


Abstract
Abediterol is a novel potent, long-acting inhaled β2-adrenoceptor agonist in development for the treatment of asthma and chronic obstructive pulmonary disease. Abediterol shows subnanomolar affinity for the human β2-adrenoceptor and a functional selectivity over β1-adrenoceptors higher than that of formoterol and indacaterol in both a cellular model with overexpressed human receptors and isolated guinea pig tissue. Abediterol is a full agonist at the human β2-adrenoceptor (Emax = 91 ± 5% of the maximal effect of isoprenaline). The potency and onset of action that abediterol shows in isolated human bronchi (EC50 = 1.9 ± 0.4 nM; t½ onset = 7–10 min) is not significantly different from that of formoterol, but its duration of action (t½ ∼ 690 min) is similar to that of indacaterol. Nebulized abediterol inhibits acetylcholine-induced bronchoconstriction in guinea pigs in a concentration-dependent manner, with higher potency and longer duration of action (t½ = 36 h) than salmeterol (t½ = 6 h) and formoterol (t½ = 4 h) and similar duration of action to indacaterol up to 48 h. In dogs, the bronchoprotective effect of abediterol is more sustained than that of salmeterol and indacaterol at doses without effects on heart rate, thus showing a greater safety margin (defined as the ratio of dose increasing heart rate by 5% and dose inhibiting bronchospasm by 50%) than salmeterol, formoterol, and indacaterol (5.6 versus 3.3, 2.2, and 0.3, respectively). In conclusion, our results suggest that abediterol has a preclinical profile for once-daily dosing in humans together with a fast onset of action and a favorable cardiovascular safety profile.
Introduction
Asthma and chronic obstructive pulmonary disease (COPD) are two of the most common chronic inflammatory diseases, affecting approximately 300 million and 80 million people, respectively, worldwide. Projections from the World Health Organization estimate that the incidence of both diseases will increase globally in the near future, placing an enormous burden on patients, families, and society (Masoli et al., 2004; Løkke et al., 2006). Both diseases are characterized by airflow obstruction, and the use of bronchodilator therapy is recommended in current international guidelines (www.ginasthma.org; www.goldcopd.org). In COPD, bronchodilators are the cornerstone of pharmacologic therapy, with inhaled β2-adrenoceptor agonists as one of the first-line therapies, together with anticholinergic drugs (Hanania and Donohue, 2007; Tashkin and Fabbri, 2010). In asthma, the use of long-acting β2-adrenoceptor agonists (LABAs) has been recently reviewed by the Food and Drug Administration, which announced that LABAs should never be used as a monotherapy in children or adults (http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2010). However, when used in combination with inhaled corticosteroids, LABAs remain critical in the symptomatic management of the disease (Lipworth, 2007; Robinson, 2010).
β-Adrenoceptors have been subdivided into three distinct subtypes: β1, β2, and β3, classically identified in cardiac, airway smooth muscle, and adipose tissues, respectively (Johnson, 2006). β2-Adrenoceptor agonists act by binding to the β2-adrenoceptor, which activates adenylate cyclase via the signal-transducing Gs protein, thereby enhancing cellular cAMP levels and activating protein kinase A. One of the consequences of this signaling is a fall in intracellular calcium and the promotion of smooth muscle relaxation (Giembycz and Newton, 2006). Apart from the affinity for β2-receptors, the potency of agonists also depends on the intrinsic efficacy of each compound. Intrinsic efficacy refers to the ability of a drug to activate its receptor, regardless of the drug concentration. Based on their intrinsic efficacy, β2-agonists are classified as full or partial agonists. Full agonists are defined as compounds that completely activate a receptor, whereas partial agonists are defined as compounds that only partially activate a receptor (Hanania et al., 2002; Kurose, 2004).
The first β2-selective adrenoceptor agonists used in respiratory diseases were the short-acting agonists salbutamol and terbutaline, with 4- to 6-h duration of bronchodilator action (Waldeck, 2002). Although salbutamol is widely used for the relief of acute bronchoconstriction, its short duration of action makes it less appropriate for regular use as maintenance medication (Beeh and Beier, 2010; Cazzola et al., 2010). The concept of LABAs was introduced with the development of twice-daily drugs such as formoterol and salmeterol, with a bronchodilator effect lasting for 12 h. Both compounds have been widely used as maintenance medication in asthma and COPD, demonstrating better efficacy than salbutamol in both diseases (Waldeck, 2002; Beeh and Beier, 2010). Because of the central role of bronchodilators in the treatment of both diseases, there is an interest in developing once-daily LABAs as an attempt to improve patient compliance and disease control (Cazzola et al., 2010). Indeed, the development of new inhaled corticosteroids, which can be used as a once-daily regimen, has provided additional rationale for the development of new ultra-LABAs that can be used in once-a-day combinations (Fitzgerald and Fox, 2007).
The most common dose-related side effects associated with β2-adrenoceptor agonists are tachycardia, muscle tremor, hypokalemia, and hyperglycemia (Rabe, 2003). Mild tachycardia is a common side effect observed in patients treated with β2-adrenoceptor agonists, which in part may result from the activation of β2-adrenoceptors present in peripheral vasculature, leading to vasodilation and reflex tachycardia, and may also be the consequence of the activation of cardiac β1-adrenoceptors, increasing heart rate directly (Sears, 2002). The stimulation of β2-adrenoceptors linked to the membrane-bound Na,K- ATPase on skeletal muscle cells leads to an influx of potassium into the cells, which may cause skeletal muscle tremor and indirectly cardiac effects (Sears, 2002). β-Agonists also may affect glucose homeostasis by modulating insulin secretion, liver metabolism, and uptake of glucose (Philipson, 2002). To reduce the potential of systemic adverse events, a new generation of β2-adrenoceptor agonists has been designed to have a higher therapeutic margin than marketed drugs, by reducing systemic compound exposure and/or having higher selectivity versus the β1-adrenoceptor.
Abediterol [5-((1R)-2-{[6-(2,2-difluoro-2-phenylethoxy)hexyl]amino}-1-hydroxyethyl)-8-hydroxyquinolin-2(1H)-one] (Fig. 1), previously known as LAS100977, is a potent and selective inhaled β2-adrenoceptor agonist with a duration of action compatible with once-daily dosing in humans, a fast onset of action, and a favorable safety margin in preclinical models. In this article, we report the pharmacological characterization of abediterol in in vitro and in vivo functional models compared with other marketed inhaled LABAs such as salmeterol, formoterol, and indacaterol.

Chemical structure of abediterol.
Materials and Methods
Materials and Drug Preparation
Abediterol, salmeterol α-hydroxynaphthoate, formoterol fumarate, indacaterol maleate, and propranolol hydrochloride were synthesized by the Department of Medicinal Chemistry of Almirall (Barcelona, Spain); 1-[2-((3-carbamoyl-4-hydroxy)phenoxy)ethylamino]-3-[4-(1-methyl-4-trifluoromethyl-2-imidazolyl) phenoxy]-2-propanol methanesulfonate (CGP20712A), (±)-erythro-(S*,S*)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3[(1-methylethyl)amino]-2-butanol hydrochloride (ICI-118551), 3-isobutyl-1-methylxanthine (IBMX), isoprenaline hemisulfate, acetylcholine (Ach) hydrochloride, carbachol chloride, salbutamol, zileuton, fexofenadine, theophylline, and phosphate-buffered saline with calcium and magnesium were purchased from Sigma-Aldrich (Tres Cantos, Spain). Alprenolol was purchased from Sigma/RBI (Natick, MA). Hanks' buffered salt solution was obtained from Invitrogen (Carlsbad, CA). Ketamine hydrochloride (Imalgene) was purchased from Merial (Barcelona, Spain), and xylazine hydrochloride (Rompun) was purchased from Bayer (Barcelona, Spain). Acepromazine maleate (Calmoneosan) was purchased from Pfizer Salud Animal (Alcobendas, Spain), and propofol (Lipuro) was obtained from B. Braun Surgical (Rubí, Spain).
Membrane preparations expressing human β1- and β2-adrenergic receptors (obtained from Sf9 cells) were purchased from Membrane Target Systems (PerkinElmer Life and Analytical Sciences, Waltham, MA). Membranes were prepared from HTB-10 cells (human SK-N-MC neurotumor cells) obtained from the American Type Culture Collection (Manassas, VA) and LGC Standards (Teddington, UK) and used as a source of β3-adrenergic receptor. U937 cells (a human leukemic monocyte lymphoma cell line from the American Type Culture Collection) were used as a source of β2-receptor. [3H]4-[3-(tert-butylamino)-2-hydroxypropoxy]-1,3-dihydrobenzimidazol-2-one (CGP12177) was obtained from PerkinElmer Life and Analytical Sciences. (−)-3-[125I]iodocyanopindolol was obtained from GE Healthcare (Chalfont St. Giles, Buckinghamshire, UK).
Human lung tissue was obtained from patients undergoing surgery for lung carcinoma. None of the patients had a history of asthma. The protocol was approved by the Ethics Committee of University Clinic Hospital (Valencia, Spain), and informed consent was obtained from all patients.
Agonists were dissolved in dimethyl sulfoxide for binding and cellular studies. For in vitro isolated organ and in vivo studies, compounds were dissolved in a maximum of 2% (v/v) HCl or 2% (v/v) NaOH, and when required, in the presence of polyethylene glycol 300 (the maximal percentage was 5%). Krebs-Henseleit solution (guinea pig trachea and left atria studies) was composed of 118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 25 mM NaHCO3, 1.2 mM KH2PO4, 5.5 mM glucose, and 2.6 mM CaCl2. Test compounds were kept under dry conditions and prepared daily.
Animals
Male Dunkin-Hartley guinea pigs (450–600 g at the time of experimental procedures) were obtained from Charles River Laboratories (Les Oncins, France). Guinea pigs were housed in groups of four or five at 20 to 24°C and 40 to 70% humidity. Room air was changed 10 times per hour, and the light/dark cycle was 12 h. Food [maintenance diet for guinea pigs with vitamin C (SAFE 114; SAFE, Augy, France)] and water were available ad libitum. Male Beagle dogs (9–20 kg at the time of experimental procedures) were supplied by guaranteed commercial suppliers [Harlan Winkelman (Barchen, Germany); Harlan France, SARL (Gannat, France); Green Hill (Montichiari, Brescia, Italy); Morini (San Polo D'enza, Italy); Centre d'elevage du domaine des souches (Domaine des Souches, Mezilles, France); Marshall Farms (North Rose, NY); and R.C. Hartelust BV (Tilburg, The Netherlands)]. Dogs were housed at 15 to 21°C and 40 to 70% humidity, under a 12-h light/dark cycle, and fed a maintenance diet (Harlan Teklad, Madison, WI) with free access to water. All experiments were carried out with the approval of the Animal Ethics Committee of Almirall (Barcelona, Spain) and conducted in accordance with guidelines approved by the European Community (Directive 86/609/CEE) and the Catalan Parliament (Decret 214/1997).
Radioligand Displacement Studies
Affinity for Human β1- and β2-Adrenergic Receptors.
The affinity of abediterol, salmeterol, formoterol, and indacaterol for human β1- and β2-adrenergic receptors was determined by measuring their ability to displace the binding of [3H]CGP12177 to cell membrane preparations expressing one of the receptor subtypes. Protein concentrations were 16 and 5 μg/well for β1- and β2-receptor membrane preparations, respectively. Radioligand concentration was 0.14 and 0.6 nM for the β1 and β2 assays, respectively. A range of agonist concentrations was tested in duplicate to generate competition curves. Nonspecific binding was measured in the presence of 1 μM propranolol. Assay reagents were dissolved in assay binding buffer (25 mM MgCl2 and 2 mM EDTA, pH 7.4) in a total volume of 250 μl. After 60 min of incubation at room temperature with gentle shaking in GF/C Multiscreen 96-well plates (Millipore Corporation, Barcelona, Spain), previously treated with assay buffer containing 0.3% polyethyleneimine (Sigma-Aldrich, St. Louis, MO), binding reactions were terminated by filtration and washing with 2.5 volumes of 50 mM Tris/HCl, pH 7.4. Filters were then dried for 30 min before the addition of 30 μl of OptiPhase Hifase II (PerkinElmer Life and Analytical Sciences), and radioactivity was quantified by using a MicroBeta Trilux microplate scintillation counter (PerkinElmer Life and Analytical Sciences).
Affinity for Human β3-Adrenergic Receptors.
The affinity for human β3-adrenergic receptors was determined by measuring the ability to displace the binding of (−)-3-[125I]iodocyanopindolol to cell membrane preparations expressing this receptor. Human SK-N-MC neurotumor cells were grown, and the membranes were prepared following methods described previously (Curran and Fishman, 1996). Membranes were incubated with 1 nM radioligand and 0.3 μM CGP20712A, the β1-antagonist. Assay buffer was 50 mM HEPES, 4 mM MgCl2, and 0.4% bovine serum albumin, pH 7.5. The final volume of each reaction was 250 μl. Nonspecific binding was determined in the presence of 100 μM alprenolol. Samples were incubated for 90 min at 30°C with shaking. Binding reactions were terminated by filtration through Whatman GF/C membranes (Brandel Inc., Gaithersburg, MD), prewet in assay buffer at 4°C, using an M-24 harvester (Brandel Inc.). Filters were washed three times with 4 ml each of 50 mM Tris/HCl and 4 mM MgCl2, pH 7.4. Filters were then dried for 30 min before the addition of 5 ml of OptiPhase Hifase 2, and radioactivity was quantified by using the LKB 1219 RackBeta counter (PerkinElmer Life and Analytical Sciences).
Data Analysis.
The affinity of each test compound for a given β-adrenoceptor was determined by using at least six different concentrations ran in duplicate. Half-maximal inhibitory concentration (IC50) values were obtained by nonlinear regression using Activity Base (IDBS Ltd, Guildford, UK) and the four parameters-log equation.
Intrinsic Efficacy
Intrinsic efficacy was determined by measuring the intracellular accumulation of cAMP induced by the interaction of agonists with the human β2-adrenoceptors present in U937 cells. Cells were recovered and washed, and the pellet was dissolved in assay buffer with the following composition: Hanks' buffered salt solution with 10 mM HEPES, 1 mM ascorbic acid, 0.1% bovine serum albumin, and 500 μM IBMX. After counting and making the appropriate dilution, 50,000 cells were dispensed in each well of a 96-well microtiter plate. cAMP was measured as an indicator of the receptor function after stimulation with 1 μM isoprenaline (100% stimulation) or the appropriate serial dilutions of the tested compounds, using the cAMP dynamic homogeneous time-resolved fluorescence kit from Cisbio International (Bagnols/Ceze, France).
The efficacy of each test compound on the receptor was determined by using at least seven different concentrations run in duplicate. Maximal (Emax) and half-maximal (EC50) stimulations of each compound relative to 1 μM isoprenaline were calculated by using Activity Base.
Selectivity Studies
cAMP Production in β1-, β2-, and β3-Overexpressing Cells.
Assays to determine selectivity for human β-adrenergic receptors were performed at Euroscreen S.A. (Brussels, Belgium). In brief, CHO-K1 cells expressing the human adrenergic β1-, β2-, or β3- receptor were detached by gentle flushing with phosphate-buffered saline-EDTA (5 mM EDTA), recovered by centrifugation, and resuspended in assay buffer (Krebs-Ringer-HEPES and 1 mM IBMX) at a concentration of 5000 cells per well. The reference compounds were epinephrine, formoterol hemifumarate, and (S)-4-[2-hydroxy-3-phenoxypropylaminoethoxy]-N-(2-methoxyethyl)phenoxyacetamide (ZD7114) for β1-, β2-, and β3-receptors, respectively.
Cells were incubated with test compounds in 96-well plates for 30 min at room temperature. During the incubation, the anti-cAMP cryptate antibody and the cAMP-D2 were added to each well according to the kit specifications (homogeneous time-resolved fluorescence kit from CisBio International). The plate was covered and incubated for at least 1 h at room temperature. The plate was then read on the RUBYstar-High-Performance Time-Resolved Fluorescence Microplate Reader (BMG Labtech GmbH, Offenburg, Germany). Data were analyzed by nonlinear regression by using a single-site model, and EC50 values for each β-adrenergic receptor subtype were reported.
Effect on Spontaneous Tone Guinea Pig Trachea.
The β2 functional activity of compounds was assessed in guinea pig tracheal rings by measuring their ability to relax tracheal smooth muscle as described previously (Cortijo et al., 1991). In brief, after the equilibration period, isoprenaline was added at a concentration of 0.1 μM to test tracheal ring relaxation. Preparations were then washed twice with Krebs-Henseleit solution and left to recover for 15 to 30 min. Cumulative concentration-response curves (CRCs) were built for each compound (0.01 nM to 0.1 μM). Incubation of each concentration did not exceed 30 min. At the end of the experiment, 0.1 μM isoprenaline was added to each preparation to obtain maximal relaxation.
Data Analysis.
β2-Adrenergic functional activity was determined through the quantification of the relaxation produced by each concentration of compound with respect to the response evoked by the initial administration of 0.1 μM isoprenaline, which was considered as maximum and therefore equal to 100%. Potency values were expressed as the concentration required to induce a 50% of maximal relaxation (EC50), calculated by using Activity Base and the four parameters-log equation. IC50 values were compared by using the t test.
Effect on Guinea Pig Left Atria.
β1-Adrenergic functional activity of each compound was assessed in guinea pig left heart atria by measuring their ability to increase force of contraction. In brief, atria were dissected and suspended in water-jacketed organ baths containing 30 ml of Krebs-Henseleit solution at 37°C and bubbled with 5% CO2 in oxygen. Isolated atria were connected with cotton thread to an isometric transducer TRI 201,202 (Panlab, Cornellà del Llobregat, Spain) under a resting tension of 1 g. This transducer was connected to a PowerLab system 8/30 (ADInstruments Ltd., Chalgrove, Oxfordshire, UK) to measure changes in tension and then paced with a field stimulator Hugo Sachs Electronic type D7806 (Harvard Apparatus, March-Hugstetten, Germany) at a frequency of 1 Hz (supramaximal voltage, 0.1 ms). Tissues were left for 45 min to stabilize for basal contractions. Initially, 0.1 μM isoprenaline was added to the bath twice to test atria responsiveness. Atria were then washed twice with Krebs-Henseleit solution and left to recover for approximately 15 min. For each compound, a range of increasing and cumulative concentrations (1 nM to 10 μM) was added every 10 to 15 min to allow the reading of a stable effect. After the last administration, atria were washed with Krebs-Henseleit solution, and 0.1 μM isoprenaline was administered to confirm that the maximal contraction was still achieved.
Data Analysis.
β1-Adrenergic functional activity was determined through the quantification of the contraction produced by each concentration of compound with respect to the response evoked by 0.1 μM isoprenaline, which was considered the maximal response and therefore equal to 100%. Potency values were expressed as the concentration required to induce a 50% of maximal contraction (EC50), calculated by using Activity Base and the four parameters-log equation.
Characterization in Human Bronchi
Relaxant Effect on Spontaneous Tone Isolated Human Bronchi.
After the resection of one or more lung lobes, a piece of macroscopically normal tissue at a distance from the malignancy was supplied by the hospital pathologist, submerged in physiological salt solution (PSS; 118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1.1 mM KH2PO4, 25.0 mM NaHCO3, and 11.7 mM glucose) at 4°C and transported to the laboratory where subsegmental bronchi were dissected free from parenchymal lung tissue and preparations were cut (3–4 mm length × 2–4 mm internal diameter) as reported previously (Cortijo et al., 2003). Bronchial rings were suspended in parallel on tissue hooks in 10-ml organ baths containing PSS and gassed with 5% CO2 in O2 at 37°C, pH 7.4. Each preparation was connected to UF1 (Harvard Apparatus) or Grass FTO3 (Grass Instruments, Quincy, MA) force displacement transducers, and isometric tension changes were recorded by using standard software (Proto5, Letica, Barcelona, Spain or PowerLab). The preparations were suspended under an initial load of 2 g and equilibrated for 60 to 90 min with changes in bath PSS every 15 to 20 min before any pharmacological intervention occurred; at the end of equilibration period, the resting load was stable between 1 and 2 g.
To assess potency, human isolated bronchial rings were first contracted with 10 μM Ach to test viability. After washing and re-equilibration, relaxation-response curves were obtained in preparations at spontaneous tone. Cumulative CRCs to tested compounds were obtained. After the maximal effect of each relaxant drug was obtained, theophylline (3 mM) was added to the bath to determine the maximal relaxation of the preparation. Only one CRC to a relaxant agonist was recorded in each ring.
To assess onset and duration of action, the preparation's viability and responsiveness was tested by adding Ach (10 μM). After washing and re-equilibration, a single concentration that produced approximately 40 to 60% of relaxation was applied. Then, after 30 min, the preparation was washed and continuously superfused, and the changes in tone were followed up to 14 to 15 h. Control preparations from the same donor received the vehicle used in each case and were followed during the same period of time to check the preparation's stability.
Onset and Duration of Action in EFS Human Bronchi.
In separate experiments, onset and duration of action of agonists was assessed by using electrical stimulation of superfused bronchial strips. Tissue was prepared by using superfusion apparatus (Coleman System HSE-HA; Harvard Apparatus Inc., Holliston, MA) at a rate of 2 ml/min with oxygenated (5% CO2 and 95% O2) PSS at 37°C. Spontaneous tone, induced by endogenous leukotrienes and histamine, was inhibited by zileuton (10 μM) and fexofenadine (10 μM), respectively. Electrical stimulation was delivered by bipolar electrodes placed parallel to the superfused tissue as 10-s trains of square-wave pulses (8 Hz; 0.5-ms duration; 40–50 V) generated every 2 min by a Grass stimulator. After the baseline contractile responses to the electrical stimulation were consistent, a concentration of agonist was added to inhibit approximately 60% of the stable baseline contractions induced by the electrical stimulation. The preparation was washed free of agonist after 30 min, and the recovery of tone was recorded for 14 to 15 h. Control preparations from the same donor received the vehicle used in each case and were followed during the same period of time to check the preparation's stability.
Human Bronchi Data Analysis.
Agonist potency was calculated from cumulative CRCs and expressed as the concentration required for inducing 50% (EC50) of the maximum relaxation caused by the highest concentration tested. To report the maximal efficacy of each compound, the effect of the highest concentration tested in spontaneous tone preparations was referred to the maximal relaxation induced by 3 mM theophylline.
Onset was defined as the time spanning from agonist addition to the attainment of 50% (t½) of the maximal relaxation produced by a single concentration. Duration of action was defined as the time spanning from agonist washout to the attainment of 50% (t½) recovery from the relaxation produced by the agonist concentration added (Coleman and Nials, 1989). Statistical analysis of results was carried out by analysis of variance (ANOVA) followed by Bonferroni post-test. All calculations and analysis were performed by using Prism (GraphPad Software, Inc., San Diego, CA). Significance was accepted when p < 0.05.
Potency and Duration of Action in Anesthetized Guinea Pigs
To assess antibronchoconstrictor effects, animals were anesthetized with an intramuscular injection of ketamine (69.8 mg/kg), xylazine (5.6 mg/kg), and acepromazine (1.6 mg/kg). If required, anesthesia was extended by additional intramuscular injections of the aforementioned anesthetic mixture. Animals were then tracheotomized, placed inside single plethysmograph chambers, and connected to a ventilator. An esophageal tube was inserted to measure transpulmonary pressure changes. The carotid artery and jugular vein were cannulated for blood pressure monitoring and intravenous administration, respectively. During the experiment, animals were artificially ventilated at the rate of 60 strokes/min and a tidal volume of 10 ml/kg. Temperature was maintained at 37°C.
A FinePointe RC System (Buxco Research Systems, Wilmington, NC) was used to register the airway resistance and dynamic compliance in the anesthetized animals. This system measures the pressure changes that drive respiration and the resultant flows in and out of the airways. These variations in flow, transpulmonary pressure, and blood pressure were registered with BioSystem XA software (version 2.10 for Windows; Buxco Research Systems). Before starting the pulmonary dynamics evaluation, animals were connected to the ventilator until their breath pattern was stabilized. Once baseline values were in the range of 0.1 to 0.3 cmH2O/ml/s of airway resistance and 0.8 to 0.3 ml/cmH2O of dynamic pulmonary compliance, the pulmonary measurement was initiated. After the stabilization period, bronchoconstriction was induced by two consecutive intravenous injections of Ach: a first dose of 10 μg/kg Ach, and a second dose of 15 μg/kg Ach, once the basal resistance level was recovered. The bronchoconstriction response achieved with the 15 μg/kg Ach bolus, which caused a bronchoconstriction representing a 3- to 5-fold increase of the baseline lung resistance, was the one used to calculate the inhibitory effect of each treated group, compared with the response of its respective control group.
Airway resistance (cmH2O/ml/s) was calculated as the ratio of the changes in pressure and flow between isovolumetric points on inspiration and expiration. The airway resistance response to the Ach challenge was calculated for the vehicle and agonists from the formula: airway resistance = (RM − RB) × 100/RB, where RM is the peak resistance after challenge (maximal value) and RB is the baseline resistance (10 breaths before challenge). The inhibitory effect of each agonist was compared with its respective control group (vehicle only).
For compounds administered by inhalation route, guinea pigs were placed in a methacrylate box (17 × 17 × 25 cm) over a period of 10 min. On the first and fifth minutes of this 10-min period, vehicle or compound was aerosolized for 1 min by using an ultrasonic nebulizer (Devilbiss Ultraneb 2000; DeVilbiss Healthcare, Somerset, PA). The nebulization was driven by a mixture of 5% CO2, 21% O2, and 74% N2 at a flow of 3 l/min. Aerosols were generated from aqueous solutions, using different concentrations for each compound (0.01–3 μg/ml for abediterol, 0.1–30 μg/ml for indacaterol, 0.3–10 μg/ml for salmeterol, 0.1–30 μg/ml for formoterol, and 10–300 μg/ml for salbutamol).
The evaluation of the bronchoprotective potency was assessed 1 h after compound administration. For the duration of action study, the concentration inhibiting ≥85% of the bronchoconstriction at 1 h postadministration was selected for each compound, and the antibronchoconstrictor effect was followed at different time points after compound exposure (2, 4, 6, 18, 24, 48, 72, or 96 h).
In preliminary experiments, plasma levels of aerosolized abediterol at 4 μg/ml, measured by high-performance liquid chromatography after 1 h of exposure, were close to the limit of quantification (0.02 ng/ml). Plasma levels at the aerosol concentrations used in the experiments (0.01–3 μg/ml) were not detectable and did not allow us to establish pharmacokinetics/pharmacodynamics assessment in the core experiments.
Data Analysis.
Potency was calculated from concentration-response values obtained at 1 h after treatment, which were fitted in a sigmoidal CRC by using Prism software to estimate bronchoprotective IC50. IC50 values were compared by using t test.
Duration of action (t½) was defined as the time taken to recover 50% of the maximal inhibitory effect achieved by the agonist, derived from time-course bronchoconstriction inhibition curves, and calculated by using a one-phase exponential decay formula. A one-way ANOVA followed by Dunnett's post-test was used to determine statistical differences in bronchoconstriction. These analyses were performed by using Prism software.
Safety Margin and Duration of Action in Anesthetized Beagle Dogs
The method of Konzett and Rössler (1940) modified according to Misawa et al. (1986) was adapted. The animals were anesthetized with propofol (6 mg/kg i.v. plus a constant infusion of 0.6–0.7 mg/kg/min i.v.) with a perfusor (BD Biosciences, Brèzin, France) through one of the cephalic veins. A cuffed endotracheal tube was inserted through the fauces. Then, the animal was connected to a respirator and mechanically ventilated with room air under a constant pressure of 15 cm of water according to the following conditions: respiratory rate of 15 breaths/min and tidal volume of 10 ml/kg, by means of a pump (model 607-A; Harvard Apparatus Inc.). ECG leads were attached to the animal to record the lead II derivative of the ECGs. Responses of the bronchial musculature were measured considering the ventilation overflow, which was continuously measured by means of a pneumotachograph (TSD127; Biopac Systems Inc., Santa Barbara, CA) attached to a highly sensitive differential pressure transducer (TSD160A; Biopac Systems) as an index of airway resistance. Bronchospasms were induced by a dose of Ach (5 μg/kg i.v.), administered as a bolus, which produced a bronchial response that returned rapidly to baseline levels, did not affect physiological parameters such as heart rate and blood pressure, and was high enough to be dose-dependently inhibited by bronchodilators agents. The compounds to be tested were administered by inhalation and aerosolized in solution by means of an ultrasonic nebulizer from Aerogen Ltd. (Galway, Ireland) or MuMed Ltd (London, UK) for safety margin and duration of action experiments, respectively, which were attached between the respirator pump and the endotracheal tube. Lead II of the ECG, heart rate, and pulmonary resistance were continuously acquired, analyzed, and stored by means of a computer-based data acquisition system and the Acknowledge III program (Biopac Systems).
Once two similar Ach basal responses at 10-min intervals were obtained, the compound to be tested was administered by inhalation. Ach was administered again at 10 and 20 min after compound aerosolization. The inhibition of the bronchospasm was calculated at 20 min. Then, the intravenous infusion of the anesthetic agent was stopped, and the animal was allowed to recover. To measure the response to Ach at 3, 6, and 24 h after compound aerosolization, the same procedure was followed, and two Ach injections were performed at 10-min intervals. The inhibition elicited at these time points was considered as the mean of both bronchospasms.
Duration of action experiments were performed with a dose that produced a submaximal inhibitory effect at 20 min after compound inhalation.
The dose of the compound inhaled by each dog was calculated by measuring the amount of liquid delivered by the aerosol in the aforementioned conditions divided by the dog's body weight in kilograms.
In preliminary experiments, plasma levels of abediterol at 2.4 μg/kg did not render detectable plasma levels (limit of quantification of 0.02 ng/ml). The low plasma levels observed following the nebulization protocol did not allow us to establish pharmacokinetics/pharmacodynamics assessment in the core experiments.
Data Analysis.
ID50 and ED5 were calculated by sigmoidal dose-response and linear regression analysis, respectively. The safety margin was defined as the quotient between the dose increasing heart rate by 5% and the dose inhibiting bronchospasm by 50%.
Statistical analysis of bronchospasm inhibition and heart rate increase data were performed by one-way ANOVA, followed by Dunnett's post-test in the case of the percentages of inhibition. ID50 values were compared by using t test. In the duration of action experiments, the statistical analysis of bronchospasm inhibition data at various time points compared with the vehicle-treated group was done by two-way ANOVA followed by Bonferroni post-test. These analyses were performed by using Prism software.
Results
In Vitro Binding, Functional Activity, and Selectivity at the Human Adrenergic Receptors.
The affinity of abediterol, salmeterol, formoterol, and indacaterol for the human β-adrenergic receptors was assessed by using membranes of Sf9 cells expressing the β1- or β2-receptor subtype and membranes of HTB-10 cells as a source of the β3-receptor subtype, and determined in radioligand binding displacement experiments. The affinities for the different β-adrenoceptor subtypes are summarized in Table 1. Abediterol affinity for the human β2-adrenoceptor was in the subnanomolar range, and abediterol was the compound that showed the greatest affinity for the β2-receptor (abediterol > salmeterol > formoterol > indacaterol) of those studied.
Binding affinity and selectivity ratios of abediterol, salmeterol, formoterol, and indacaterol for the human β1-, β2-, and β3-adrenergic receptors
Affinities are expressed as IC50 (inhibitory concentration at which 50% of the total binding was inhibited). Each value represents the mean ± S.E.M. of at least three independent experiments (n = 3–7).
Selectivity for β2 over the other β-adrenoceptor subtypes was also determined by calculating β1/β2 and β3/β2 selectivity ratios using the IC50 values (Table 1). Salmeterol was the most selective compound over the human β1-receptor, followed by abediterol, formoterol, and indacaterol. Both salmeterol and abediterol showed high selectivity over the human β3-receptor, with higher ratios than formoterol and indacaterol (Table 1).
Functional selectivity for β2- over β1- and β3-adrenoceptors was determined by measuring agonist concentrations required to achieve 50% of maximal cAMP production (EC50) versus reference agonists in CHO-K1 cell lines stably expressing the human β1-, β2-, and β3-adrenergic receptors. β1/β2 and β3/β2 selectivity ratios were then calculated with the EC50 values obtained in the different cell lines (Table 2). Salmeterol was the most selective compound for the human β2-receptor over the human β1- and β3-receptor, followed by abediterol, formoterol, and indacaterol (Table 2).
Functional β2 selectivity of agonists against the different human β-adrenergic receptor subtypes
CHO cells selectively expressing the human adrenergic β1-, β2-, and β3-receptors were stimulated with increasing concentrations of agonists (0.03–10,000 nM), and cAMP levels were quantified as described under Materials and Methods. Data are reported as mean ± S.E.M. of three independent experiments. EC50 is the concentration required to do 50% of the maximal effect (versus the reference agonists epinephrine, formoterol hemifurate, and ZD7114 for the β1, β2, and β3 assays, respectively).
Intrinsic Efficacy in U937 Human Monocytic Cells.
The intrinsic efficacy of the compounds was studied by measuring the stimulation of cAMP production in U937 human monocytic cells. This cellular model was first characterized by using selective β1- and β2-adrenoceptor antagonists. The β2-antagonist (ICI-118551) was able to completely block the isoprenaline-induced cAMP production, whereas no effect was observed when the cells were incubated with the β1- adrenoceptor antagonist CGP20712 (data not shown), indicating that in U937 cells the β2-receptor is the subtype responsible for cAMP production induced by β-agonists. Figure 2 shows the efficacy of abediterol, formoterol, salmeterol, and indacaterol, expressed as percentage of the maximal effect of each compound on cAMP production with respect to 1 μM isoprenaline. Abediterol, formoterol, and indacaterol behaved as full agonists with maximal efficacies of 91 ± 5, 100 ± 3, and 99 ± 5% at the highest concentration tested, respectively, whereas salmeterol behaved as partial agonist with a maximal efficacy of 41 ± 3% (Fig. 2).

Intrinsic efficacy of β2-adrenergic receptor agonists. Effect of agonists on intracellular cAMP levels were quantified in U937 cells. Data represent percentages of cAMP production with respect to the maximal production induced by isoprenaline. Maximal efficacies of abediterol, salmeterol, formoterol, and indacaterol were 91 ± 5% (n = 6), 41 ± 0.3% (n = 6), 100 ± 3% (n = 6), and 99 ± 5% (n = 4), respectively. Data are shown as mean ± S.E.M.
Functional β1/β2 Selectivity in Guinea Pig Tissue.
All agonists (0.001–30 nM) produced concentration-dependent relaxations of the spontaneous tone in isolated guinea pig trachea (a β2-adrenoceptor-containing preparation; Fig. 3A). Maximal efficacy, expressed as percentage of relaxation versus 0.1 μM isoprenaline, was achieved at 0.3, 1, 10, and 30 nM for formoterol, abediterol, indacaterol, and salmeterol, respectively, corresponding to the highest concentration tested for each compound (Fig. 3A). The potency (EC50) of abediterol (0.02 nM) was comparable with that of formoterol and significantly higher than that of indacaterol and salmeterol (p < 0.01; Table 3).

Functional β1/β2 selectivity in guinea pig tissues. Cumulative concentration-response curves for the β2-adrenergic receptor agonists were performed in the guinea pig trachea model with preparations at spontaneous tone (A) and in guinea pig left atria (B) to assess relaxant and inotropic effects, respectively. Data are shown as percentages of relaxation (A) and percentages of contraction (B) relative to 0.1 μM isoprenaline, expressed as mean ± S.E.M., of at least four independent experiments (n = 4–12).
Functional β1/β2 selectivity in guinea pig tissues
β1 and β2 activities of abediterol, salmeterol, formoterol, and indacaterol were assessed in guinea pig left atria and trachea, respectively. Values are expressed as EC50 and represent the mean and confidence interval (95% CI) of 4 to 12 independent preparations. Functional β1/β2 selectivity ratio was calculated. EC50 is the concentration required to induce 50% of the maximal effect relative to 0.1 μM isoprenaline.
β1-Adrenergic activity was assessed in electrically stimulated guinea pig left atria. All compounds (0.001–10 μM) produced concentration-dependent contractions (Fig. 3B). The maximal efficacy observed at the highest concentration tested (10 μM) expressed as percentage of contraction versus 0.1 μM isoprenaline was 103.0 ± 5.9, 57.8 ± 3.6, 32.5 ± 5.2, and 4.4 ± 5.6% for formoterol, indacaterol, abediterol, and salmeterol, respectively. Because of methodological reasons it was not possible to test concentrations higher than 10 μM. When potency was expressed as EC50, compounds followed the potency rank of formoterol > indacaterol > abediterol ∼ salmeterol (Table 3).
β1/β2 functional selectivity was calculated as the ratio between the β1 and β2 potency in both guinea pig assays. Abediterol showed a high functional selectivity versus β1 atria activity and a higher ratio than formoterol and indacaterol (Table 3).
Potency, Onset, and Duration of Action in Spontaneous Tone Human Bronchi.
The pharmacological behavior of abediterol, salmeterol, formoterol, and indacaterol was assessed in isolated human bronchi at spontaneous tone. All of the agonists (0.0001–10 μM) produced concentration-dependent relaxation. The rank of potency (EC50) was formoterol > abediterol > indacaterol ≫ salmeterol (Table 4). When efficacy was expressed as the maximal effect versus the relaxation achieved by 3 mM theophylline, formoterol was the compound that showed the highest efficacy (92 ± 2%), followed by abediterol (82 ± 4%), indacaterol (76 ± 4%), and salmeterol (68 ± 5%).
Potency, onset, and duration of action of β-adrenergic receptor agonists in isolated spontaneous tone human bronchi
EC50 values were calculated from percentages of relaxation obtained from cumulative concentration-response curves in preparations at spontaneous tone. Onset and duration of action were determined by applying a single concentration of agonist that produced a 40 to 60% of effect with respect to theophylline. Onset of action was defined as the time spanning from agonist addition to the attainment of 50% of the maximal relaxation produced by the concentration added. Duration of action was defined as the time spanning from agonist washout to the attainment of 50% recovery from the relaxation produced by the concentration added. Potency data are reported as mean and confidence interval (95% CI). Onset and duration of action data are reported as mean ± S.E.M. n/p experiments involve the number of rings (n) and the number of patients (p).
The onset and duration of action were also studied in preparations at spontaneous tone, using agonist concentrations that produced between 40 and 60% of relaxation with respect to theophylline. As shown in Fig. 4 and Table 4, formoterol was the compound with the fastest onset of action, followed by abediterol, whereas indacaterol and salmeterol showed significantly slower onset of relaxant activity than formoterol. Abediterol was the compound that demonstrated the most sustained duration of action (t½ = 669 min), followed by indacaterol (t½ = 449 min), whereas salmeterol (t½ = 230 min) and formoterol (t½ = 76 min) showed shorter duration of the relaxant effect (Fig. 4; Table 4).

Onset and duration of action of β2-adrenergic receptor agonists in isolated human bronchi. Onset and duration of action were determined in preparations at spontaneous tone by applying a concentration of agonist that produced 40 to 60% of maximal relaxation induced by 3 mM theophylline. Onset was defined as the time spanning from agonist addition to the attainment of 50% of the maximal relaxation produced by the concentration added. Duration of action was defined by the time spanning from agonist washout to the attainment of 50% recovery from the relaxation produced by the agonist concentration added. Data are reported as mean ± S.E.M. of 5 to 12 experiments from three to five patient samples.
Onset and Duration of Action in EFS Human Bronchi.
The onset and duration of action of abediterol and reference compounds were studied in preparations contracted by electrical stimuli by using agonist concentrations that caused approximately 60% inhibition. Formoterol was the compound with the fastest onset of action (t½ = 3.1 min), followed by indacaterol (t½ = 6.5 min) and abediterol (t½ = 7.4 min), whereas salmeterol (t½ = 15.7) showed significantly slower onset (Table 5). Abediterol, salmeterol, and indacaterol showed similar duration of action that was significantly longer than that of formoterol (Table 5).
Onset and duration of action of β-adrenergic receptor agonists in EFS isolated human bronchi
Onset and duration of action were determined by applying a single concentration of agonist that produced approximately 60% inhibition of the EFS-induced contraction. Onset of action was defined as the time spanning from agonist addition to the attainment of 50% of the maximal inhibition produced by the concentration added. Duration of action was defined as the time spanning from agonist washout to the attainment of 50% recovery from the inhibition produced by the concentration added. Onset and duration of action data are reported as mean ± S.E.M. n/p experiments involve the number of rings (n) and the number of patients (p).
Potency and Duration of Bronchoprotection in Guinea Pigs.
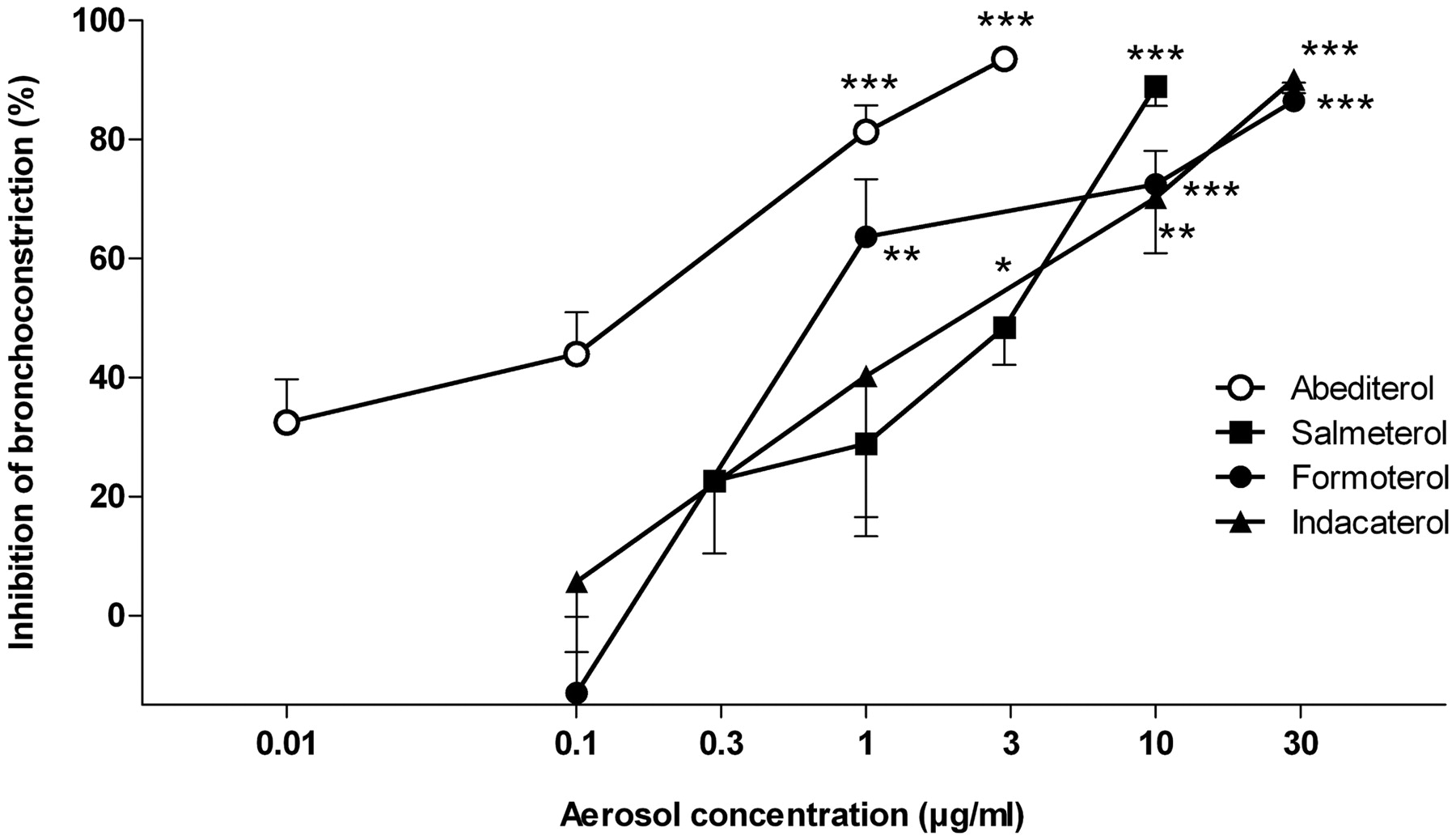
To evaluate the in vivo bronchoprotective potency of inhaled abediterol and comparators, four to five concentrations of each agonist were administered to different groups of guinea pigs (4–12 animals per group) 1 h before Ach-induced bronchoconstriction. All of the compounds inhibited the bronchoconstriction in a concentration-response manner (Fig. 5), achieving an inhibitory effect of 85 to 94% at the highest concentration tested. Abediterol showed the highest potency with an IC50 value of 0.12 μg/ml (Table 6). Formoterol, indacaterol, and salmeterol showed significantly lower potency with IC50 values of 1.1, 2.2, and 2.7 μg/ml, respectively.

Concentration-response curves of inhaled abediterol, salmeterol, formoterol, and indacaterol in the Ach-induced bronchoconstriction model in guinea pigs. Aerosols were generated from aqueous solutions by using different concentrations of each compound. Then, animals were anesthetized, and bronchoconconstriction was induced by Ach (15 μg/kg i.v.) 1 h after aerosol administration. Data are shown as mean ± S.E.M. of 4 to 12 experiments per dose. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus baseline bronchospasm (one-way ANOVA, followed by Dunnett's post-test).
Potency of inhaled abediterol, salmeterol, formoterol, and indacaterol inhibiting the Ach-induced bronchoconstriction in anesthetized guinea pigs and anesthetized dogs
Nebulized agonists were administered to conscious guinea pigs and, 1 h later, maximal bronchoconstriction was induced by Ach (15 μg/kg i.v.) in anesthetized animals. Antibronchoconstrictor effects were determined as a percentage of inhibition of response to Ach versus the vehicle group. In anesthetized dogs, maximal bronchoconstriction was induced with Ach (5 μg/kg i.v.). Then, compounds were administered by aerosol, and 20 min later animals received a further Ach challenge. Antibronchoconstrictor effects were determined as a percentage of inhibition of response to Ach versus baseline values. Potency was expressed as IC50 and ID50 in the guinea pig and dog model, respectively (n = 4–2 guinea pigs; n = 2–4 dogs).
To assess the duration of bronchoprotective effects, submaximal concentrations of inhaled agonists were studied up to 96 h (Fig. 6). At the concentrations selected, an equieffective inhibition of Ach-induced bronchoconstriction (85–94%) was observed in concentration-response curve experiments at 1 h after compound inhalation (Fig. 5). Abediterol and indacaterol demonstrated sustained duration of action, showing 57 to 60% of bronchospasm inhibition at 24 h after treatment, maintained up to 48 h for both compounds, whereas in the case of formoterol and salmeterol there was a significant loss of the bronchoprotective effect at 6 h, with complete loss of effect by 24 h. When the duration of action was expressed as the time taken to reduce the maximal bronchoconstriction achieved at 1 h by 50% (t½), abediterol and indacaterol showed values of 36 h (28–51) and 51 h (39–73), expressed as mean [95% confidence interval (CI)], respectively. This duration of action was considerably longer than that of salmeterol and formoterol, with a t½ of 6 h (4–13) and 4 h (3–7), expressed as mean (95% CI), respectively (Fig. 6). The model was validated with the short-acting β-agonist (SABA) salbutamol, showing a t½ of 2 h (1–4), expressed as mean (95% CI).

Duration of action of inhaled abediterol, salmeterol, formoterol, and indacaterol in the Ach-induced bronchoconstriction model in guinea pigs. Conscious animals were exposed to aerosol solutions of agonists, and then, at various time points, they were anesthetized and bronchoconstriction was induced by Ach (15 μg/kg i.v.). The bronchoprotective effect of each agonist was assessed and expressed as inhibition percentage of control (vehicle) response. Data are represented as mean ± S.E.M. (n = 5–12). *, p < 0.05; **, p < 0.01; ***, p < 0.005 compared with first observational time point (one-way ANOVA followed by Dunnett's post-test).
Safety Margin in Anesthetized Beagle Dogs.
These experiments were performed to assess the efficacy and safety ratio of abediterol with respect to salmeterol, formoterol, and indacaterol when administered by inhalation. Efficacy was determined as the ability to revert the acetylcholine-induced bronchospasm in Beagle dogs, and safety was assessed as the effect on heart rate in the same animals.
Compounds inhibited the acetylcholine-induced bronchospasm in a concentration-dependent manner (Fig. 7). At the concentration range tested for each compound, the range of calculated doses was 0.04 to 0.6, 1.7 to 13, 0.2 to 5, and 0.5 to 11 μg/kg for abediterol, salmeterol, formoterol, and indacaterol, respectively. Abediterol and formoterol were the most potent compounds with ID50 values of 0.16 and 0.24 μg/kg, respectively, followed by indacaterol and salmeterol, with significantly lower potency than abediterol (ID50 values of 1.2 and 4.4 μg/kg, respectively) (Table 6).

Effect of abediterol, salmeterol, formoterol, and indacaterol on acetylcholine-induced bronchoconstriction and heart rate in anesthetized Beagle dogs. Nebulized compounds were administered to anesthetized animals, and bronchoconstriction was induced by Ach (5 μg/kg) at 20 min. Inhibition of bronchoconstriction was recorded as a percentage of the baseline response to Ach (straight lines). Effect on heart rate was recorded as a percentage of increase from baseline values (dotted lines). Data are shown as mean ± S.E.M. of two to four experiments per dose. *, p < 0.05; **, p < 0.01 versus baseline bronchospasm (one-way ANOVA, followed by Dunnett's post-test).
All of the tested compounds demonstrated increases in heart rate, with salmeterol and indacaterol showing the highest percentages of increase at the concentrations tested (Fig. 7). The ED5 and the safety margin, expressed as the ratio between ED5 increasing heart rate and the ID50 inhibiting acetylcholine-induced bronchospasm, were calculated. Abediterol had the highest safety margin (5.6), followed by formoterol (3.3), indacaterol (2.2), and salmeterol (0.3).
Duration of Action in Anesthetized Beagle Dogs.
To assess the duration of bronchoprotection in Beagle dogs, the effects of single concentrations of inhaled agonists were studied up to 24 h in the acetylcholine-induced bronchoconstriction model. The effect on heart rate limited the agonist dose to be tested, thus in the case of salmeterol and indacaterol this effect prevented achievement of submaximal inhibitions (∼80%). At the concentrations tested (Fig. 8), the calculated doses of abediterol, salmeterol, and indacaterol were 1, 9, and 18 μg/kg, respectively, and achieved a peak effect inhibiting the acetylcholine-induced bronchoconstriction by 76 ± 7, 56 ± 5, and 45 ± 2%, respectively, at 20 min after inhalation (Fig. 8). The effect on heart rate was also recorded just before the first Ach challenge, with percentages of heart rate increasing by 4 ± 2, 26 ± 2, and 17 ± 2% for abediterol, salmeterol, and indacaterol, respectively.

Duration of action of aerosolized abediterol, salmeterol, and indacaterol in acetylcholine-induced bronchoconstriction in anesthetized Beagle dogs. Nebulized compounds or vehicle were administered to anesthetized animals, and bronchoconstriction was induced by Ach (5 μg/kg) at 20 min and 3, 6, and 24 h. Inhibition of bronchoconstriction was recorded as percentage of the baseline response to acetylcholine. Data are reported as mean ± S.E.M. (n = 2- 5). **, p < 0.01; ***, p < 0.001 versus the vehicle-treated group (two-way ANOVA followed by Bonferroni post-test).
When the bronchoprotective effect of agonists was assessed at different time points after inhalation, abediterol showed a sustained effect up to 6 h with percentages of inhibition of the acetylcholine-induced bronchospasm of 83 ± 2 and 71 ± 4% at 3 and 6 h, respectively, whereas the bronchoprotective effect of the other agonists was markedly decreased after 3 h of administration, with percentages of inhibition of 28 ± 12 and 20 ± 10% for salmeterol at 3 and 6 h, respectively, and percentages of inhibition of 30 ± 6 and 21 ± 5% for indacaterol at 3 and 6 h, respectively (Fig. 8). When the 24-h effects were compared, abediterol was the only compound showing some trend of inhibition of the acetylcholine-induced bronchospasm (38 ± 19%), whereas the effect of salmeterol and indacaterol was negligible (Fig. 8).
Discussion
The aim of this study was to establish the pharmacological profile of abediterol, a novel long-acting inhaled β2-adrenoceptor agonist in development as a once-daily bronchodilator for maintenance treatment of asthma and COPD.
In radioligand binding displacement studies, abediterol demonstrated high affinity for the human β2-adrenoceptor and great selectivity over the other human β-adrenoceptor subtypes. These data agree with the cAMP production data obtained in CHO transfected cells, where abediterol and salmeterol showed higher β1/β2 and β3/β2 selectivity ratios than formoterol and indacaterol. However, when β1/β2 selectivity was calculated with the activity observed in functional models using relevant tissues (atria and trachea) the selectivity was not an issue for any of the agonists tested. There was a trend toward higher selectivity for abediterol and salmeterol than for formoterol and indacaterol; the comparison between abediterol and salmeterol selectivity ratios was difficult because of the low β1 efficacy observed for both compounds in guinea pig atria. Our results are consistent with those reported previously, indicating that the β1/β2 and β3/β2 selectivity ratios of formoterol and indacaterol are inferior to those of salmeterol (Ball et al., 1991; Battram et al., 2006; Bouyssou et al., 2010). In humans, it has been reported that activity at the β1-adrenoceptor might be responsible for some of the cardiac side effects associated with β-adrenergic agonism (Levine and Leenen, 1989). For this reason, discovery programs of new-generation once-daily β2 adrenergic agonists have sought compounds with β2 selectivity over β1. The relevance of the high β3/β2 selectivity is less clear, although it has been suggested that the β3-adrenoceptor might have a role in lipolysis (Philipson, 2002).
The intrinsic efficacy of β-adrenergic agonists is also an important parameter to consider because partial agonists may act as β2-antagonists in the presence of an agonist with higher efficacy at the same receptor (Källström et al., 1994). Abediterol behaved as a full or near-full agonist in both U937 cells and human bronchi. The results obtained with salmeterol and formoterol were consistent with those reported previously (Jeppsson et al., 1992; Naline et al., 1994) where the compounds behaved as partial and full agonists, respectively. It is noteworthy that the different intrinsic efficacies of β2-agonists measured in preclinical models have not translated in humans, where formoterol and salmeterol have reached comparable bronchodilator efficacies in both COPD and asthma (Kottakis et al., 2002). It has been suggested that β2 intrinsic efficacy, together with physicochemical properties, contribute to the clinical onset of bronchodilation of β2-adrenoceptor agonists, with full agonists more prone to exhibit faster onset of action (Rosethorne et al., 2010). Accordingly, abediterol showed a fast onset of action relaxing human bronchi, not significantly different from that of formoterol. The results obtained with the reference compounds were consistent with the rates of onset of action in humans; formoterol was an effective bronchodilator within 5 min, salmeterol took 15 to 30 min to achieve significant bronchodilation over baseline, and indacaterol showed an onset of action faster than salmeterol (Kottakis et al., 2002; Sugihara et al., 2010). The onset of action of abediterol in preclinical models was in line with the first results in humans, where abediterol (5, 10, and 25 μg), at 5 min postdose, significantly improved lung function compared with placebo (p < 0.0001) and salmeterol (p < 0.05) in patients with persistent asthma, suggesting a rapid onset of action (Beier et al., 2010). Although a fast-acting profile is not essential for inhaled drugs used as maintenance medication, a rapid relief of symptoms could increase patient's confidence and reduce the risk of overdosing.
Potency of abediterol and reference compounds was assessed in isolated human bronchi and anesthetized guinea pigs. In human bronchi, abediterol demonstrated high potency in relaxing preparations at spontaneous tone, with an effect comparable with that of formoterol and higher than that of salmeterol and indacaterol. These results are in-line with published data of β2-adrenoceptor agonists in human bronchi, where formoterol was more potent than salmeterol and indacaterol (Naline et al., 2007).
The guinea pig has been widely used to assess the bronchodilator activity of β2-agonists (Battram et al., 2006; Bouyssou et al., 2010). In the acetylcholine-induced bronchoconstriction model in anesthetized guinea pigs, abediterol exhibited the highest potency in comparison to reference compounds, in agreement with the potency observed in guinea pig tracheal preparations and human bronchi. The high potency in preclinical models was confirmed in patients with mild to moderate asthma, where single inhaled doses of abediterol (5, 10, and 25 μg) induced significant increases in trough forced expiratory volume in 1 s compared with placebo (Beier et al., 2010). These results suggest that the therapeutic dose of abediterol in humans will be lower than that of salmeterol, formoterol, and indacaterol.
A number of factors may drive the duration of action of β2-adrenoceptor agonists, for example, affinity for the β2-adrenoceptor or long dissociation half-life, local binding near the receptor, metabolic stability or low clearance, and membrane diffusion kinetics. These hypotheses have been proposed to explain different duration of action of SABAs, twice-daily and once-daily LABAs (Casarosa et al., 2011). To compare the duration of action of abediterol with that of other β2-agonists, compounds were tested in human bronchial preparations and anesthetized guinea pigs and dogs. In human bronchi, our results showed that abediterol had similar duration to indacaterol, both in spontaneous tone and electrical field-stimulated preparations. Similar models in human bronchi and guinea pig trachea have been reported to study the duration of action of LABAs (Nials et al., 1993; Naline et al., 1994). In line with our results, some discrepancies have been reported for the duration of action of formoterol and salmeterol in isolated tissue, showing shorter and longer duration of relaxant activity, respectively, based on what was expected according to their clinical profile (Naline et al., 2007). In our model, the duration of action of abediterol was similar to that of indacaterol, a compound that has demonstrated a profile compatible with once-daily dosing in patients with asthma and COPD (LaForce et al., 2008; Vogelmeier et al., 2010).
Abediterol also demonstrated long duration of action in anesthetized guinea pigs, showing a half-life of 36 h, which was 6- and 9-fold longer than that of salmeterol and formoterol, respectively. Moreover, the bronchoprotective effect of abediterol was similar to that of indacaterol up to 48 h, with a slight difference in the effect at 72 h. The effects of abediterol in anesthetized dogs also suggest that the compound has a long-lasting bronchoprotective activity, showing longer duration of action compared with salmeterol and indacaterol at doses without any impact on heart rate. Overall, this preclinical evidence is in line with the results obtained in asthma patients treated with abediterol, indicating that abediterol is suitable for once-daily dosing in humans (Beier et al., 2010).
Tachycardia is a side effect typically associated with β2-adrenoceptor agonists, which may result from the activation of both β1- and β2-adrenoceptors; dose-response effects on heart rate have been reported for SABAs and LABAs (Rabe, 2003). In anesthetized dogs, inhaled abediterol had a lesser effect on heart rate compared with the other reference compounds at the doses needed to produce equivalent bronchodilation. Under these experimental conditions, abediterol showed a safety margin higher than that of the comparators. Indeed, in duration of action experiments in dog, abediterol showed the lowest effect on heart rate at the concentration tested, although it was not possible to test equipotent bronchoprotective doses of salmeterol and indacaterol because of marked effects on heart rate at the highest dose tested. The modest effects of abediterol on heart rate might be partially explained by its selectivity for β2- over β1-receptors, shown in both binding and functional studies; however, the differences observed between the four agonists could also be explained by other factors such as a direct effect on the β2-receptor in the peripheral vasculature and the cardiac muscle itself. In the clinic, formoterol, salmeterol, and indacaterol show a therapeutic margin of 2- to 4-fold, with tachycardia being a transient side effect in most cases (Sears, 2002; LaForce et al., 2008). This might represent a concern in case of overdosing or in susceptible patients with COPD that have concomitant cardiac diseases such as chronic heart failure (Tashkin and Fabbri, 2010). In healthy subjects and asthmatic patients, abediterol was well tolerated and did not demonstrate clinically relevant effects on heart rate at doses <10 μg, suggesting a safety margin at least equivalent to that of currently licensed LABAs (Timmer et al., 2010).
In summary, the preclinical data reported in this study show that abediterol is a potent, selective, and full β2-adrenoceptor agonist with a rapid onset of action and long-lasting effects. Furthermore, abediterol has a favorable preclinical safety margin, showing reduced effects on heart rate. Hence, these data suggest that abediterol may provide a valuable treatment option for patients with asthma or COPD, with the potential to be combined with inhaled corticosteroids, long-acting anticholinergics, or novel anti-inflammatory therapies.
Authorship Contributions
Participated in research design: Aparici, Gómez-Angelats, Vilella, Gavaldà, De Alba, Gras, Cortijo, Morcillo, Ryder, Beleta, and Miralpeix.
Conducted experiments: Vilella, Otal, Carcasona, Viñals, Ramos, Cortijo, and Morcillo.
Contributed new reagents or analytic tools: Puig.
Performed data analysis: Aparici, Gómez-Angelats, Vilella, Otal, Carcasona, Viñals, Ramos, Gavaldà, Cortijo, and Morcillo.
Wrote or contributed to the writing of the manuscript: Aparici and Miralpeix.
Acknowledgments
We thank Enric Villanova, Mireia Verdú, Núria Torán, Joan Mañé, and Manuel Aznar (Almirall R&D Center) for significant technical support; Beatriz Seoane for statistical analysis assistance (Almirall R&D Center); Carol Astbury (Almirall R&D Center) for review and excellent suggestions on the manuscript; and Cath Bryant of Complete Medical Communications (Cheshire, UK), which provided medical editing support funded by Almirall S.A, Barcelona, Spain.
Footnotes
This work was supported by Almirall SA, Barcelona, Spain; the Ministry of Science and Innovation, Spanish Government [Grants SAF2008-03113 (to J.C.), SAF2009-08913 (to E.J.M.)]; European Funds for Regional Development; Centro de Investigación en Red de Enfermedades Respiratorias, Health Institute Carlos III (Spanish Government) [Grant CB06/06/0027]; the Consorcios Estratégicos Nacionales de Investigación Tecnológica Programme (Genius Pharma; Spanish Government); and Regional Government (Generalitat Valenciana) [Grant Prometeu 2008/045].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
ABBREVIATIONS:
- COPD
- chronic obstructive pulmonary disease
- Ach
- acetylcholine
- ANOVA
- analysis of variance
- CHO
- Chinese hamster ovary
- CI
- confidence interval
- CRC
- concentration-response curve
- ED5
- dose increasing heart rate by 5%
- EFS
- electrical field stimulation
- IBMX
- 3-isobutyl-1-methylxanthine
- LABA
- long-acting β-adrenergic agonist
- PSS
- physiological salt solution
- SABA
- short-acting β-agonist
- CGP12177
- 4-[3-(tert-butylamino)-2-hydroxypropoxy]-1,3-dihydrobenzimidazol-2-one
- CGP20712A
- 1-[2-((3-carbamoyl-4-hydroxy)phenoxy)ethylamino]-3-[4-(1-methyl-4-trifluoromethyl-2-imidazolyl)phenoxy]-2-propanol methanesulfonate
- ZD7114
- (S)-4-[2-hydroxy-3-phenoxypropylaminoethoxy]-N-(2-methoxyethyl)phenoxyacetamide
- ICI-118551
- (±)-erythro-(S*,S*)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol hydrochloride.
- Received March 1, 2012.
- Accepted May 14, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}