


Abstract
The high-affinity sodium glucose cotransporter (SGLT1) plays a critical role in glucose absorption from the gastrointestinal tract. We have developed 3-(3-{4-[3-(β-d-glucopyranosyloxy)-5-isopropyl-1H-pyrazol-4-ylmethyl]-3-methylphenoxy}propylamino)propionamide (KGA-2727), which has a pyrazole-O-glucoside structure, as the first selective SGLT1 inhibitor. KGA-2727 inhibited SGLT1 potently and highly selectively in an in vitro assay using cells transiently expressing recombinant SGLTs. In a small intestine closed loop absorption test with normal rats, KGA-2727 inhibited the absorption of glucose but not that of fructose. After oral intake of starch along with KGA-2727 in normal rats, the residual content of glucose in the gastrointestinal tract increased. In the oral glucose tolerance test with streptozotocin-induced diabetic rats, KGA-2727 attenuated the elevation of plasma glucose after glucose loading, indicating that KGA-2727 improved postprandial hyperglycemia. In Zucker diabetic fatty (ZDF) rats, chronic treatments with KGA-2727 reduced the levels of plasma glucose and glycated hemoglobin. Furthermore, KGA-2727 preserved glucose-stimulated insulin secretion and reduced urinary glucose excretion with improved morphological changes of pancreatic islets and renal distal tubules in ZDF rats. In addition, the chronic treatment with KGA-2727 increased the level of glucagon-like peptide-1 in the portal vein. Taken together, our data indicate that the selective SGLT1 inhibitor KGA-2727 had antidiabetic efficacy and allow us to propose KGA-2727 as a candidate for a novel and useful antidiabetic agent.
Introduction
Type 2 diabetes mellitus is characterized by hyperglycemia caused by pancreatic β-cell dysfunction and is a lifestyle-related disease connected to obesity and lack of exercise. Large clinical trials have confirmed that it is important to practice long-term tight control of blood glucose levels to avoid the development of diabetic complications [The Diabetes Control and Complications Trial Research Group, 1993; UK Prospective Diabetes Study (UKPDS) Group, 1998]. Diet, exercise, and drugs are treatment modalities used on patients with diabetes to control their blood glucose levels. For this control, it is important to attenuate postprandial hyperglycemia and reduce fasting hyperglycemia (Woerle et al., 2007). Epidemiology studies have shown that postprandial glycemia has a strong association with cardiovascular risks (DECODE Study Group and the European Diabetes Epidemiology Group, 2001), endothelial dysfunction (Williams et al., 1998), and retinopathy (Shiraiwa et al., 2005).
Although many clinical agents improve overall glycemic control including postprandial hyperglycemia, several therapeutics specifically target postprandial blood glucose. For example, α-glucosidase inhibitors, such as acarbose, voglibose, and miglitol, are prescribed in clinical practice to control postprandial blood glucose. Those inhibitors inhibit the digestion of carbohydrates, delay the absorption of carbohydrates from the gastrointestinal tract, and consequently suppress the postprandial elevation of the blood glucose level (Göke and Herrmann-Rinke, 1998). It has also been reported that acarbose is effective in preventing or delaying the incidence of diabetes when applied to patients with impaired glucose tolerance (Chiasson et al., 2002). Recently, the International Diabetes Federation (2011) launched a new global guideline for the management of postmeal glucose. From these viewpoints, modification of postprandial carbohydrate absorption is a target for the development of new antidiabetic agents.
The high-affinity sodium glucose cotransporter (SGLT1) plays a critical role in the absorption of glucose (Wright et al., 2007) and is expressed in the small intestines and encoded by the SLC5A1 gene. Patients who have defective mutations in SLC5A1 suffer from gastrointestinal symptoms caused by the impaired absorption of monosaccharides (Turk et al., 1991). Based on pathogenetic studies on diabetes, it was reported that SGLT1 played an important role in the accelerated absorption of glucose. It was confirmed that the mRNA and protein levels of SGLT1 increased, and the absorption of glucose was accelerated in Otsuka Long Evans Tokushima Fatty and streptozotocin-induced diabetic rats (Dyer et al., 1997; Fujita et al., 1998). Also in patients with diabetes, the mRNA and protein levels of SGLT1 are highly increased in the small intestine (Dyer et al., 2002). Therefore, it is reasonable to expect that delaying glucose absorption by SGLT1 inhibition would be effective in normalizing postprandial hyperglycemia.
To date, several groups of researchers have reported that SGLT inhibitors can be considered a novel class of antidiabetic drugs. However, most of them inhibit SGLT2, the low-affinity sodium glucose cotransporter, which plays a role in renal glucose reabsorption (Isaji, 2011; Kinne and Castaneda, 2011). Some of the SGLT2 inhibitors are now under investigation in clinical trials. Sergliflozin and remogliflozin, reported from our laboratory at the Kissei Pharmaceutical Co., Ltd., are also SGLT2 inhibitors (Katsuno et al., 2007, 2009; Fujimori et al., 2008, 2009). Those SGLT2 inhibitors reduce the blood glucose level by inhibiting renal glucose reabsorption and increasing urinary glucose excretion. However, no inhibitor selective for SGLT1 had been reported. In this article, we describe the efficiency of a novel selective SGLT1 inhibitor for the treatment of diabetes.
Materials and Methods
Chemicals.
KGA-2727 [3-(3-{4-[3-(β-d-glucopyranosyloxy)-5-isopropyl-1H-pyrazol-4-ylmethyl]-3-methylphenoxy}propylamino)propionamide] was synthesized by Kissei Pharmaceutical Co., Ltd. Methyl-α-d-glucopyranoside (AMG) and phlorizin dihydrate were purchased from Sigma-Aldrich (St. Louis, MO). Methyl-α-d-[U14C]glucopyranoside was obtained from GE Healthcare (Little Chalfont, Buckinghamshire, UK). Starch was purchased from Nacalai Tesque (Kyoto, Japan). Acarbose was prepared by extraction from Glucobay purchased from Bayer Yakuhin (Osaka, Japan). Dipeptidyl peptidase IV inhibitor was purchased from Linco Research (St. Charles, MO). Other chemicals were purchased from Wako Pure Chemicals (Osaka, Japan).
Animals.
Male Wistar, Zucker diabetic fatty (ZDF) fa/fa (ZDF/Gmi Crl-fa/fa), and ZDF-lean (ZDF/Gmi Crl-lean) rats were purchased from Charles River Japan, Inc. (Yokohama, Japan). All rats were housed under a 12-h light cycle (lights on 8:00 AM to 8:00 PM) under controlled room conditions (room temperature, 20–26°C; humidity, 35–65%), fed a laboratory chow diet (CE-2 pellets; CLEA Japan, Tokyo, Japan), and provided water ad libitum. All animal experiments were performed in accordance with the guidelines approved by the Laboratory Animal Committee of Kissei Pharmaceutical Co., Ltd.
Inhibitory Effects of KGA-2727 on Human and Rat SGLTs.
Human and rat SGLT expression plasmids were constructed as described previously (Katsuno et al., 2007; Fujimori et al., 2008). Cell culture, the transfection procedure, and the methyl-α-d-[U14C]glucopyranoside uptake experiments were performed as described previously (Fujimori et al., 2008). In these experiments, 0.3 and 1 mM AMG in the uptake buffer were used to calculate Ki values.
Absorption Rate of Glucose and Fructose in Rat Small Intestine.
After overnight fasting, the intestine was exposed through a midline abdominal incision under urethane anesthesia. To form a closed loop, the small intestine 2 cm distal to the bile duct opening was ligated with a cannula for injection, and then a 15-cm distal site was also ligated. Sucrose (30 mM in phosphate-buffered saline; 0.75 ml) was injected into the loop with or without the test compound. After 15 min, the loop contents were collected with 0.75 ml of ice-cold saline, and the total volume of collected contents was measured. In the initial control group, the sucrose solution was injected after the ligated loop had been isolated and then was immediately collected with ice-cold saline. After heat inactivation of enzyme-mediated digestion, the concentrations of glucose and fructose were measured with a d-glucose/d-fructose UV test (R-Biopharm GmbH; Darmstadt, Germany). The amounts of sugars and absorption rate were calculated by using the following equation: absorption rate (nmol/min/cm) = {(sucrose in initial control) − (residual sucrose) − (residual glucose or fructose)} (nmol)/15 (min)/length of loop (cm).
Effects of KGA-2727 on Residual Carbohydrate in the Gastrointestinal Tract after Oral Administration of Starch and the Inhibitor to Normal Rats.
Overnight fasted rats were orally administered the test compound solution and starch solution (2 g/kg body weight). After exsanguination 3 h after starch administration, their gastrointestinal tracts were removed immediately and divided into five parts: the stomach, upper small intestine (lower part 20 cm from the pylorus), middle small intestine (part between upper and lower small intestine), lower small intestine (upper part 20 cm from the ileocecal junction), and cecum. The contents of these gastrointestinal segments were collected with 10 ml of ice-cold saline, and the volume of collected contents was measured. After heat inactivation of enzyme-mediated digestion, subsets of collected samples were hydrolyzed with H2SO4 and then neutralized with NaOH. Glucose concentrations were measured before and after hydrolysis. For the calculation of a dosage of starch, the starch solution was hydrolyzed, and the glucose concentration was measured in the same way. Residual carbohydrate was calculated from the glucose concentration of the hydrolyzed sample. This value indicates the ratio of polysaccharides and monosaccharides to the dosage of starch. Residual glucose was calculated from the nonhydrolyzed sample and indicates the ratio of monosaccharide glucose to the starch dosage.
Effects of KGA-2727 on Plasma Glucose Concentration after Oral Glucose Loading in Streptozotocin-Induced Diabetic Rats.
Diabetic rats were induced by intravenous injection of streptozotocin (45 mg/kg body weight). For selection of diabetic rats, an oral glucose tolerance test was performed 7 days after streptozotocin injection. After overnight fasting, blood was obtained via the caudal artery 1 h after glucose loading (2 g/kg body weight), and the plasma glucose concentration was measured with a Glucose CII-Test Wako kit (Wako Pure Chemicals). The rats with plasma glucose concentrations more than 300 mg/dl after glucose loading were selected and divided into several groups so that the average of the plasma glucose concentration in each group was approximately equal. The oral glucose tolerance test for evaluating the test compound was performed at 10 days after streptozotocin injection. Overnight fasted rats were orally administered a test compound solution and glucose (2 g/kg body weight) solution. Blood was collected via the caudal artery immediately before and 0.25, 0.5, 1, 2, 3, and 4 h after dosing, and then the plasma glucose concentration was measured.
Effects of Chronic Treatment with KGA-2727 in Zucker Diabetic Fatty Rats.
Starting from 7 weeks of age, ZDF fa/fa rats, which are obese-type animals, were fed a powder diet (5008 Formulab; Purina LabDiet, Richmond, IN) containing the test compound for 48 days. The test compound concentrations were as follows: KGA-2727, 10, 30, and 100 ppm; and acarbose, 100 ppm. ZDF-lean rats of the same age served as the lean control group. The day when the chronic treatment was started was defined as day 0. Body weight, plasma glucose, and glycated hemoglobin (GHb) were measured periodically in the fed state. The plasma glucose concentration was determined as described above. GHb was determined with an HLC-723GHbV device (Tosoh, Tokyo, Japan). The weight of powder diet was measured periodically.
On day 37, the oral glucose tolerance test was performed for the evaluation of insulin secretion. After overnight fasting, the plasma glucose and insulin concentrations were measured at different time points after oral glucose loading (2 g/kg body weight). The plasma insulin concentration was measured with an insulin enzyme-linked immunosorbent assay kit (Seikagaku Corporation, Tokyo, Japan).
On days −2, 19, and 40, the rats were relocated to metabolic cages, and urine samples were collected for 24 h. The urine volume and glucose concentration were measured.
At the end of the chronic treatment study, the rats were anesthetized with ether, and blood was collected via the portal vein with syringes containing aprotinin and a dipeptidyl peptidase IV inhibitor. The plasma concentration of glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) were measured with a glucagon-like peptide-1 (active) enzyme-linked immunosorbent assay kit (Linco Research) and GIP radioimmunoassay kit (Peninsula Laboratories, Belmont, CA), respectively.
The pancreas and kidney were fixed with 10% phosphate-buffered formalin and then embedded in paraffin to prepare tissue sections. The pancreas sections were stained with azan stain and immunostaining for insulin and quantitatively analyzed with an image processor (LUZEX-3; Nikon, Tokyo, Japan) as follows. For assessment of the insulin index (Ins-I) or fibrosis index (Fib-I), 10 islets larger than 1 × 104 mm2 were randomly selected from each section, and the percentage of insulin-positive area or fibrous area within each selected islet was calculated, respectively. The kidney sections were stained with hematoxylin-eosin, periodic acid-Schiff, and periodic acid methenamine silver stain and morphologically analyzed. For the assessment of glomerulosclerosis, 50 glomeruli were randomly selected, and the percentage of sclerotic area within glomerular area was scored as follows: normal, 0; ∼0 to 25%, 1; ∼25 to 50%, 2; ∼50 to 75%, 3; > 75%, 4. The average of these scores was used as a glomerulosclerosis score of each animal. Glycogen deposition in distal tubules was assessed by counting the number of periodic acid-Schiff -positive tubules in 20 microscopic fields (total area = approximately 14.8 mm2) and was scored as follows: 0, negative, −; 1–20, minimal, ±; 21–60, slight, +; >60, moderate, ++. A change in dilatation of distal tubules was scored as follows: negative, −; there were a few changes, minimal, ±; the changes were focally observed, slight, +; the changes were diffusely observed, moderate, ++.
Statistical Analysis.
Data were presented as the mean ± S.E.M. for each group. Statistical analyses were performed with SAS Systems version 8.2 (SAS Institute, Cary, NC). In numerical data analysis, statistical significance was determined with one-way analysis of variance, Dunnett's multiple comparison, nonparametric multiple comparison, univariate repeated-measures analysis as a split-plot design, multiplex comparison by each time period, or t test as appropriate. In categorical data analysis, statistical significance was determined with Wilcoxon test, Kruska-Wallis test, and nonparametric Dunnett's multiple comparison test as appropriate. Differences assessed by each test were considered statistically significant at p < 0.05.
Results
Structure of KGA-2727.
The structure of KGA-2727 is shown in Fig. 1A, and the structure of phlorizin, a nonselective SGLT inhibitor, is shown in Fig. 1B. KGA-2727 has an O-glucoside structure similar to that of phlorizin. The aglycon portion of KGA-2727 is a pyrazole derivative.

Chemical structures of KGA-2727 (A) and phlorizin (B).
Potency and Selectivity of KGA-2727 in Inhibiting SGLT1.
A Dixon plot analysis for KGA-2727 displayed good linearity for human SGLT1 and SGLT2 (Fig. 2, A and B, respectively). The results of the Dixon plot show that KGA-2727 inhibited these SGLTs in a competitive manner. KGA-2727 dose-dependently inhibited AMG uptake by SGLT1 and SGLT2. The Ki values for KGA-2727 and phlorizin against human and rat SGLTs are shown in Table 1. KGA-2727 more potently inhibited SGLT1 than did phlorizin and vice versa for SGLT2. The selectivity ratios (Ki for SGLT2/Ki for SGLT1) of KGA-2727 were 140 (human) and 390 (rat), whereas those of phlorizin were 0.084 (human) and 0.195 (rat). These data suggest that KGA-2727 was a potent and selective SGLT1 inhibitor.

Inhibitory effects of KGA-2727 on recombinant human SGLT1 and SGLT2. Dixon plots of KGA-2727 action against human SGLT1 (A) and human SGLT2 (B). For Dixon plots, AMG uptake at 0.3 or 1 mM was measured in the presence of various concentrations of KGA-2727. Data are presented as the means ± S.E.M. (n = 6 or 7).
Ki values for KGA-2727 and phlorizin against human and rat SGLTs
Data are presented as the means ± S.E.M. (n = 6 or 7).
Effects of KGA-2727 on Saccharide Absorption Rate in Rat Small Intestine.
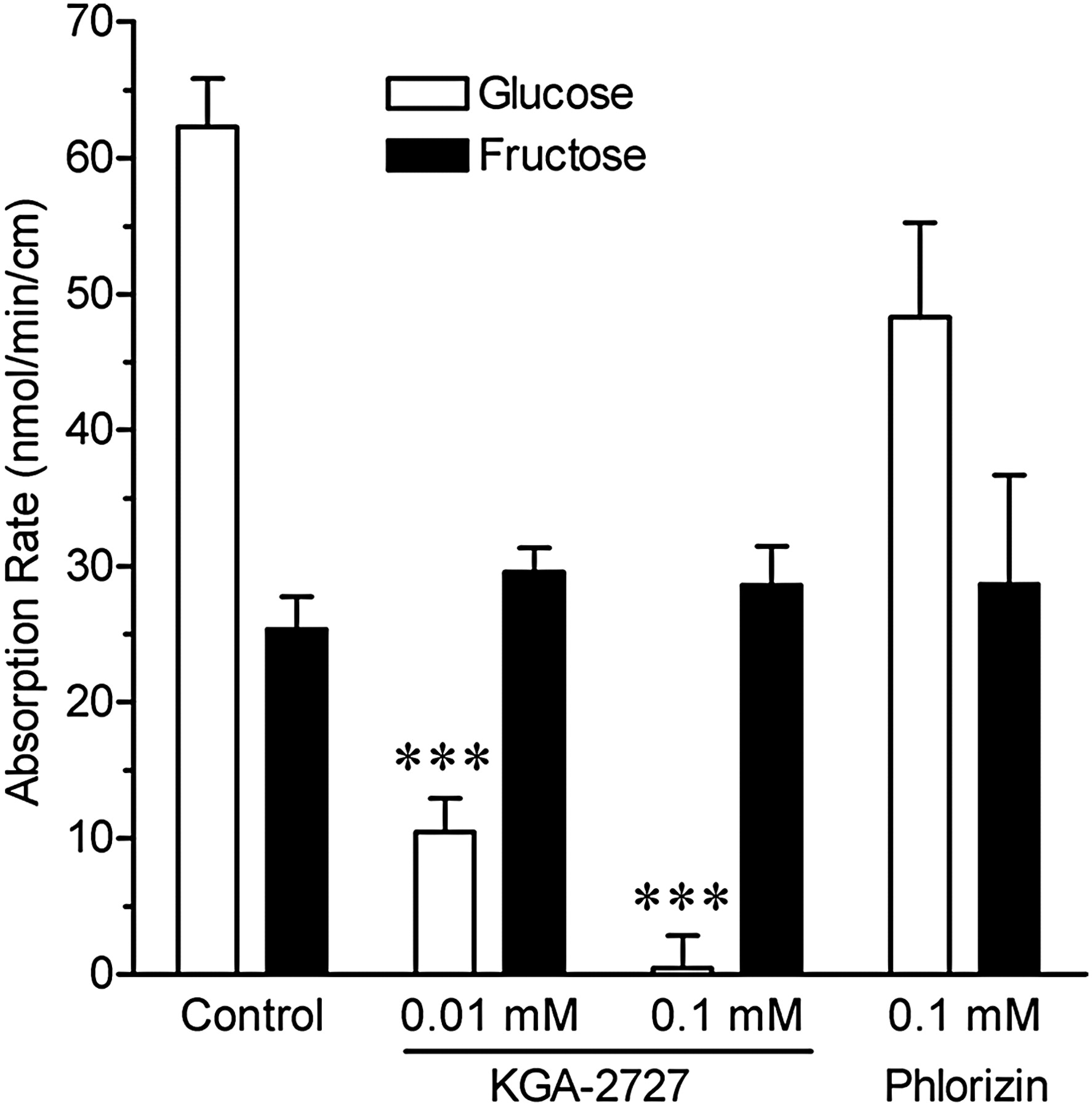
The percentage of sucrose recovery was approximately 78%, as calculated from the initial control value. In the control group, 80.6% of the sucrose was digested after 15 min. The effects of KGA-2727 on glucose and fructose absorption rates are shown in Fig. 3. KGA-2727 decreased the absorption rate of glucose in a dose-dependent manner. Phlorizin tended to decrease this rate, but its effect was not statistically significant. On the other hand, KGA-2727 and phlorizin had no effect on the absorption rate of fructose after sucrose injection. Thus, KGA-2727 inhibited the absorption of only glucose.

Effects of KGA-2727 or phlorizin on saccharide absorption rates in the closed loop of the rat small intestine. Absorption rates of glucose (empty bars) and fructose (filled bars) were calculated from the amounts of glucose and fructose in the closed loop contents after the injection of sucrose and test compound solution. Data are presented as the means ± S.E.M. (n = 5). ***, p < 0.001 versus control.
Effects of KGA-2727 on Gastrointestinal Carbohydrate Content after Administration of Starch and Inhibitor to Normal Rats.
Figure 4 shows the effects of KGA-2727 (A and C) and acarbose (B and D) on carbohydrate and glucose contents in the gastrointestinal tract. Residual carbohydrate (Fig. 4, A and B) was calculated from the amount of glucose in the hydrolyzed sample and indicates the content of carbohydrate (i.e., monosaccharide plus polysaccharides). Residual glucose (Fig. 4, C and D) was calculated from the amount of glucose in the nonhydrolyzed sample and indicates the content of monosaccharide glucose. The amount of residual carbohydrate in the control was approximately 5% at 3 h after oral starch administration. KGA-2727 increased the total amount of residual carbohydrate in a dose-dependent manner. Three hours after administration, the total amount of residual carbohydrate was 6.7% in the control group but 14.0, 26.2, and 45.5% in the presence of 0.1, 0.3, and 1.0 mg/kg KGA-2727, respectively (Fig. 4A). More than half of the residual carbohydrate in the KGA-2727-treated groups was monosaccharide glucose (Fig. 4C). KGA-2727 increased the residual glucose in the lower intestine. Acarbose also increased the content of residual carbohydrate (Fig. 4B); but in this case, the residual carbohydrate was not glucose (Fig. 4D). The amount of residual carbohydrate in ∼0.1 to 0.3 mg/kg KGA-2727-treated groups was similar to that in the 2 mg/kg acarbose-treated group. These data indicate that KGA-2727 delayed carbohydrate absorption in the gastrointestinal tract and increased the amount of glucose in the lower intestine.

Effects of KGA-2727 (A and C) or acarbose (B and D) on residual gastrointestinal carbohydrate and glucose after oral starch administration to normal rats. A and B, the residual carbohydrate indicates the ratio of polysaccharides and monosaccharides to the dosage of starch. C and D, the residual glucose indicates the ratio of monosaccharides glucose to the dosage of starch. Data are presented as the means ± S.E.M. (n = 4). *, p < 0.05; **, p < 0.01 versus control. Int., intestine.
Effects of KGA-2727 on Plasma Glucose Concentration after Oral Glucose Loading in Streptozotocin-Induced Diabetic Rats.
Streptozoticin-induced diabetic rats showed hyperglycemia after glucose loading compared with normal rats. In those diabetic rats, KGA-2727 significantly inhibited, in a dose-dependent manner, the elevation of plasma glucose after glucose loading (Fig. 5). The plasma glucose elevation in diabetic rats in the presence of 0.3 mg/kg KGA-2727 was similar to that in the normal group. Acarbose at a dose of 30 mg/kg did not inhibit the rise in plasma glucose after glucose loading (Fig. 5).

Effects of KGA-2727 or acarbose on postprandial hyperglycemia in the oral glucose tolerance test in streptozotocin-induced diabetic rats. KGA-2727 or acarbose and glucose (2 g/kg body weight) were orally administered to streptozotocin-induced diabetic rats. The plasma glucose (A) was measured, and the AUC0–4 h for plasma glucose (B) was calculated. Data are presented as the means ± S.E.M. (n = 5 for normal group; n = 6 for other groups). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus vehicle. ††, p < 0.01; †††, p < 0.001 versus normal.
Effects of Chronic Treatment of ZDF Rats with KGA-2727.
The changes in body weight of ZDF rats chronically treated with KGA-2727 or acarbose are shown in Table 2. The body weight of the obese rats was significantly higher than that of the lean rats throughout the observation period. The increase in body weight of the obese control slowed down after day 38, but not that of the KGA-2727 or acarbose-treated obese groups. As a result, rats treated with KGA-2727 or acarbose gained more weight than the obese control rats at the endpoint (Table 2). KGA-2727 reduced food consumption, and acarbose tended to reduce food consumption on day 44 (Table 2). The amounts of drug intake, calculated from food consumption from days 23 to 44, were as follows: KGA-2727 at 10, 30, and 100 ppm, 0.7, 1.8, and 6.0 mg/kg/day, respectively and acarbose at 100 ppm, 6.7 mg/kg/day. The levels of alanine aminotransferase and aspartate aminotransferase, markers of hepatic damage, were not elevated in the KGA-2727-treated groups or the acarbose-treated group (data not shown).
Chronic effects of KGA-2727 or acarbose on body weight and food consumption in Zucker diabetic fatty rats
Data are presented as the means ± S.E.M. (n = 8).
The plasma glucose level (Fig. 6A) and GHb (Fig. 6B) in the obese control group were significantly higher than that in the lean control group and increased with time. KGA-2727 reduced the plasma glucose and GHb in a dose-dependent manner (Fig. 6). KGA-2727 almost normalized the plasma glucose level at 30 ppm or higher. Acarbose (100 ppm) also attenuated the increases in plasma glucose and GHb to the same levels as did 10 ppm of KGA-2727.
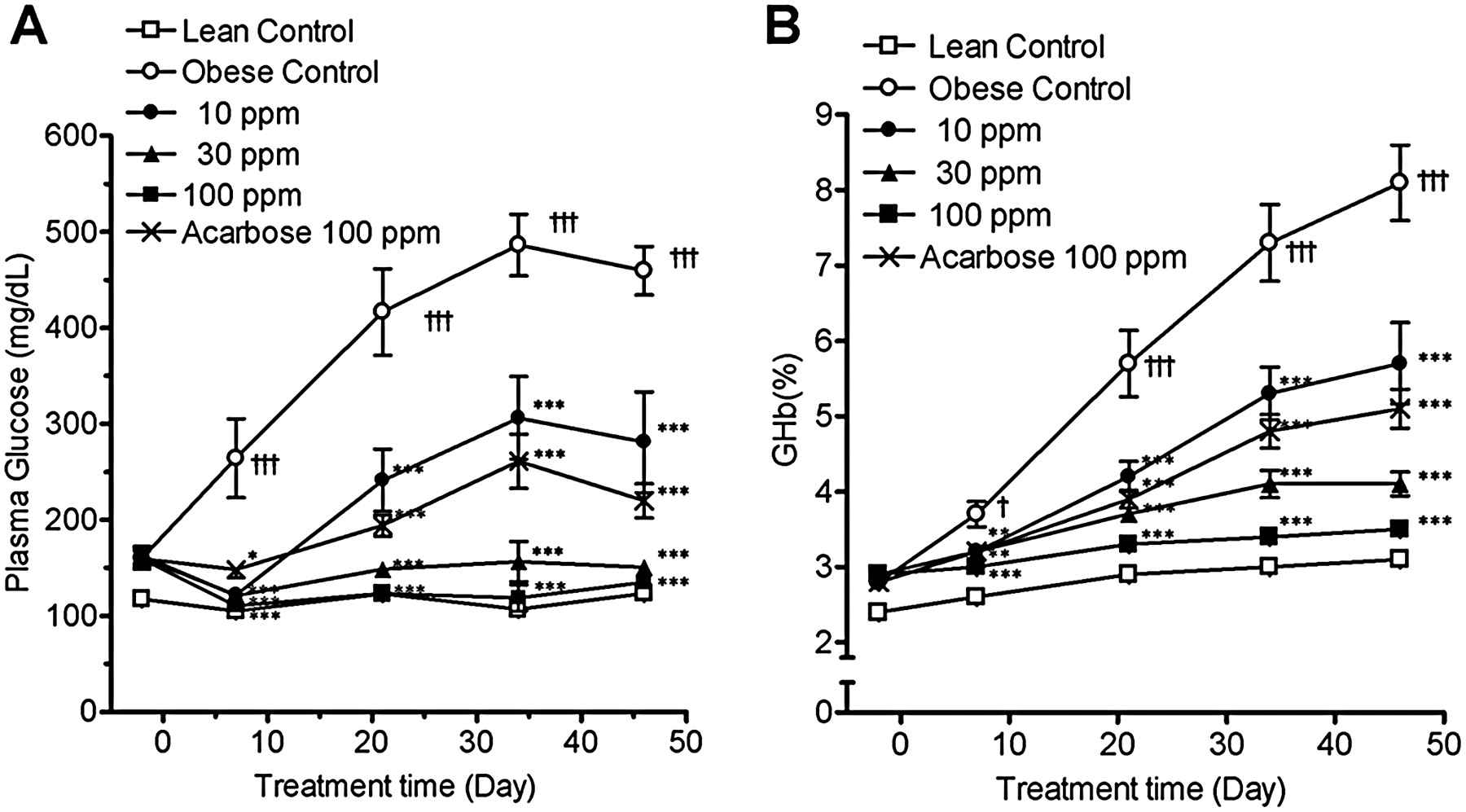
Changes in plasma glucose and GHb during chronic treatment of Zucker diabetic fatty rats with KGA-2727 or acarbose. The plasma glucose concentration (A) and GHb (B) were measured in the ZDF rats and the ZDF-lean rats fed a powder diet containing the indicated test compound in the fed state. Data are presented as the means ± S.E.M. (n = 8). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus the obese control group. †, p < 0.05; †††, p < 0.001 versus the lean control group.
In urinalysis, urinary glucose excretion (Fig. 7A) and urine volume (Fig. 7B) of the obese control group were significantly increased compared with those of the lean control group on day 19. KGA-2727 inhibited both increases in a dose-dependent manner at day 19 (Fig. 7). In addition, KGA-2727 at 30 or 100 ppm significantly reduced urinary glucose excretion on day 19 or 40 compared with that on day −2 (Fig. 7A).

Changes in urinary glucose excretion and urine volume in Zucker diabetic fatty rats during chronic treatment with KGA-2727 or acarbose. On days −2, 19, and 40, the urine samples were collected for 24 h. The urinary glucose excretion (A) was calculated from the urine volume (B) and glucose concentration. Data are presented as the means ± S.E.M. (n = 8). **, p < 0.01; ***, p < 0.001 versus the obese control. #, p < 0.05; ##, p < 0.01; ###, p < 0.001 versus day −2. †††, p < 0.001 versus the lean control group.
To evaluate insulin secretion, we performed an oral glucose tolerance test on day 37. The fasting plasma glucose (0 h) in the obese control group was significantly higher than that in the lean control group (Fig. 8A). Plasma glucose after glucose administration in the obese control group was significantly higher than that in the lean control group. KGA-2727 or acarbose significantly reduced both the fasting and the postprandial plasma glucose compared with those in the obese control group (Fig. 8, A and C). The plasma insulin was transiently elevated after glucose administration in the KGA-2727 and acarbose groups but not in the lean or obese control groups (Fig. 8, B and D).

Change in plasma glucose and insulin levels in Zucker diabetic fatty rats subjected to the oral glucose tolerance test after chronic treatment with KGA-2727 or acarbose. On day 38, the rats were orally administered only glucose (2 g/kg body weight) after an overnight fast. The plasma glucose (A) and insulin (B) were measured, and the AUC0–4 h for plasma glucose (C) and the AUC0–2 h for plasma insulin (D) were calculated. Data are presented as the means ± S.E.M. (n = 8). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus the obese control group. †, p < 0.05; ††, p < 0.01; †††, p < 0.001 versus the lean control group.
In morphometric analyses of the pancreas, Ins-I indicates the insulin content of the islets, whereas Fib-I indicates the extent of fibrosis in them. A decrease in Ins-I and an increase in Fib-I were observed in the obese control group compared with the lean control group (Table 3). KGA-2727 improved these indices in a dose-dependent manner. Acarbose did not cause significant improvement of either index. In morphometric analysis of kidney, the sclerotic changes of renal glomerulus were almost mild, and there were not marked difference in all groups except for the lean control (Table 4). Therefore, there were no effects of drugs on glomerular changes. KGA-2727 or acarbose decreased endothelial glycogen deposition and dilatation of the distal renal tubules (Table 4). This beneficial change in glycogen deposition was observed in all rats treated with 100 ppm of KGA-2727.
Chronic effects of KGA-2727 or acarbose on Ins-I and Fib-I of the pancreas in Zucker diabetic fatty rats
Data are presented as the means ± S.E.M. (n = 8).
Chronic effects of KGA-2727 or acarbose on morphometric changes in the kidneys of Zucker diabetic fatty rats
Data are presented as the means ± S.E.M. or as the number of animals in each grade (n = 8).
The portal concentration of GLP-1 was not significantly different between the lean control and obese control groups (Fig. 9A). KGA-2727 (30 and 100 ppm) significantly increased the plasma GLP-1 concentration compared with that in the obese control group (Fig. 9A). Acarbose significantly did so as well. On the other hand, the concentration of GIP was not significantly different in all groups (Fig. 9B).

Effect of KGA-2727 or acarbose on portal concentration of GLP-1 and GIP in Zucker diabetic fatty rats. After chronic treatment for 48 days, the blood was collected via the portal vein in the fed state. The plasma GLP-1 (A) and GIP (B) concentrations were measured. Data are presented as the means ± S.E.M. (n = 8). *, p < 0.05; ***, p < 0.001 versus the obese control group.
Discussion
In the present work, we described the efficacy of a novel selective SGLT1 inhibitor, KGA-2727, as an antidiabetic drug. Recently, SGLTs have attracted attention as targets for antidiabetic drugs. Many of the reported SGLT inhibitors have high affinity for SGLT2, which is the predominant transporter that mediates renal glucose reabsorption in the proximal tubule. However, there are no reports characterizing SGLT1-selective inhibitors. We focused on SGLT1, which plays a critical role in glucose absorption from the gastrointestinal tract, as a molecular target based on the concept that inhibition of accelerated glucose absorption by SGLT1 would lead to control of the blood glucose level.
To develop a selective SGLT1 inhibitor, we selected a pyrazole-O-glucoside structure as the basal scaffold structure. We evaluated the potency of KGA-2727 against human and rat SGLTs in cells transiently expressing these transporters and observed dose-dependent inhibition by the compound. Based on Ki values, we confirmed KGA-2727 to be a potent and highly selective inhibitor of SGLT1. The selectivity ratios of KGA-2727 were far higher than those of phlorizin (Table 1). According to Oku et al. (1999), the selectivity ratio (human SGLT2/human SGLT1) of 3-(1-benzofuran-5-yl)-1-(2-hydroxy-4-methyl-6-{[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl]oxy}phenyl)propan-1-one (T-1095A), a nonselective SGLT inhibitor, is 0.25, and that of phlorizin is 1. Other SGLT inhibitors reported so far are selective SGLT2 inhibitors. For instance, dapagliflozin and remogliflozin, respectively, have 1200- and 365-fold specificity for SGLT2 versus SGLT1 (Fujimori et al., 2008; Meng et al., 2008). These results indicate that KGA-2727 is a novel selective SGLT1 inhibitor.
Carbohydrates are an important component of the diet and range from simple monosaccharides to complex polysaccharides. The ingested carbohydrates are hydrolyzed by digestive enzymes in the gastrointestinal tract. α-Glucosidase cleaves the α-glucoside bonds of polysaccharide to yield monosaccharides. The resulting monosaccharides are then absorbed via different types of transporters in the small intestine. The origin of glucose within the gastrointestinal tract is carbohydrates such as starch, sucrose, and glucose in foods and drinks. It is known that glucose and galactose absorption occurs via SGLT1, whereas fructose absorption occurs via GLUT5,which is located on epithelial cells in the small intestine (Levin, 1994). α-Glucosidase inhibitors inhibit the digestion of complex carbohydrates to monosaccharides.
KGA-2727 decreased the absorption rate of glucose but not that of fructose (Fig. 3). Therefore, these results indicate that KGA-2727 inhibits SGLT1 but not GLUT5. In cells transiently expressing GLUT5, KGA-2727 had no effect on fructose uptake (data not shown). Although phlorizin showed SGLT inhibition potency (Table 1), it caused only a nonsignificant decreasing trend in glucose absorption from the small intestine (Fig. 3). This is probably because phlorizin is rapidly hydrolyzed to phloretin and glucose in this tissue (Ehrenkranz et al., 2005).
The residual glucose in the gastrointestinal tract was elevated in the KGA-2727-treated groups, but not in the acarbose-treated group (Fig. 4). In the oral glucose tolerance test, KGA-2727 suppressed postprandial hyperglycemia in streptozotocin-induced diabetic rats, but acarbose could not (Fig. 5). These differences between KGA-2727 and acarbose are accounted for by the above-mentioned mechanisms; KGA-2727 inhibits glucose absorption, whereas acarbose inhibits metabolism to glucose in the lumen of the gastrointestinal tract. In the oral glucose tolerance test, the loading carbohydrate is glucose. Therefore, acarbose could not inhibit the absorption of glucose, and the plasma glucose was elevated after glucose loading (Fig. 5). High-fructose corn syrup and other forms of sugar that are rich in glucose are routinely added to processed food and many types of beverages (Bray et al., 2004; Malik et al., 2010). Thus, diabetic patients have many opportunities to take in carbohydrates as glucose. Therefore, KGA-2727, which suppresses postprandial hyperglycemia after glucose intake, may have an advantage over α-glucosidase inhibitors as an antidiabetic medication.
The most common adverse effects of α-glucosidase inhibitors are abdominal distension and flatulence (Chiasson et al., 2002; Kawamori et al., 2009). If the dosage is too high relative to the amount of carbohydrate in the meal, undigested carbohydrates pass in to the large bowel. Carbohydrates fermented by the intestinal flora cause the abdominal symptoms. In view of this, KGA-2727 also may cause the abdominal symptoms as an adverse effect as well as α-glycosidase inhibitors. However, the obvious adverse effects were not observed at the dose that used in this study.
In diabetes, hyperglycemia on its own contributes to the development of disturbed insulin secretion by pancreatic β-cells and insulin resistance (Rossetti et al., 1987). Hyperglycemia causes progressive damage to pancreatic β-cells through endoplasmic reticulum stress and oxidative stress and consequently diminishes insulin secretion (Robertson et al., 2003; Prentki and Nolan, 2006; Eizirik et al., 2008). The blood insulin level in the ZDF rat increases to compensate for insulin resistance until 10 weeks of age and then decreases along with the progressive dysfunction of pancreatic β-cells (Sugimoto et al., 2008). Several studies have shown that pharmacological treatment to control hyperglycemia results in preservation of pancreatic β-cells in rodent models (Koyama et al., 2000; Fukaya et al., 2009). For example, an α-glucosidase inhibitor preserves pancreatic β-cells in Goto-Kakizaki rats, a model for type 2 diabetes (Goda et al., 2007). In our results, chronic treatment with KGA-2727 also exhibited protective effects on the insulin contents or fibrotic changes of pancreatic β-cells (Table 3) through the improvement of hyperglycemia (Fig. 6) in the ZDF rats. Furthermore, the chronic treatment of ZDF rats with KGA-2727 preserved their ability to secrete insulin from pancreatic β-cells through improving hyperglycemia in the ZDF rats (Fig. 8). The lean rats, without insulin resistance, would be able to control their plasma glucose with a much lower amount of insulin than the obese rats with insulin resistance. Therefore, no increase in the plasma insulin after glucose loading was observed in the lean control group (Fig. 8). Our results suggest that the amelioration of hyperglycemia with KGA-2727 prevents the development of pancreatic β-cells exhaustion and improves glucose metabolism.
Hyperglycemia causes damages to the kidneys and pancreatic β-cells (Yamagishi et al., 2007). According to Coimbra et al. (2000), glomerulosclerosis was first noted in 18-week-old ZDF rats. In our study, the assessment of glomerulosclerosis was performed in 13-week-old ZDF rats. Therefore, the sclerotic changes of renal glomerulus were almost mild (Table 4). On the other hand, the morphometric changes in the distal tubules were observed in the obese control group (Table 4). KGA-2727 ameliorated these changes in the distal tubules and decreased urinary glucose and volume (Table 4; Fig. 7). Therefore, the decreases in urinary glucose and volume induced by the chronic treatment with KGA-2727 were associated with the morphometric changes in the distal tubules rather than the glomerulosclerosis.
Thus, like chronic treatment with acarbose, that with KGA-2727 suppressed the development of diabetic conditions such as dysfunction of pancreatic β-cells and the kidneys, and that suppression was a consequence of attenuation of the postprandial hyperglycemia. These results show that KGA-2727 had efficacy in the treatment of diabetes comparable with that of acarbose, a widely used antidiabetic agent.
It has been reported that α-glucosidase inhibitors enhance and prolong GLP-1 secretion in human and rodent models (Qualmann et al., 1995; Moritoh et al., 2009). In our results, chronic treatment with KGA-2727 increased the portal GLP-1 concentration in ZDF rats (Fig. 9A). GLP-1 is secreted from intestinal endocrine L-cells, which are located mainly in the distal ileum and colon, and exerts glucoregulatory actions. GLP-1 acts on pancreatic β-cells, and that action leads to glucose-dependent insulin secretion, induction of β-cell proliferation, and enhanced resistance to apoptosis (Baggio and Drucker, 2007). GLP-1 also reduces food intake by direct action on central nervous systems that regulate ingestive behavior and by inhibiting gastric emptying (Baggio and Drucker, 2007). In our results, the reduction of food consumption was observed in the drug-treated groups (Table 2) that showed higher concentrations of GLP-1 (Fig. 9A). This enhanced GLP-1 secretion may have contributed in part to the antidiabetic effects of KGA-2727. According to Moritoh et al. (2009), delayed absorption of carbohydrates caused by α-glucosidase inhibitors might have been responsible for the increased GLP-1 observed. GLP-1 release can be stimulated by nutrients including glucose and other sugars (Baggio and Drucker, 2007; Gorboulev et al., 2012). KGA-2727 increased the amount of glucose in the distal part of the small intestine (Fig. 4), which possesses many GLP-1-secreting cells. The glucose increased in the distal intestine could thus stimulate GLP-1 secretion.
In clinical studies, pharmacological interventions have been used to investigate whether drugs delay the onset of type 2 diabetes. α-Glucosidase inhibitors (acarbose and voglibose) prevent the progression of impaired glucose tolerance to type 2 diabetes by improving postprandial hyperglycemia (Chiasson et al., 2002; Kawamori et al., 2009). Likewise, KGA-2727 may delay the onset of type 2 diabetes. We suggest that KGA-2727 represents an attractive therapeutic candidate for the treatment of diabetes and impaired glucose tolerance.
Authorship Contributions
Participated in research design: Itoh and Isaji.
Conducted experiments: Shibazaki, Tomae, Ishikawa-Takemura, and Itoh.
Contributed new reagents or analytic tools: Fushimi.
Performed data analysis: Shibazaki, Tomae, Ishikawa-Takemura, and Itoh.
Wrote or contributed to the writing of the manuscript: Shibazaki, Tomae, Ishikawa-Takemura, Fushimi, Itoh, Yamada, and Isaji.
Footnotes
This work was supported by the Kissei Pharmaceutical Co., Ltd.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
ABBREVIATIONS:
- SGLT1
- high-affinity sodium glucose cotransporter
- SGLT2
- low-affinity sodium glucose cotransporter
- AMG
- methyl-α-d-glucopyranoside
- AUC
- area under the curve
- Fib-I
- fibrosis index
- GHb
- glycated hemoglobin
- GIP
- glucose-dependent insulinotropic polypeptide
- GLP-1
- glucacon-like peptide-1
- GLUT5
- glucose transporter type 5
- Ins-I
- insulin index
- ZDF
- Zucker diabetic fatty
- KGA-2727
- 3-(3-{4-[3-(β-d-glucopyranosyloxy)-5-isopropyl-1H-pyrazol-4-ylmethyl]-3-methylphenoxy}propylamino)propionamide
- T-1095A
- 3-(1-benzofuran-5-yl)-1-(2-hydroxy-4-methyl-6-{[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl]oxy}phenyl)propan-1-one.
- Received February 10, 2012.
- Accepted April 23, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}