycline Cmax than DOX rapid infusions; however, this is due to the low distribution volume and prolonged plasma half-life of DOX that is sequestered within the liposomal vesicle and eventually extravasates in tumors (Drummond et al., 1999). In this study, we simulated myocardial exposure to that limited amount of free DOX that leaks from the liposomal vesicle in the bloodstream (Swenson et al., 2003). Such defined conditions diminished cardiac accumulation of DOX and increased the 5% risk dose to 800 mg/m2.
Role of DOXOL and HF Risk Aggravation by CBR3 Polymorphism.
DOXOL is many times more potent than DOX at inhibiting ion transporters and channels that govern cardiac contraction and relaxation (e.g., the Ca2+-Mg2+ ATPase and Ca2+ release channel of sarcoplasmic reticulum, the Na+-K+ ATPase and Na+-Ca2+ exchanger of sarcolemma) (Olson and Mushlin, 1990). Moreover, DOXOL is more polar than DOX, does not diffuse through the plasma membrane, and accumulates in cardiomyocytes (Menna et al., 2008). It follows that intramyocardial formation of DOXOL, mediated by cytoplasmic reductases, represents an important determinant of anthracycline cardiotoxicity, as evidenced by the 40-fold higher potency of DOXOL compared with DOX in suppressing systolic contraction and diastolic relaxation in preclinical models (Boucek et al., 1987; Mushlin et al., 1993). In this study, the size of cardiac anthracycline pools was calculated by correcting anthracycline levels for the greater toxicity of endogenous DOXOL. Corrections were not required for exogenous DOXOL, which was too polar to diffuse from plasma in myocardial strips.
Only DOX rapid infusions were associated with formation of endogenous DOXOL. DOX slow infusions produced a cytoplasmic anthracycline pool that was too small for endogenous DOXOL to be formed, whereas liposomal DOX produced a cytoplasmic pool that was cleared toward plasma before it reached steady state levels permissive to metabolization. DOX movements from the cytoplasmic fraction of the strips toward the membrane fraction or plasma were important determinants of DOXOL formation after one modality of DOX administration or another.
After DOX rapid infusion, the net levels of endogenous DOXOL were only 0.5% compared with unmodified DOX (∼0.02 versus ∼4 μM, see Table 2). Following correction of DOXOL levels for its 40-fold higher toxicity, the contribution of endogenous DOXOL to the size of infusion-related anthracycline pools can be approximated to no more than ∼20%. Differences between DOX rapid infusion and DOX slow infusion or liposomal DOX are therefore determined primarily by the different levels of accumulation of unmodified DOX. However, changes in DOXOL formation may accentuate differences between DOX rapid infusion and DOX slow infusion or liposomal DOX. Previous studies showed that gain-of-function homozygosis for the CBR3 V244M G allele caused cancer patients to develop cardiomyopathy after low cumulative anthracycline doses (Blanco et al., 2012). In this study, we simulated that CBR polymorphism increased DOXOL formation and expanded the cardiac anthracycline pool induced by DOX rapid infusions but not those induced by DOX slow infusions or liposomal. We in fact suggest that the higher catalytic activity of CBR variants would be balanced by the low or altered cytoplasmic distribution of DOX associated with slow infusions or liposomal DOX. Consequently, simulations of CBR polymorphism caused no effect on the 5% risk dose of DOX slow infusion or liposomal DOX but decreased the 5% risk dose of DOX rapid infusion from 400 to 335 mg/m2. This latter finding introduces concerns, as 330 mg/m2 is reported to be safe in low-risk patients.
Study Limitations and Strengths.
Our model cannot provide information about the time elapsing between DOX administration and clinical HF, which is an important issue in modern cardio-oncology (Drafts et al., 2013; Cardinale et al., 2015; Murtagh et al., 2016). Our experiments can only quantify the potential for different DOX schedules to cause high or low HF risk. Moreover, we assumed that CBR polymorphism increased DOXOL formation by a factor of 2, but this was solely based on the catalytic characteristics of CBR variants (Blanco et al., 2008, 2012). We cannot exclude that disease- or treatment-related upregulation of CBR or other reductases induced higher levels of metabolite formation, causing HF to occur after lower cumulative doses of DOX than calculated by us.
We next acknowledge that myocardial samples were obtained primarily from males ≥70 years of age. This may have biased our calculations of HF risk for males and females <65 years of age. We compared the results of selected experiments that adopted myocardial samples from relatively young women (52–62 years) or men (50–54 years) and old women (78–85 years) or men (78–82 years). These samples produced highly comparable patterns of anthracycline uptake, metabolization, and efflux (data not shown). We therefore conclude that under pharmacokinetically controlled conditions, such as those adopted in this study, age and gender of sample donors did not significantly influence cardiac anthracycline accumulation and HF risk assessment.
Critical appraisal is needed also for the equation that we used to build risk versus dose curves. Equation 2 incorporated a variable, the correction constant, that was specific to each modality of DOX administration; however, eq. 2 also incorporated a variable, the 5% risk pool, that was common to all experimental conditions and was bound to the known 5% risk dose of DOX rapid infusion. For DOX slow infusion or liposomal DOX, correction constants made the equation suitable for unbiased calculations of 5% risk doses. For DOX rapid infusion, all variables were inferred from its known 5% risk dose, which may have biased revisions of HF risk. The experimental and clinical risk versus dose curves of DOX rapid infusion were in fact very similar, and the minor difference between 5% risk doses (380 versus 400 mg/m2) might very well be due to experimental noise. Interestingly, however, the experimental curve identified a range of low–moderate doses over which HF risk was significantly higher than in the clinical curve. We can calculate that HF risk was 1–2% over 220–280 mg DOX/m2 in the experimental curve as opposed to 300–340 mg/m2 in the clinical curve. For patients with CBR polymorphism, 1–2% HF risk occurred after 180–230 mg DOX/m2, a dose range over which the clinical curve showed <0.5% risk. Thus, eq. 2 was flexible enough to revise HF risk from DOX rapid infusion and provided information to support concerns about low–moderate doses of DOX causing HF in 1–2% of patients. This concept is summarized in Fig. 8.

Conclusions
Cancer patients usually receive DOX by rapid infusion. In this study, we modeled human myocardium exposure to DOX rapid infusion and showed that concerns about HF risk from safe cumulative doses are justified. These pharmacological findings have obvious clinical implications. Further dose reductions would limit the oncologic efficacy of DOX; therefore, HF risk should be managed by other cardioprotective measures.
Liposomal formulations were shown to be safer than DOX in adult cancer patients (van Dalen et al., 2010). Our studies illuminate the pharmacological foundations of clinical facts, showing that liposomal DOX induces cardiac anthracycline pools of a limited size, does not generate DOXOL, and therefore prevents risk aggravation by CBR polymorphism. Also, slow infusions should be considered. A Cochrane analysis showed that an infusion duration of 6 hours or longer reduced HF risk in adult patients (van Dalen et al., 2016). In this work, we simulated that an infusion duration of 4 hours, which was the longest possible in our model, caused the same effects as those of liposomal DOX. Unfortunately, however, liposomal formulations are approved for use in very limited indications, whereas slow infusions are considered as too laborious and demanding for doctors and patients, and their protective efficacy has not been demonstrated in some pediatric settings (Lipshultz et al., 2012). These limitations having been recognized, our results provide pharmacological correlates to encourage a wider use of DOX slow infusions or liposomal DOX in adult patients.
Authorship Contributions
Participated in research design: Salvatorelli, Menna, Minotti.
Conducted experiments: Salvatorelli, Minotti.
Contributed new reagents or analytic tools: Menna, Chello, Covino.
Performed data analysis: Salvatorelli, Menna, Minotti.
Wrote or contributed to the writing of the manuscript: Salvatorelli, Menna, Chello, Covino, Minotti.
Footnotes
- Received April 27, 2017.
- Accepted May 26, 2017.
This work was supported by University Campus Bio-Medico Special Project “Cardio-Oncology” (to G.M.).
Abbreviations
- CBR
- carbonyl reductase
- Cmax
- peak concentration
- DOX
- doxorubicin
- DOXOL
- doxorubicinol
- HF
- heart failure
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}