


Abstract
Many acute and chronic conditions, such as acute kidney injury, chronic kidney disease, heart failure, and liver disease, involve mitochondrial dysfunction. Although we have provided evidence that drug-induced stimulation of mitochondrial biogenesis (MB) accelerates mitochondrial and cellular repair, leading to recovery of organ function, only a limited number of chemicals have been identified that induce MB. The goal of this study was to assess the role of the 5-hydroxytryptamine 1F (5-HT1F) receptor in MB. Immunoblot and quantitative polymerase chain reaction analyses revealed 5-HT1F receptor expression in renal proximal tubule cells (RPTC). A MB screening assay demonstrated that two selective 5-HT1F receptor agonists, LY334370 (4-fluoro-N-[3-(1-methyl-4-piperidinyl)-1H-indol-5-yl]benzamide) and LY344864 (N-[(3R)-3-(dimethylamino)-2,3,4,9-tetrahydro-1H-carbazol-6-yl]-4-fluorobenzamide; 1–100 nM) increased carbonylcyanide-p-trifluoromethoxyphenylhydrazone–uncoupled oxygen consumption in RPTC, and validation studies confirmed both agonists increased mitochondrial proteins [e.g., ATP synthase β, cytochrome c oxidase 1 (Cox1), and NADH dehydrogenase (ubiquinone) 1β subcomplex subunit 8 (NDUFB8)] in vitro. Small interfering RNA knockdown of the 5-HT1F receptor blocked agonist-induced MB. Furthermore, LY344864 increased peroxisome proliferator–activated receptor coactivator 1-α, Cox1, and NDUFB8 transcript levels and mitochondrial DNA (mtDNA) copy number in murine renal cortex, heart, and liver. Finally, LY344864 accelerated recovery of renal function, as indicated by decreased blood urea nitrogen and kidney injury molecule 1 and increased mtDNA copy number following ischemia/reperfusion-induced acute kidney injury (AKI). In summary, these studies reveal that the 5-HT1F receptor is linked to MB, 5-HT1F receptor agonism promotes MB in vitro and in vivo, and 5-HT1F receptor agonism promotes recovery from AKI injury. Induction of MB through 5-HT1F receptor agonism represents a new target and approach to treat mitochondrial organ dysfunction.
Introduction
Mitochondrial dysfunction is linked to diverse acute insults such as surgery, trauma, ischemia/reperfusion (I/R), and drug toxicity, as well as chronic conditions, such as metabolic syndrome, diabetes, neurodegenerative diseases, and aging (Robertson, 2004; Monsalve et al., 2007; Legrand et al., 2008; Pessayre et al., 2012; Bayeva et al., 2013). Within the body, the organs most affected by mitochondrial dysfunction include those with the highest energy demand that primarily rely on mitochondrial aerobic respiration for ATP generation (e.g., kidney, heart, and brain) (Beeson et al., 2010). Decreased mitochondrial function may not only cause cell death, but may also decrease cellular functions and reduce the ability of a cell to respond to increased cellular energy demand.
Induction of mitochondrial biogenesis (MB) has been shown to alleviate mitochondrial dysfunction following injury in several pathophysiological model systems (Funk et al., 2010; Dam et al., 2013; Funk and Schnellmann, 2013; Whitaker et al., 2013). Requiring coordination of both the nuclear and mitochondrial genomes, MB is the synthesis of new mitochondria which is mediated by the master regulator peroxisome proliferator–activated receptor coactivator 1-α (PGC1α) (Scarpulla et al., 2012), and replaces defective or deficient mitochondria and/or supplements existing mitochondria to increase respiratory capacity. MB is an integral component of mitochondrial homeostasis that also includes mitophagy, fission, and fusion.
We have proposed that MB is a good drug target for diseases with mitochondrial dysfunction (Funk et al., 2010). Unfortunately, very few nontoxic chemicals or drugs suitable for pharmacological development have been identified that promote MB. For example, resveratrol (Csiszar et al., 2009; Mudo et al., 2012), catecholamines (Frier et al., 2012), AMP-activated protein kinase activators, such as N1-(β-D-ribofuranosyl)-5-aminoimidazole-4-carboxamide (AICAR) (Kukidome et al., 2006), the thiazolidinedione class of antidiabetic drugs including rosiglitazone and pioglitazone (Pagel-Langenickel et al., 2008; Pardo et al., 2011), pyrroloquinoline quinine moieties of quinoproteins (Chowanadisai et al., 2010), and the dietary supplement β-guanidinopropionic acid (Williams et al., 2009) have been shown to upregulate PGC1α. Mixed results have been obtained with the fibrate drug bezafibrate, a pan–peroxisome proliferator–activated receptor agonist (Wenz et al., 2008; Romanino et al., 2011; Yatsuga and Suomalainen, 2012). A recent high-throughput screen in murine primary skeletal muscle cells elucidated several distinct classes of drugs promoting MB; however, several of these, including glucocorticoids and inhibitors of microtubules and protein synthesis, would not be suitable for clinical administration due to toxicity and side effects (Arany et al., 2008).
We previously identified isoflavones and isoflavone derivatives as promoters of MB, characterized the minimal pharmacophore, determined the mechanism of action, and demonstrated that isoflavone treatment following oxidant exposure accelerated recovery of mitochondrial and cellular function (Rasbach and Schnellmann, 2008). Additionally, the reported sirtuin 1 activator SRT-1720 (N-[2-[3-(piperazin-1-ylmethyl)imidazo[2,1-b][1,3]thiazol-6-yl] phenyl]quinoxaline-2-carboxamide) promoted MB and expedited recovery of mitochondria following oxidant injury (Funk et al., 2010). More recently, β2-adrenergic receptor agonists, such as formoterol, were also identified as potent promoters of MB in vitro and in vivo, and a pharmacophore was developed (Wills et al., 2012; Peterson et al., 2013). Interestingly, not all β2-adrenoceptor agonists were capable of inducing MB (Peterson et al., 2013). Finally, specific inhibition of phosphodiesterases 3 and 5 increased cGMP to produce MB in renal proximal tubule cells (RPTC) and mice, and accelerated the recovery of renal function following acute kidney injury (Whitaker et al., 2013).
As part of our drug discovery program in MB, we explored the agonism of 5-hydroxytryptamine (5-HT; serotonin) receptors in MB. Agonism of the 5-HT2 receptor with DOI [1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane; 1–10 μM] promoted MB in vitro as evidenced by increased FCCP (carbonylcyanide-p-trifluoromethoxyphenylhydrazone)-uncoupled respiration, ATP, PGC1α promoter activity, and the mitochondrial proteins ATP synthase β and NADH dehydrogenase (ubiquinone) 1β subcomplex subunit 8 (NDUFB8) (Rasbach et al., 2010). Additionally, DOI accelerated restoration of mitochondrial function following oxidant injury (Rasbach et al., 2010). Considering that micromolar concentrations of DOI were needed to promote MB in vitro, and that micromolar concentrations of DOI also activate the 5-HT1F receptor, we examined the role of the 5-HT1F receptor in MB.
Materials and Methods
In Vitro Studies.
Renal proximal tubules were isolated from kidneys of female New Zealand white rabbits (1.5–2 kg) using an iron oxide perfusion method and cultured under improved conditions, resulting in normal aerobic metabolism comparable to that found in vivo (Nowak and Schnellmann, 1995). We have previously developed a high-throughput screening assay which identifies compounds that exhibit elevated FCCP-induced uncoupled oxygen consumption rates (OCRs), indicative of increased mitochondrial capacity (Beeson et al., 2010). OCRs were measured using a Seahorse Bioscience analyzer (North Billerica, MA) before (basal) and after addition of 1 μM FCCP, an ionophore that uncouples electron transport from ATP generation. The selective 5-HT1F receptor agonists LY334370 (4-fluoro-N-[3-(1-methyl-4-piperidinyl)-1H-indol-5-yl]benzamide) and LY344864 (N-[(3R)-3-(dimethylamino)-2,3,4,9-tetrahydro-1H-carbazol-6-yl]-4-fluorobenzamide), and the nonselective 5-HT receptor agonist α-methyl 5-HT were purchased from Tocris (Ellisville, MO).
For 5-HT1F receptor knockdown, RPTC were treated with scrambled small interfering RNA (siRNA) (siGENOME nontargeting siRNA #5; Dharmacon RNAi Technologies, Lafayette, CO) or siRNA designed against the 5-HT1F receptor using RNAiFect transfection reagent (Qiagen, Valencia, CA). siRNA knockdown was accomplished using a 1:1 mixture of siRNA recognizing the following sequences: siRNA-1, 5′-CCT TCA GCA TTG TGT ATA T-3′ and siRNA-2, 5′- CCA CAT TGT TTC CAC TAT T-3′. Following a 72-hour exposure, cells were scraped and analyzed for changes in protein levels.
In Vivo Studies.
Nonfasted naïve 8- to 10-week-old male C57BL/6NCr mice (National Cancer Institute, Frederick, MD) weighing 25–30 g received an intraperitoneal injection of 250 μl of vehicle (0.9% saline) or test compound every 8 hours for a total of three doses in a 24-hour period (1 mg/kg × 3) or one dose of test compound (2 or 10 mg/kg). At the time of euthanasia, organs were harvested, snap-frozen in liquid nitrogen, and stored at −80°C. For the I/R model of acute kidney injury (AKI), mice were subjected to bilateral renal pedicle ligation as described previously (Zhuang et al., 2009; Jesinkey et al., 2014). In brief, renal artery and vein were isolated and blood flow was occluded with a vascular clamp for 18 minutes. Mice were maintained at 37°C throughout the procedure using a homeothermic heating system. Sham surgery mice received all manipulations with the exception of clamping of the renal pedicles. Mice were treated once daily beginning at 24 hours after reperfusion with saline vehicle or LY344864 (2 mg/kg). Blood was collected from mice via retro-orbital bleed at 24 and 144 hours after surgery. Blood urea nitrogen (BUN) levels were measured using a QuantiChrom Urea Assay Kit (BioAssay Systems, Hayward, CA) according to the manufacturer's protocol. Mice were euthanized at 144 hours after the procedure, at which time kidneys were harvested for molecular analyses. Animal protocol was approved by and procedures completed in compliance with Institutional Use and Care of Animals Committee guidelines.
Nucleic Acid Isolation and Quantitative Polymerase Chain Reaction.
RPTC were scraped in TRIzol (Life Technologies, Grand Island, NY), and RNA was isolated using a phenol-based centrifugation method. DNA was isolated using a DNeasy Blood and Tissue Kit (Qiagen). cDNA was reverse transcribed from 2 μg RNA using a RevertAid First Strand cDNA Synthesis kit (Thermo Fisher Scientific, Waltham, MA), diluted 1:5, and 5 μl added to a real-time Maxima SYBR green quantitative polymerase chain reaction master mix containing 6-carboxy-X-rhodamine (Thermo Fisher Scientific). The following primers were used: actin forward 5′-GGG ATG TTT GCT CCA ACC AA-3′, actin reverse 5′-GCG CTT TTG ACT CAA GGA TTT AA-3′; apolipoprotein B (ApoB) forward 5′-CGT GGG CTC CAG CAT TCT-3′, ApoB reverse 5′-TCA CCA GTC ATT TCT GCC TTT G-3′; ATP synthase β forward 5′-GAG ACC AAG AAG GTC AAG ATG-3′, ATP synthase β reverse 5′-GAA GGG ATT CGG CCC AAT AAT GCA G 3′; cytochrome c oxidase 1 (Cox1) forward 5′-TAA TGT AAT CGT CAC CGC ACA-3′, Cox1 reverse 5′-ATG TGA GGA GCC CCA ATT ATC-3′; D loop forward 5′-CCCAAG CAT ATA AGC TAG TA-3′, D loop reverse 5′-ATA TAA GTC ATA TTT TGG GAA CTA C-3′; NDUFB8 forward 5′-ACC CAA TCC AAC CGC CTT CA-3′, NDUFB8 reverse 5′-CTA GGA CCC CAG AGG AAC GC 3′; PGC1α forward 5′-AGG AAA TCC GAG CTG AGC TGA ACA-3′, and PGC1α reverse 5′-GCA AGA AGG CGA CAC ATC GAA CAA-3′. Changes in gene expression were calculated based on the δ-δ threshold cycle method. Mitochondrial DNA (mtDNA) copy number was calculated based on comparison of mitochondrial D loop to nuclear ApoB.
Protein Isolation and Western Blotting.
Freshly isolated renal proximal tubules or RPTC (cultured until confluent, about 6 days) were rinsed with ice-cold phosphate-buffered saline, pelleted, and subjected to membrane fractionation (Subcellular Protein Fractionation Kit; Pierce Biotechnology, Rockford, IL). For nonfractionated samples, RPTC were scraped in radioimmunoprecipitation assay buffer containing protease inhibitors and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO). Following sonication, protein was quantified using a bicinchoninic acid assay, subjected to SDS-PAGE, transferred onto nitrocellulose membranes, and incubated with primary and secondary antibodies [glyceraldehyde-3-phosphate dehydrogenase from Fitzgerald (Acton, MA); Cox1 and NDUFB8 from Invitrogen (Frederick, MD); kidney injury molecule 1 (KIM-1) from R&D Systems Inc. (Minneapolis, MN); ATP synthase β, 5-HT1F receptor, and rabbit and mouse secondary antibodies from Abcam (Cambridge, MA)]. Images were acquired with AlphaEase software (Protein Simple, Santa Clara, CA) and processed using ImageJ (NIH, Bethesda, MD) software.
Statistics.
One-way analysis of variance (ANOVA) or Student’s t test was used, as appropriate, to analyze data for significance (P < 0.05). Significance in ANOVA was scrutinized for multiple comparisons using the Fisher least significant difference post-hoc test. When normality failed, a one-way ANOVA on Rank-sum was performed.
Results
The 5-HT1F Receptor Is Present in RPTC and 5-HT1F Receptor Agonism Leads to an Increase in FCCP-Uncoupled OCR.
To verify the expression of the 5-HT1F receptor in our model system, RNA and protein were isolated from renal proximal tubules and RPTC. The 5-HT1F receptor mRNA (362 bp) was observed in RPTC (Fig. 1A), and the 5-HT1F receptor protein (44 kDa) was present in freshly isolated renal tubules, tubule membrane fraction, RPTC, and RPTC membrane fraction (Fig. 1B).
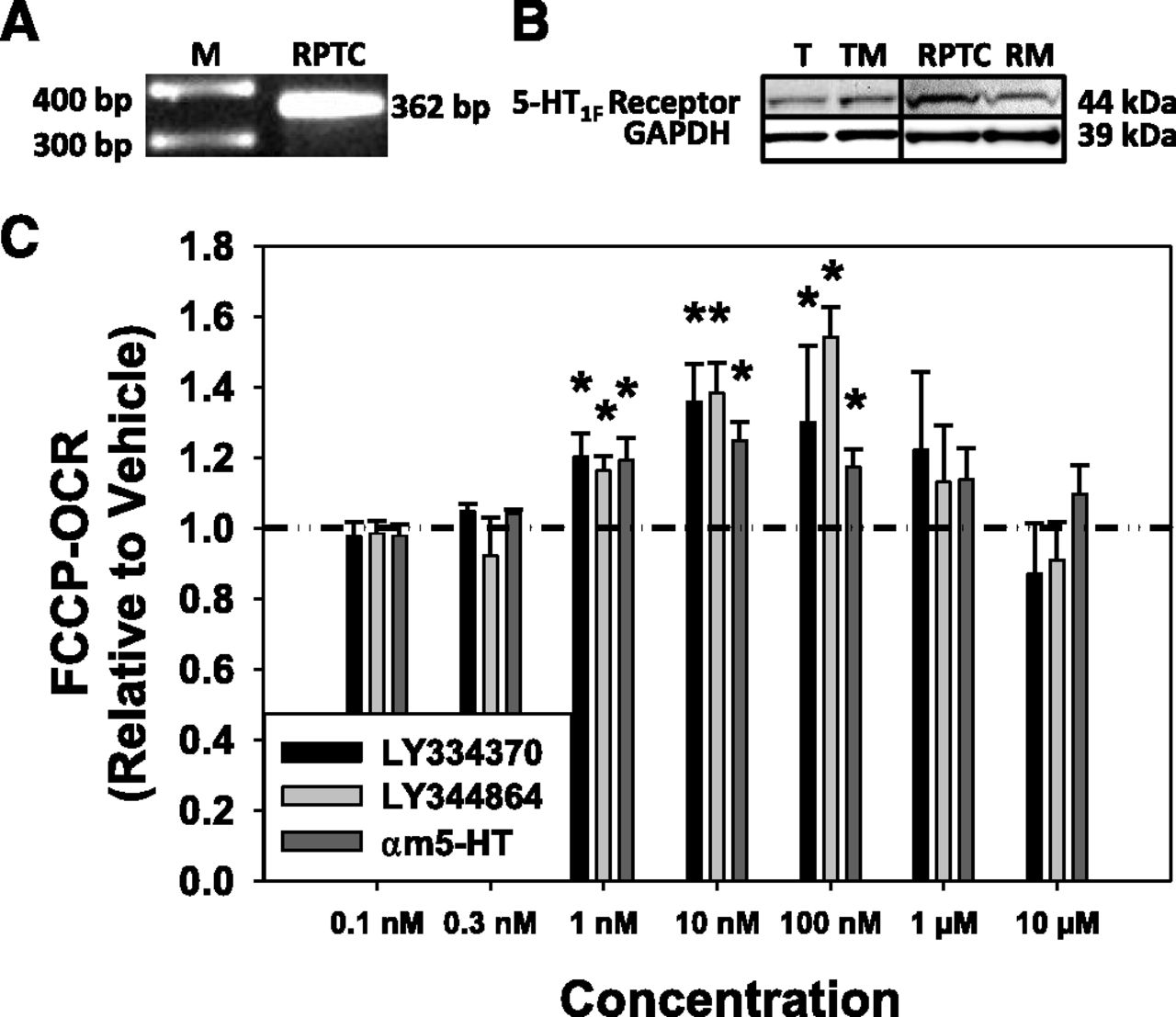
Htr1f is expressed in RPTC (A), and the 5-HT1F receptor protein is expressed in freshly isolated renal proximal tubules (T), tubule membranes (TM), RPTC, and RPTC membranes (RM) (B). (C) Various concentrations of α-methyl 5-HT (αm5-HT), a nonselective 5-HT receptor agonist, and selective 5-HT1F receptor agonists LY334370 and LY344864 stimulate FCCP-OCR in RPTC at 24 hours. Data are X + S.E.M., n ≥ 3. *Significantly different from vehicle (P < 0.05). GAPDH, glyceraldehyde 3-phosphate dehydrogenase; M, marker.
Previous studies have shown that LY334370 and LY344864 are specific agonists exhibiting high affinity for the 5-HT1F receptor (Ramadan et al., 2003; http://www.iuphar-db.org/index.jsp). LY334370 and LY344864 increased FCCP-uncoupled OCR, a screening assay readout for MB (Beeson et al., 2010), 1.2- to 1.4-fold and 1.2- to 1.5-fold, respectively, between 1 and 100 nM, but not at lower (0.1, 0.3 nM) or higher (1, 10 μM) concentrations compared with control cells treated with vehicle (<0.5% dimethylsulfoxide) in RPTC after a 24-hour exposure (Fig. 1C). Comparatively, the nonselective 5-HT receptor agonist α-methyl 5-HT increased FCCP-OCR 1.2- to 1.3-fold relative to control cells in RPTC at 24 hours. These results demonstrate that the 5-HT1F receptor is present in RPTC, and that the 5-HT1F receptor agonists LY334370 and LY344864 increase FCCP-uncoupled OCR.
5-HT1F Receptor Agonists Increase Mitochondrial Proteins in RPTC.
MB increases the quantity of electron transport chain proteins and copies of mtDNA, requiring coordination of both the nuclear and mitochondrial genomes. Three representative oxidative phosphorylation (OXPHOS) proteins were measured to confirm MB: ATP synthase β (ATP Synth), a portion of the F0-F1 ATP synthase enzyme (nuclear-encoded); Cox1, a constituent of complex IV (mitochondrial-encoded); and NDUFB8, a nuclear-encoded component of complex 1. The 5-HT1F receptor agonist LY334370 increased OXPHOS protein expression 1.4- to 1.6-fold at 1–100 nM in RPTC at 24 hours (Fig. 2A). Likewise, LY344864 (1–100 nM) similarly increased OXPHOS protein expression 1.4- to 2.1-fold in RPTC at 24 hours (Fig. 2B). Thus, the 5-HT1F receptor agonists LY334370 and LY344864 increased the levels of ATP Synth, Cox1, and NDUFB8 proteins at concentrations that also increased uncoupled respiration, consistent with MB.

5-HT1F receptor agonists increase mitochondrial proteins in RPTC at 24 hours. Representative immunoblots and quantification of concentration response for LY334370 (A) and LY344864 (B). Data are X + S.E.M., n ≥ 5. *Significantly different from vehicle (P < 0.05). GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
siRNA Knockdown of the 5-HT1F Receptor Blocks MB in RPTC.
To confirm that the MB actions of LY334370 and LY344864 were mediated through the 5-HT1F receptor, siRNA transfection was used to knockdown 5-HT1F receptor protein expression. Transfection of RPTC with siRNA targeted against the 5-HT1F receptor decreased 5-HT1F receptor protein 38% after 72 hours compared with scramble-treated RPTC (Fig. 3A). Interestingly, 5-HT1F receptor knockdown alone led to a significant diminution in ATP Synth, Cox1, and NDUFB8 protein levels (Fig. 3B). Knockdown of the 5-HT1F receptor protein blocked LY334370- and LY344864-induced upregulation of Cox1 and NDUFB8 proteins (Fig. 3C). These data reveal that the 5-HT1F receptor may regulate MB under basal conditions, and that post-transcriptional silencing of the 5-HT1F receptor blocked the agonist-stimulated MB response.

Knockdown of 5-HT1F receptor protein in RPTC by siRNA transfection at 72 hours (A) Representative immunoblot and quantification of 5-HT1F receptor protein knockdown. (B) Mitochondrial protein levels after 5-HT1F receptor knockdown. (C) Decreased 5-HT1F receptor levels block LY334370- and LY344864-induced MB. RPTC were treated with scramble or siRNA for 72 hours and treated with LY334370 or LY344864 for 24 hours. Data are X + S.E.M., n ≥ 3. *Significantly different from scrambled or 5-HT1F receptor agonist treatment (P < 0.05). GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
5-HT1F Receptor Agonism Increases OXPHOS Gene Expression and Mitochondrial DNA Copy Number in Renal Cortex.
A published study showed that rat brain cortex levels of LY344864 remained constant for 6 hours, whereas plasma levels declined over time following an intravenous dose of 1 mg/kg LY344864 (Phebus et al., 1997). Thus, an in vivo time course experiment (1, 8, 24 hours) was performed to determine gene expression and mtDNA copy number changes in the renal cortex induced by vehicle (0.9% saline) or LY344864 (2 mg/kg i.p.) (Fig. 4A). PGC1α and Cox1 mRNA were significantly increased at 1 and 8 hours, but not at 24 hours. NDUFB8 gene expression was unchanged after 1 hour, but was upregulated 8–24 hours. Although mtDNA copy number was unchanged at 1–8 hours, it was increased at 24 hours. These results indicate promotion of MB through agonism of the 5-HT1F receptor in vivo.

Gene expression of PGC1α, Cox1, NDUFB8, and mitochondrial copy number after selective 5-HT1F receptor agonist LY344864 (2 mg/kg) in the kidney cortex at 1, 8, and 24 hours (A) and after selective 5-HT1F receptor agonist LY344864 at 1 mg/kg every 8 hours (1 mg/kg × 3), 2 mg/kg, and 10 mg/kg in the kidney cortex at 24 hours (B). Data are X + S.E.M., n ≥ 3. *Significantly different from vehicle (P < 0.05).
Subsequent to the initial time course experiment, multiple dosing regimens were assessed. Mice were injected intraperitoneally with vehicle (0.9% saline), 1 mg/kg LY344864 every 8 hours for a total of 3 doses in a 24-hour period (1 mg/kg × 3), one bolus dose of 2 mg/kg LY344864, or one bolus dose of 10 mg/kg LY344864, and sacrificed at 24 hours (Fig. 4B). Gene expression of PGC1α and Cox1 in the renal cortex was significantly upregulated at 24 hours after 1 mg/kg × 3, unchanged with 2 mg/kg, and decreased after 10 mg/kg LY344864 compared with vehicle. NDUFB8 mRNA was increased with 1 mg/kg × 3 and 2 mg/kg but not 10 mg/kg LY344864 at 24 hours. Finally, mtDNA copy number was unchanged after 1 mg/kg × 3 and was increased after 2 mg/kg and 10 mg/kg LY344864 at 24 hours in the renal cortex. These data reveal that agonism of 5-HT1F receptor promotes MB in vivo in the kidney.
The 5-HT1F Receptor Is Present in Murine Cardiac and Hepatic Tissues and 5-HT1F Receptor Agonism Increases OXPHOS Expression and Mitochondrial DNA Copy Number.
To determine whether 5-HT1F receptor–mediated MB was selective for the kidney or also stimulated MB in other tissues, OXPHOS gene expression and mtDNA copy number were evaluated in the heart and the liver. 5-HT1F receptor mRNA (362 bp) was observed in both cardiac and hepatic tissues (Fig. 5A). In the heart, LY344864 (2 mg/kg) increased PGC1α mRNA at 1–8 hours but not at 24 hours (Fig. 5B). Cox1 gene expression was unchanged at 1 hour, but was upregulated at 8–24 hours. NDUFB8 mRNA was increased at 8 hours but not at 1 or 24 hours. Although unchanged at 1–8 hours, mtDNA copy number increased with LY344864 treatment at 24 hours in the heart. In the liver, PGC1α, Cox1, and NDUFB8 gene expression was significantly upregulated at 8–24 hours but not at 1 hour (Fig. 5C). Hepatic tissue mtDNA copy number was unchanged at 1–8 hours but was significantly increased at 24 hours. Taken together, these data indicate that LY344864 promotes MB through upregulation of PGC1α and other OXPHOS genes culminating in an increase in mtDNA copy number in extrarenal heart and liver tissues within 24 hours.
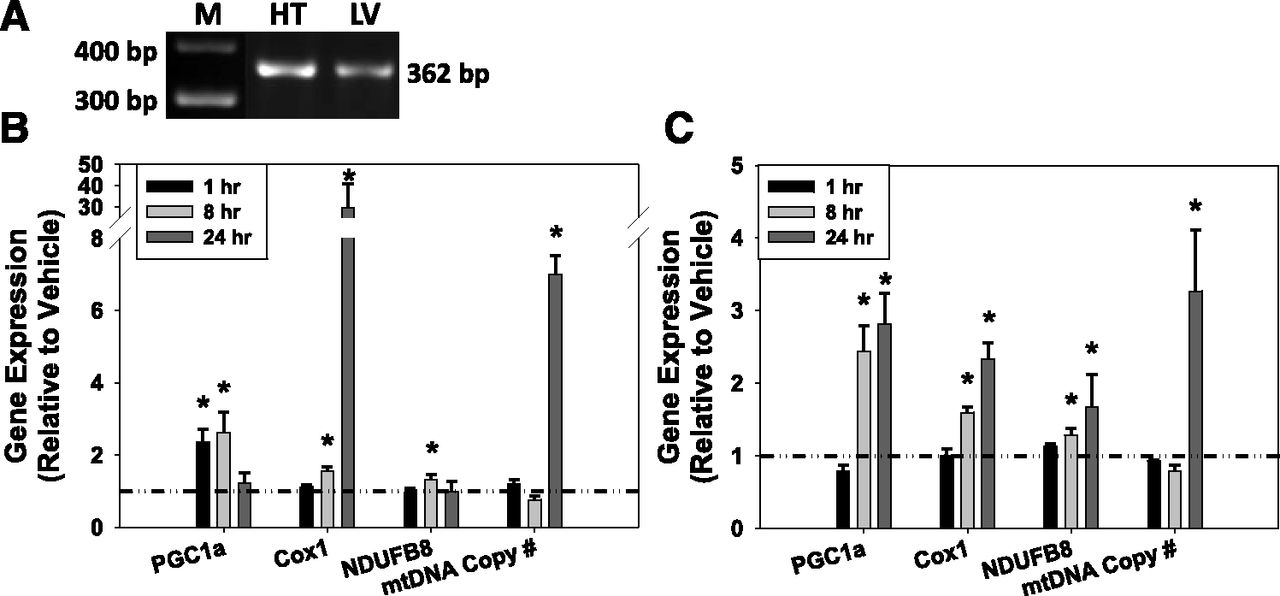
(A) Htr1f is expressed in murine heart (HT) and liver (LV). Gene expression of PGC1α, Cox1, NDUFB8, and mtDNA copy number after 5-HT1F receptor agonist LY344864 (2 mg/kg) in heart (B) and liver (C) at 1, 8, and 24 hours. Data are X + S.E.M., n ≥ 3. *Significantly different from vehicle (P < 0.05). M, marker.
LY344864 Increases Mitochondrial DNA Copy Number and Promotes Recovery from Ischemia/Reperfusion-Induced Acute Kidney Injury.
Stimulation of MB has been previously reported to accelerate the recovery of renal structure and function after AKI (Whitaker et al., 2013; Jesinkey et al., 2014). We examined the ability of LY344864 to stimulate MB and promote renal recovery in an I/R-induced AKI model. Mice were subjected to bilateral renal ischemia and treated daily with saline vehicle or LY344864 (2 mg/kg) over the course of 144 hours following surgery. All I/R-AKI mice had equal initial injury (BUN levels of 93 ± 15 mg/dl at 24 hours), but vehicle-treated mice failed to recover normal renal function as demonstrated by persistently elevated BUN levels (75 ± 6 mg/dl) (Fig. 6A). Mice treated with LY344864 showed accelerated recovery of renal function as evidenced by a decrease in BUN levels from initiation of treatment at 24 hours (99 ± 11 mg/dl) to near-control levels at the completion of treatment at 144 hours (45 ± 7 mg/dl).
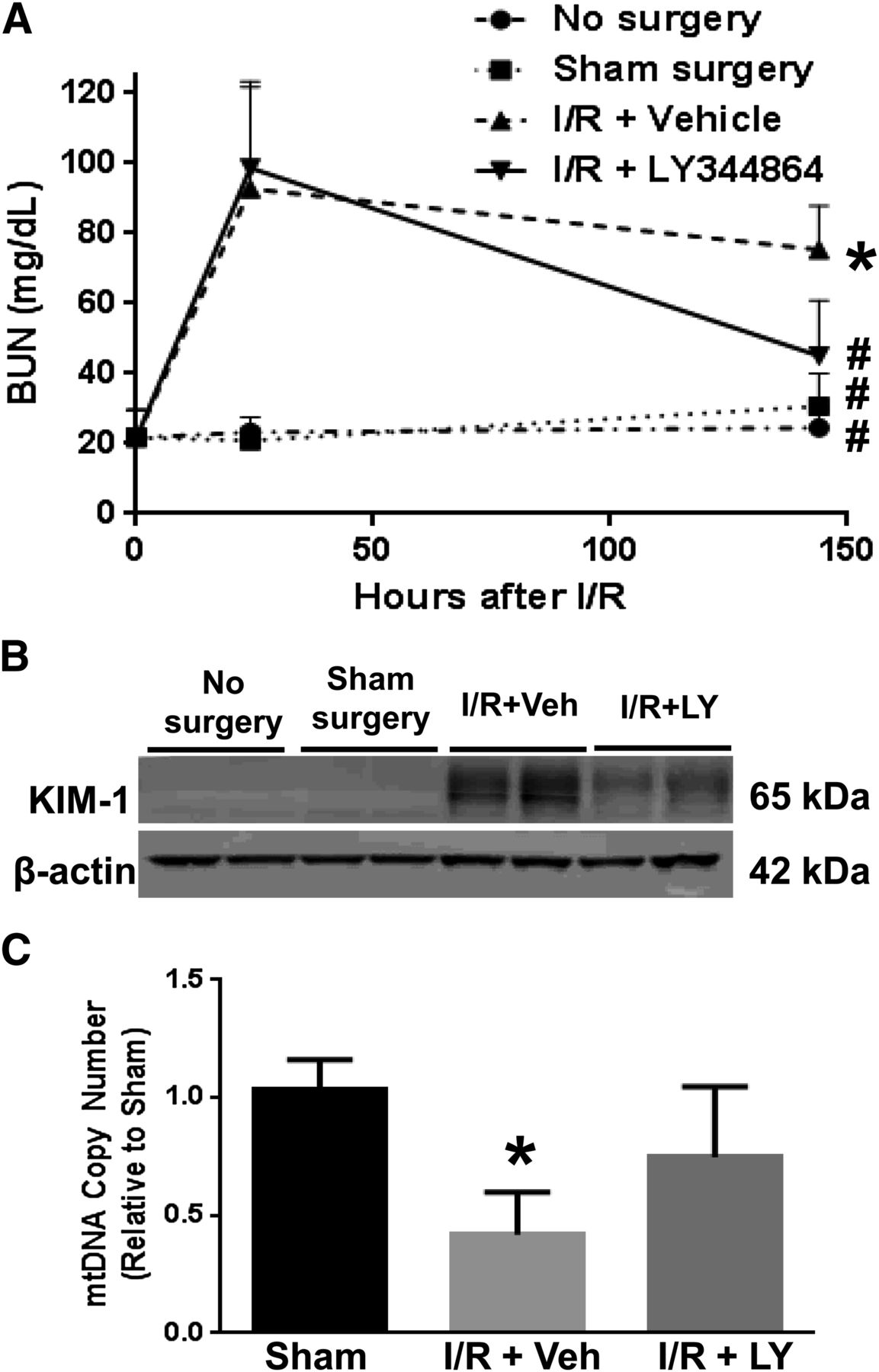
Blood urea nitrogen (24 and 144 hours) (A), renal cortical KIM-1 (144 hours) (B), and renal mtDNA copy number (144 hours) (C) in a murine I/R-AKI model. Data are X + S.E.M., n ≥ 4. *P < 0.05 versus sham; #P < 0.05 versus I/R + vehicle.
To assess renal tubular recovery, renal cortical KIM-1 levels were measured by immunoblot analysis (Fig. 6B). KIM-1 levels were upregulated in mice 144 hours after I/R injury compared with mice receiving no surgery or sham surgery. However, mice treated with LY344864 exhibited reduced KIM-1 protein expression in the renal cortex compared with vehicle-treated mice, demonstrating an accelerated recovery of the proximal tubular epithelium. Finally, the observed recovery of renal function was associated with recovery of mtDNA copy number. Renal cortical mtDNA copy number was 42% of sham surgery control levels at 144 hours after surgery in vehicle-treated I/R mice, and treatment with LY344864 promoted the recovery of the mtDNA copy number to 75% of sham surgery control levels (Fig. 6C). These data provide strong evidence that 5-HT1F receptor agonism is a viable strategy to stimulate recovery of renal function in the setting of AKI, and that recovery is correlated with a restoration of mtDNA copy number and function.
Discussion
Historically, the 5-HT1F receptor has been reported to be a neuronal receptor in the central nervous system mediating pain and lacking vasoactive properties, which has led to the development of 5-HT1F receptor agonists for the treatment of migraines (Mitsikostas and Tfelt-Hansen, 2012). However, despite the potential role of 5-HT1F receptor in migraine pathogenesis, other physiologic functions for 5-HT1F receptors have yet to be established.
In a high-throughput MB screening assay incorporating FCCP-OCR and the Seahorse Bioscience analyzer (Beeson et al., 2010), the selective 5-HT1F receptor agonists LY334370 and LY344864 potently induced uncoupled oxygen consumption in RPTC. Validation assays revealed increased levels of both mitochondrial-encoded (Cox1) and nuclear-encoded (ATP Synth and NDUFB8) proteins with 5-HT1F receptor agonist treatment, consistent with MB. Since 5-HT1F receptor antagonists have not been reported, siRNA was used to knockdown 5-HT1F receptors. A modest 38% knockdown of the 5-HT1F receptor not only blocked the MB effects of LY334370 and LY344864, but also significantly decreased basal levels of mitochondrial proteins in the kidney up to 50%. Time- and dose-dependent changes in OXPHOS genes and mitochondrial copy number were found in vivo in the kidney, heart, and liver in response to 5-HT1F receptor agonism. This is the first study to report that 5-HT1F receptors are associated with mitochondrial function, including MB, and that agonists to this receptor promote MB in multiple tissues.
Both LY334370 and LY344864 have been reported to be selective and efficacious agonists at the 5-HT1F receptor, with reported pKd values of 8.7 and 8.2 for LY334370 and LY344864, respectively (Ramadan et al., 2003; http://www.iuphar-db.org/index.jsp). LY334370 also has affinity for the 5-HT1A receptor (7.8 pKd), whereas LY344864 binds with 100-fold greater affinity at the 5-HT1F receptor than other 5-HT1 receptors. Although LY334370 showed no overt adverse effects in our studies, preclinical toxicology studies led to identification of the liver as a potential target of injury in beagle dogs when administered for longer than 1 month (Ramadan et al., 2003). However, no toxicity was shown in rats, and there was no increase in liver enzymes when administered to humans through phase II clinical trials (Ramadan et al., 2003). There are no reports that LY344864 is toxic. Therefore, although both agonists performed equally in vitro, only LY344864 was used for in vivo studies in this report.
5-HT receptors are grouped into seven families of either ligand-gated ion channels (5-HT3 receptors) or class A (rhodopsin-like) guanine nucleotide-binding protein (G protein)–coupled receptors (5-HT1,2,4-7 receptors), which are further divided into multiple subtypes. The majority of the 5-HT receptors are located in the central nervous system, platelets, and the gastrointestinal tract, where they mediate diverse physiologic processes, including anxiety, sleep, and appetite (Nichols and Nichols, 2008). The 5-HT1F receptor shares 42–57% amino acid identity with the other 5-HT1 receptors (Barnes and Sharp, 1999), although its localization to various sites within the brain (e.g., cortex, hippocampus, and cerebellum) as well as outside of the central nervous system, including reproductive tissues (uterus, testes), mesentery tissue, retina/whole eye, small intestine, kidney, liver, and heart (Lovenberg et al., 1993; Bouchelet et al., 2000; Su et al., 2004; Lucaites et al., 2005; Xu et al., 2007), suggests that agonism of the 5-HT1F receptor has the potential to promote MB across a wide range of tissues. The exact signaling mechanism mediating MB through 5-HT1F receptor agonism in RPTC still needs to be determined.
PGC1α mediates MB through activation of transcription factors such as the nuclear respiratory factors 1 and 2 and the estrogen-related receptors in the nuclear genome; the nuclear respiratory factors trigger the nuclear-encoded mitochondrial transcription factor A, which drives the mitochondrial gene transcription and genome replication, whereas the estrogen-related receptors and other transcription factors activate transcription of nearly a thousand mitochondrial genes of nuclear origin (Ventura-Clapier et al., 2008). In the renal cortex, 5-HT1F receptor agonism led to a rapid induction of PGC1α (e.g., 1 hour), promoting MB and resulting in an increase in mtDNA copy number within 24 hours. Timely responsiveness of PGC1α levels has also been reported after exercise and experimentally induced sepsis, confirming the importance of bioenergetics in response to environmental stimuli (Mathai et al., 2008; Sweeney et al., 2010). Addition of 5-HT1F receptor agonist (1 mg/kg × 3) led to an increase in PGC1α, Cox1, and NDUFB8 transcript levels, but not a change in mtDNA copy number at 24 hours; these increased gene expression levels are most likely due to a more recent exposure to agonist (8 hours prior to sacrifice) and an incomplete MB program that perhaps takes longer than 24 hours to complete. A single dose of 2 mg/kg agonist led to a complete MB program in 24 hours as evidenced by early increases in PGC1α and OXPHOS expression followed by an increase in mtDNA copy number at 24 hours. Dosing at a higher concentration (10 mg/kg) also led to a complete MB program with increased mtDNA copy number at 24 hours; however, downregulation of MB gene expression (PGC1α, Cox1) at 24 hours presumes an earlier robust upregulation of the MB program with subsequent negative feedback inhibition occurring at 24 hours. Thus, activation of PGC1α precedes increases in gene expression of target OXPHOS genes, ultimately culminating in greater quantities of mtDNA and increased mtDNA copy number at the end of the MB program.
5-HT1F receptor agonism also led to MB in extrarenal tissues. Within the liver, there was a robust induction of PGC1α and OXPHOS gene expression at 8–24 hours with an increase in mtDNA copy number at 24 hours. Promotion of MB was even more pronounced in the heart as 5-HT1F receptor agonism led to a 7-fold increase in mtDNA copy number at 24 hours. Differences in the amplitude of the MB response among tissues indicate greater specificity of agonist at the tissue or greater 5-HT1F receptor density/expression.
We have proposed that drugs inducing MB have the potential to improve therapeutic outcomes in the treatment of acute and chronic diseases in which mitochondrial dysfunction occurs (Funk et al., 2010). Mitochondrial dysfunction is commonly observed in acute organ injury of the kidney, heart, and liver (Di Lisa et al., 2007; Havasi and Borkan, 2011; Jaeschke et al., 2012). Recovery of renal structure and function following AKI has been reported to be accelerated through stimulation of MB by agonism of the β2-adrenergic receptor or by inhibition of cGMP-selective phosphodiesterases (Whitaker et al., 2013; Jesinkey et al., 2014). Likewise, stimulation of MB via agonism of the 5-HT1F receptor with LY344864 herein was similarly able to promote accelerated renal recovery in an I/R-induced AKI model. Thus, accelerated recovery of AKI, and possibly other acute or chronic diseases with mitochondrial dysfunction, can be induced through stimulation of MB through different upstream targets (e.g., 5-HT1F receptor agonism, β2-adrenergic receptor agonism, phosphodiesterase inhibition), which is therapeutically beneficial due to differential patient diseases and responses. In addition to renal AKI, mitochondrial dysfunction is also involved in numerous neurodegenerative diseases, including Alzheimer’s disease, Huntington disease, Parkinson’s disease, and other non-neurologic pathologies, such as obesity and type 2 diabetes (Hojlund et al., 2008; Lezi and Swerdlow, 2012). Upregulation of PGC1α has successfully been shown to increase OXPHOS and ameliorate several in vivo models of diseases, including age-related pathogenesis of Alzheimer’s disease, Huntington disease, and Parkinson’s disease (Wenz, 2009; Mudo et al., 2012; Tsunemi et al., 2012; Tsunemi and La Spada, 2012). Collectively, the ability of 5-HT1F receptor agonism to mitigate pathologic mitochondrial damage through upregulation of PGC1α and promotion of MB should be explored as a potential therapeutic strategy to ameliorate many diseases featuring mitochondrial dysfunction in a variety of tissues.
Authorship Contributions
Participated in research design: Garrett, Whitaker, Beeson, Schnellmann.
Conducted experiments: Garrett, Whitaker.
Performed data analysis: Garrett, Whitaker, Schnellmann.
Wrote or contributed to the writing of the manuscript: Garrett, Whitaker, Beeson, Schnellmann.
Footnotes
- Received March 17, 2014.
- Accepted May 16, 2014.
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM084147]; the National Institutes of Health National Center for Research Resources [Grant UL1-RR029882]; the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs [1BX000851]; the South Carolina Clinical and Translational Research Institute, with an academic home at the Medical University of South Carolina; and the National Institutes of Health Ruth L. Kirschstein National Research Service Award [Grant 5T32-DK083262-03]. Animal facilities were funded by the National Institutes of Health National Center for Research Resources [Grant C06-RR015455].
Portions of this work have been presented previously: Garrett SM, Wills LP, and Schnellmann RG (2012) Serotonin (5-HT) 1F receptor agonism as a potential treatment for acceleration of recovery from acute kidney injury. American Society of Nephrology Annual Meeting; 2012 Nov 1–4; San Diego, CA.
Abbreviations
- AKI
- acute kidney injury
- ANOVA
- analysis of variance
- ApoB
- Apolipoprotein B
- ATP Synth
- ATP synthase β
- BUN
- blood urea nitrogen
- Cox1
- cytochrome c oxidase 1
- DOI
- 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane
- FCCP
- carbonylcyanide-p-trifluoromethoxyphenylhydrazone
- 5-HT
- 5-hydroxytryptamine, serotonin
- I/R
- ischemia/reperfusion
- KIM-1
- kidney injury molecule 1
- LY334370
- 4-fluoro-N-[3-(1-methyl-4-piperidinyl)-1H-indol-5-yl]benzamide
- LY344864
- N-[(3R)-3-(dimethylamino)-2,3,4,9-tetrahydro-1H-carbazol-6-yl]-4-fluorobenzamide
- MB
- mitochondrial biogenesis
- αm5-HT
- α-methyl 5-hydroxytryptamine
- mtDNA
- mitochondrial DNA
- NDUFB8
- NADH dehydrogenase (ubiquinone) 1β subcomplex subunit 8
- OCR
- oxygen consumption rate
- OXPHOS
- oxidative phosphorylation
- PGC1α
- peroxisome proliferator–activated receptor coactivator 1-α
- RPTC
- renal proximal tubule cells
- siRNA
- small interfering RNA
- SRT-1720
- N-[2-[3-(piperazin-1-ylmethyl)imidazo[2,1-b][1,3]thiazol-6-yl] phenyl]quinoxaline-2-carboxamide
- U.S. Government work not protected by U.S. copyright





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}