


Abstract
Oxidative stress is central to the pathology of several neurodegenerative diseases, including multiple sclerosis, and therapeutics designed to enhance antioxidant potential could have clinical value. The objective of this study was to characterize the potential direct neuroprotective effects of dimethyl fumarate (DMF) and its primary metabolite monomethyl fumarate (MMF) on cellular resistance to oxidative damage in primary cultures of central nervous system (CNS) cells and further explore the dependence and function of the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) pathway in this process. Treatment of animals or primary cultures of CNS cells with DMF or MMF resulted in increased nuclear levels of active Nrf2, with subsequent up-regulation of canonical antioxidant target genes. DMF-dependent up-regulation of antioxidant genes in vivo was lost in mice lacking Nrf2 [Nrf2(−/−)]. DMF or MMF treatment increased cellular redox potential, glutathione, ATP levels, and mitochondrial membrane potential in a concentration-dependent manner. Treating astrocytes or neurons with DMF or MMF also significantly improved cell viability after toxic oxidative challenge in a concentration-dependent manner. This effect on viability was lost in cells that had eliminated or reduced Nrf2. These data suggest that DMF and MMF are cytoprotective for neurons and astrocytes against oxidative stress-induced cellular injury and loss, potentially via up-regulation of an Nrf2-dependent antioxidant response. These data also suggest DMF and MMF may function through improving mitochondrial function. The clinical utility of DMF in multiple sclerosis is being explored through phase III trials with BG-12, which is an oral therapeutic containing DMF as the active ingredient.
Introduction
Free radicals exert significant oxidative stress on tissues and cells and are implicated as contributing factors to the pathology of diverse neurodegenerative disorders such as multiple sclerosis (MS), amyotrophic lateral sclerosis, and Huntington's, Parkinson's, and Alzheimer's diseases. The postmitotic cells that reside in the central nervous system (CNS) have an inherently low capacity for mitigating oxidative stress and are highly susceptible to its damaging effects on DNA, proteins, and lipids (Hardingham and Lipton, 2010). Current therapeutic approaches typically target molecular pathways associated with individual diseases or, in the case of MS, broadly affect immune function. However, little has been done to mitigate the common pathological feature of oxidative stress.
All cells have an intrinsic mechanism for combating reactive oxygen species (ROS) that is dynamically controlled through the actions of Nrf2, a transcription factor that is the principal regulator of the phase II cellular antioxidant response (Nguyen et al., 2003). Nrf2 is normally sequestered in the cytoplasm through interaction with kelch-like erythroid cell-derived protein with CNC homology-associated protein 1 (Keap1), which results in constitutive protein ubiquitination and proteosomal degradation. However, excessive ROS, or treatment with electrophilic compounds, results in the modification of Keap1 such that Nrf2 is no longer constitutively degraded (Itoh et al., 2004; Li and Kong, 2009; Linker et al., 2011). This results in the accumulation of Nrf2 in cellular nuclei and enhanced transcription of a host of antioxidant and detoxification genes (Kwak et al., 2003; Lee et al., 2003).
Electrophilic compounds have been shown to induce the activation of the Nrf2 pathway and many of them, including carnosine (Calabrese et al., 2005), tert-butylhydroquinone (Li et al., 2005), the synthetic triterpenoid methyl-2-cyano-3,12-dioxooleana-1,9-dien-28-oate amide (Tran et al., 2008; Yang et al., 2009), sulforaphane (Soane et al., 2010), and 1,5-dicaffeoylquinic acid (Cao et al., 2010), have demonstrated in vitro cytoprotective responses against oxidative and inflammatory stress in treated neuronal cells. By using these types of compounds or genetic overexpression, the neuroprotective effects of Nrf2 pathway activation have been investigated in animal models of various neurodegenerative disorders, such as Alzheimer's disease (Dumont et al., 2009), Parkinson's disease (Jakel et al., 2007; Chen et al., 2009; Yang et al., 2009), amyotrophic lateral sclerosis (Vargas et al., 2008; Neymotin et al., 2011), and Huntington's disease (Stack et al., 2010).
Recent studies have demonstrated that the fumaric acid ester dimethyl fumarate (DMF) and its primary metabolite monomethyl fumarate (MMF) (Table 1) were able to activate the Nrf2 pathway and induce expression of antioxidant proteins (Lin et al., 2011) and administration of MMF could protect motor neurons and astrocytes against H2O2-induced oxidative stress (Linker et al., 2011). These results may point to an underlying functional cellular substrate for the neuroprotective response observed in mouse experimental autoimmune encephalomyelitis, in which DMF treatment, in comparison with vehicle controls, reduced oxidative damage and consequential nerve fiber demyelination, resulting in greater axonal preservation and improved motor function (Schilling et al., 2006; Linker et al., 2011). The therapeutic relevance of the Nrf2 pathway has been shown by the lack of response to DMF in Nrf2(−/−) experimental autoimmune encephalomyelitis models (Linker et al., 2011). DMF has also been shown to improve lifespan, reduce behavioral deficits, and preserve striatal and motor cortex neurons in two different genetic models of Huntington's disease (Ellrichmann et al., 2011), suggesting it may have broad neuroprotective properties.
Structure and basic properties of DMF and MMF
The results from the present study have demonstrated that DMF induced a broad pharmacodynamic response after oral dosing in mice, and the up-regulation of this response depended on Nrf2. Primary cultures of neurons, astrocytes, and oligodendrocyte precursor cells were used to demonstrate the DMF- and MMF-dependent stabilization of Nrf2 in different CNS cell types, indicating the relevance of these compounds in treating diseases that result in the degeneration of CNS cells. The functional consequence of Nrf2 activation was characterized in astrocytes and neurons challenged with toxic oxidative stress, in which DMF and MMF promoted improved viability in an Nrf2-dependent manner. A potential functional mechanism by which cytoprotection may be conferred was identified through up-regulation of the cellular antioxidant response and preservation of mitochondrial function. These studies expand on the potential mechanism of action for BG-12, a second-generation fumarate containing DMF, which has demonstrated promising efficacy in phase II and phase III clinical trials for MS.
Materials and Methods
All materials, unless otherwise specified, were purchased from Sigma-Aldrich (St. Louis, MO). All procedures involving animals were performed in accordance with standards established in the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996) as adopted by the National Institutes of Health. All animal protocols were approved by the Biogen Idec Inc. Institutional Animal Care and Use Committee, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Animal Handling and Tissue RNA Extraction.
Wild-type C57BL/6 mice and Nrf2(−/−) (Itoh et al., 1997) were dosed with a suspension of 0, 50, or 200 mg/kg DMF in 0.8% hydroxypropyl methyl cellulose vehicle (Sigma-Aldrich). Drug was delivered by oral gavage at 10 μl/g. Tissues were removed 4 h after dosing and snap-frozen in liquid nitrogen then stored at −80°C until use. RNA was prepared from frozen tissues according to the manufacturer's protocol (RNeasy 96 Universal Tissue Protocol; QIAGEN GmbH, Hilden, Germany). In brief, frozen tissues were placed in 2-ml 96-well block plates with QIAzol (QIAGEN GmbH) and a 3.2-mm stainless-steel bead (BioSpec Products, Bartlesville, OK). Tissues were disrupted for four cycles of 45 s in a Mini-Beadbeater (Biospec Products). RNA was extracted in chloroform, and the aqueous phase was mixed with 70% ethanol. Extracted RNA was applied to RNeasy96 plates (QIAGEN GmbH) and purified by the spin method.
Gene Expression Profiling and Quantitative Polymerase Chain Reaction.
RNA was analyzed for purity and integrity by capillary electrophoresis on an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). Global transcript profiling was done by using the Affymetrix GeneChip Mouse 430A 2.0 array (Affymetrix, Santa Clara, CA). Hybridization probe synthesis, microarray hybridization, and scanning were performed according to the manufacturers' protocols. Probeset level data were normalized by using the robust microarray average algorithm and analyzed with GeneSpring 7.3 data mining software (Agilent Technologies). Quantitative polymerase chain reaction (qPCR) was performed from total mRNA isolated from tissues or from cells extracted by using an RNeasy kit (QIAGEN GmbH) and reverse-transcribed into cDNA according to the manufacturer's protocols (Applied Biosystems, Carlsbad, CA). Target gene primers [mouse nicotinamide adenine dinucleotide phosphate dehydrogenase (quinone 1) (NQO1), Mm00500821_m1; mouse aldo-keto reductase family 1, member B8 (AKR1B8), Mm00484314_m1, human heme-oxygenase 1 (HO1), Hs01110250_m1; human Nrf2, Hs00232352_m1; human NQO1, Hs00168547_m1; and glutamate-cysteine ligase catalytic subunit (GCLC), Hs00155249_m1] and 6-carboxyfluorescein dye-labeled TaqMan MGB probes (Applied Biosystems) were used for qPCR. Reactions containing 100 ng of cDNA, 900 nM of each primer, and 250 nM TaqMan probes were cycled on a Stratagene Mx3005P (Agilent Technologies) once for 10 min at 95°C, followed by 40 cycles of 95°C for 10 s and 60°C for 1 min. All samples were measured in duplicate by using glyceraldehyde 3-phosphate dehydrogenase as a normalizing gene. Final analysis was performed by using the comparative CT method to calculate fold changes. Samples were normalized relative to vehicle or dimethyl sulfoxide (DMSO) control conditions within each data set.
Cell Culture.
Primary cultures of human spinal cord astrocytes, human oligodendrocyte precursor cells (hOPCs), human hippocampal neurons (hNeurs), and appropriate growth media were purchased from ScienCell Research Laboratories (San Diego, CA) and maintained using supplier specifications. Cells for plate-based assays were seeded into clear-bottom tissue culture 24- or 96-well plates (Corning Life Sciences, Lowell, MA). Primary cultures of rat cortical neurons and rat oligodendrocyte precursor cells (rOPCs) were prepared and maintained as described previously (Mi et al., 2009) except for use of the gentleMACS system (Miltenyi Biotec, Bergisch Gladbach, Germany) for dissociation.
Compound Handling.
DMF and MMF were prepared as 10 mM solutions in DMSO, titrated in DMSO, and then diluted into normal growth media for cell treatments. The final concentration of DMSO (0.3%) was consistent for all treated cells. After DMF or MMF treatment, cells for use in all assays were washed with Hanks' balanced salt solution + 20 mM HEPES, pH 7.4.
Cellular Extract Preparation, Nrf2 Activity Assay, and Western Blotting.
Cytosolic and nuclear extracts were prepared by using a nuclear extract kit from Active Motif Inc. (Carlsbad, CA). Whole-cell extracts for Western blotting were prepared in 150 mM NaCl, 1.0% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0. Antibodies for Western blotting and the TransAM Nrf2 assay (Active Motif Inc.) were used according to the manufacturer's instructions. The following antibodies were used: Nrf2 (Epitomics, Burlingame, CA or Santa Cruz Biotechnology, Inc., Santa Cruz, CA), NQO1 (Epitomics), HO-1 (Epitomics), GCLC (Proteintech Group, Chicago, IL), and actin (MP Biomedical, Solon, OH).
Immunostaining.
Immunostaining was performed on cells fixed in 4% paraformaldehyde as described previously (Mi et al., 2009). To increase the visualization of nuclear Nrf2 immunostaining, astrocytes were also incubated for 10 min with 0.1% SDS in Tris-buffered saline. Primary antibodies (Nrf2, Epitomics; βIII-tubulin, Millipore Corporation, Billerica, MA; glial fibrillary acidic protein, Sigma-Aldrich) and secondary antibodies (Alexa Fluor 488-labeled, Invitrogen, Carlsbad, CA) were used according to the manufacturers' instructions. Nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI) or Hoechst dye, which was included with the secondary antibody incubation. Cells were imaged on a Zeiss Observer Z1 microscope using a 20× LD Plan Neofluar objective (Carl Zeiss Inc., Thornwood, NY). Astrocyte Nrf2 immunostaining was quantified by random selection and imaging of 10 fields for each condition and captured by using identical gain, offset, and accumulation time. Nuclear immunostaining intensity for all nuclei in each field was quantitated in AxioVision (Carl Zeiss Inc.). For quantitation of cortical neurons, cells grown in 24-well plates were imaged and counted on the Cellomics ArrayScan VTi (Thermo Fisher Scientific, Pittsburgh, PA) using a neuronal counting protocol to identify intact neurons. Five fields per condition were counted.
Plate-Based Cellular Assays.
CellTiter-Blue (Promega, Madison, WI), fluorescent glutathione (GSH) (Sigma Aldrich), LIVE/DEAD (Invitrogen), and ATPLite (PerkinElmer Life and Analytical Sciences, Waltham, MA) assays were performed according to the manufacturers' instructions. In general, human astrocytes were plated into 96-well plates, incubated for 4 h at 37°C with 5% CO2, with DMSO or titrations of DMF or MMF added, and incubated for an additional 20 h at 37°C with 5% CO2. For CellTiter-Blue, fluorescent GSH, mitochondrial membrane potential, and ATP assays, cells were washed with Hanks' balanced salt solution + 20 mM HEPES, pH 7.4, then incubated with kit reagents as directed. For oxidative challenge experiments, after 20-h compound incubation, cells were washed and then challenged with 0, 50, or 100 μM H2O2 for 2 h at 37°C, and then allowed to recover in normal growth media for an additional 20 h at 37°C with 5% CO2. Cellular viability was assessed by using a LIVE/DEAD viability stain quantified by fluorescence intensity from calcein AM fluorescence in live cells (excitation wavelength, 488 nM; emission wavelength, 525 nM) and in parallel by counting Hoechst-labeled nuclei using automated imaging and counting. In both assay formats, a subset of DMSO-treated control cells were treated with 0.1% saponin 10 min before labeling or fixation to generate negative controls on the plate. In the LIVE/DEAD assay, dead cells were labeled with ethidium homodimer and appeared red in fluorescent micrographs (excitation wavelength, 550 nM; emission wavelength, 575 nM). Live images from LIVE/DEAD-labeled cells were imaged as above. Cell nuclei from Hoechst dye-labeled cells were quantitated in an automated fashion on the Cellomics ArrayScan VTi platform (Thermo Fisher Scientific). Mitochondrial membrane potential was assessed through a ratiometric approach incubating astrocytes with MMF for 20 h as above, followed by incubation with 100 nM tetramethyl rhodamine ester (TMRE) and 200 nM MitoFluor Green (Invitrogen) for 30 min at room temperature. The accumulation of TMRE in mitochondria is based on the membrane potential driving intraorganelle accumulation of the anionic probe (Farkas et al., 1989). MitoFluor Green was included as a control for total mitochondrial content and to normalize TMRE levels. Signal intensities in the green (excitation wavelength, 488 nm; emission wavelength, 525 nm) and red (excitation wavelength, 550 nm; emission wavelength, 575 nm) channels were measured. Assays were quantitated on EnVision (PerkinElmer Life and Analytical Sciences) or FlexStation (Molecular Devices, Sunnyvale, CA) plate readers. Intracellular calcium levels were measured in astrocytes that were preincubated with a titration of MMF for 20 h as described, washed, and loaded with a calcium-sensitive dye (Calcium 4; Molecular Devices). Baseline fluorescence values were detected using a FLIPR TETRA system (Molecular Devices), then H2O2 was added to all wells simultaneously to achieve a final concentration of 50 μM, and fluorescence intensities were recorded for a total of 120 min. Relative fluorescence units (RFUs) were graphed against time, and the net change in fluorescence over baseline (ΔRFU) was calculated by subtracting the maximum value from the minimum value over the entire recording period.
Small Interfering RNA Analysis.
Human spinal cord astrocytes were transfected by using the Lonza (Basel, Switzerland) Amaxa T-020 nucleofaction protocol and the Primary Neurons Solution Kit (Lonza). Cells were transfected with 200 nM Nrf2-specific small interfering RNA (siRNA) (Dharmacon RNA Technologies, Lafayette, CO) or nonspecific siRNA (Dharmacon RNA Technologies). Cells were incubated for 18 to 24 h after transfection, then treated with a titration of MMF and analyzed in viability assays as described.
Results
DMF-Induced Antioxidant Response in the CNS Is Nrf2-Dependent.
Previous studies have demonstrated that fumaric acid esters such as DMF and MMF can activate genes typically associated with the Nrf2 antioxidant response pathway (Thiessen et al., 2010; Linker et al., 2011). To ascertain the necessity of Nrf2 in this activation, the global transcriptional response of genes modulated by DMF was examined in both wild-type and Nrf2(−/−) mice. As demonstrated here in the spleen, wild-type mice responded to DMF with a significant up-regulation of 738 genes compared with only seven transcripts in Nrf2(−/−) mice (changes with p < 10−5; Fig. 1A). Analysis of Akr1b8 expression, a known Nrf2 target gene, provided further evidence that Nrf2 was essential for DMF-induced antioxidant response in the spleen (Fig. 1B). The transcriptional changes in response to DMF treatment were somewhat tissue type-specific. In colon, both NQO1 and Akr1b8 were up-regulated, whereas in other tissues, such as liver and duodenum, Akr1b8 was significantly up-regulated, whereas NQO1 exhibited modest to no induction, and mesenteric lymph nodes exhibited an opposite pattern of increased NQO1 and little change in Akr1b8 (Supplemental Fig. 1). In brain, NQO1 message levels were significantly increased, also in an Nrf2-dependent fashion (Fig. 1C).

DMF induction of an antioxidant response in the CNS was Nrf2-dependent. Wild-type (WT) or Nrf2(−/−) (KO) mice were treated with 0, 50, or 200 mg/kg DMF by oral gavage. Tissues were harvested 4 h after DMF dosing. A, global transcriptional profiling of spleen tissue revealed induction of 738 specific genes with p < 10−5 in wild-type mice, whereas in Nrf2(−/−) mice only seven transcripts were found to have changed in response to 200 mg/kg DMF treatment. B, analysis of spleen tissue using qPCR to characterize expression level differences of Akr1b8 between wild-type mice (filled bars) and Nrf2(−/−) mice (empty bars). C, similar analysis as in B, using brain tissue and quantitation of NQO1 transcript levels. **, p < 0.01; ***, p < 0.001, based on two-way ANOVA with Dunnett's post-test for multiple-sample comparison against the relevant WT or KO vehicle group. PD, pharmacodynamic.
Fumarates Activate the Nrf2 Pathway in CNS Cells.
Recent studies have described a potential neuroprotective mechanism of action for DMF and MMF demonstrated from in vitro and in vivo data (Ellrichmann et al., 2011; Linker et al., 2011). To discern direct molecular events around this mechanism, primary cultures of spinal cord astrocytes were exposed to DMF or MMF for 6 h and assessed for Nrf2 expression by using immunofluorescent microscopy (Fig. 2A). Mean nuclear densiometric immunostaining intensity was increased relative to DMSO controls, by 63.9% with DMF and 37.5% with MMF (Fig. 2B). Nrf2 activity in these cells was confined mostly to the nuclear fraction and was 2- to 5-fold greater than that seen with DMSO controls, as determined by both Nrf2 DNA binding assays and Western immunoblots (Fig. 2C). The fidelity of the nuclear and cytoplasmic preparations was confirmed by Western immunoblots for nuclear-specific protein (HDAC; Fig. 2C). Immunoblotting for Nrf2 in hOPCs and rOPCs, as well as hNeurs treated with 10 μM DMF or MMF, confirmed that all CNS cell types showed increased Nrf2 levels over time (Fig. 2D). In several of the cell types a doublet of Nrf2 immunoreactivity was observed on Western immunoblots, with only the upper band exhibiting sensitivity to modulation by DMF or MMF and enrichment in nuclear fractions (Fig. 2, C and D). Replicate immunoblots for these cell types were probed with NQO1 antibody to examine a prototypical Nrf2 target gene and showed that after 24 h there was a robust increase in NQO1 in hOPCs with DMF and MMF treatment, but this increase was only obvious with DMF in rOPCs. Human neurons or rOPCs treated with MMF had a milder response, with relatively minor increases at 24 h (Fig. 2D).

DMF and MMF increased Nrf2 in CNS cells. A, human astrocytes were treated with DMSO or 10 μM DMF or MMF for 6 h, then fixed, permeabilized, and immunostained for Nrf2. Left, images of DMSO-treated samples. Right, top, image shows cells treated with DMF, and bottom, image shows cells treated with MMF. Images have identical histogram lookup tables for display comparison. B, mean nuclear immunostaining intensity was quantitated for every cell in five fields from each condition. ***, p < 0.001 based on one-way ANOVA with Dunnett's multiple comparison post-test against DMSO control. C, astrocytes were treated with either DMSO, 3 μM DMF, 30 μM DMF, 3 μM MMF, or 30 μM MMF for 6 h. Extracts from harvested cells were separated into nuclear or cytoplasmic fractions. Equal protein amounts from replicate aliquots of each fraction were tested in an Nrf2 DNA binding assay, and Western immunoblots were probed with antibodies for Nrf2 or HDAC. D, Nrf2 and NQO1 Western immunoblots of extracts from hOPCs, hNeurs, and rOPCs were treated with 10 μM DMF or MMF, then harvested after 1, 2, 4, 6, or 24 h of treatment. The 0-h time point represents the DMSO control sample.
Our previous work and that of others have demonstrated that fumarates can activate downstream transcription of a number of canonical Nrf2 target genes; however, the majority of these studies have been performed in cultured cells lines or assessed in non-CNS tissues (Linker et al., 2011). Activation of the Nrf2 transcriptional pathway by DMF and MMF was confirmed from analysis of Nrf2 mRNA levels in primary astrocyte cultures. A modest, but significant, approximately 2-fold transient increase in Nrf2 mRNA was observed after 2 and 4 h of DMF treatment, but no changes were observed with MMF treatment, although robust stabilization of the Nrf2 protein was observed (Fig. 3A). There was a greater accumulation of Nrf2 with DMF treatment; however, the levels of Nrf2 seemed more consistent at the later 24-h time point with MMF treatment (Fig. 3A). The expression profiles of several canonical Nrf2-responsive genes were also probed at the mRNA and protein levels. NQO1 (Fig. 3B), HO1 (Fig. 3C), and GCLC (Fig. 3D) all demonstrated significant induction by MMF or DMF, although the temporal aspect varied between analytes. NQO1 seemed to gradually accumulate mRNA and protein over time (Fig. 3B), whereas in contrast GCLC exhibited a transient induction in mRNA between 4 and 6 h that translated into increased protein from 6 to 24 h, by which time message levels had returned to baseline (Fig. 3D). HO1 also demonstrated a fairly transient response with mRNA and protein levels peaking from 4 to 6 h then declining out to 24 h (Fig. 3C). Although not a comprehensive profile of Nrf2-responsive genes, these data suggest that human astrocytes are capable of responding to MMF and DMF treatment, leading to induction of a prototypical phase II antioxidant response.
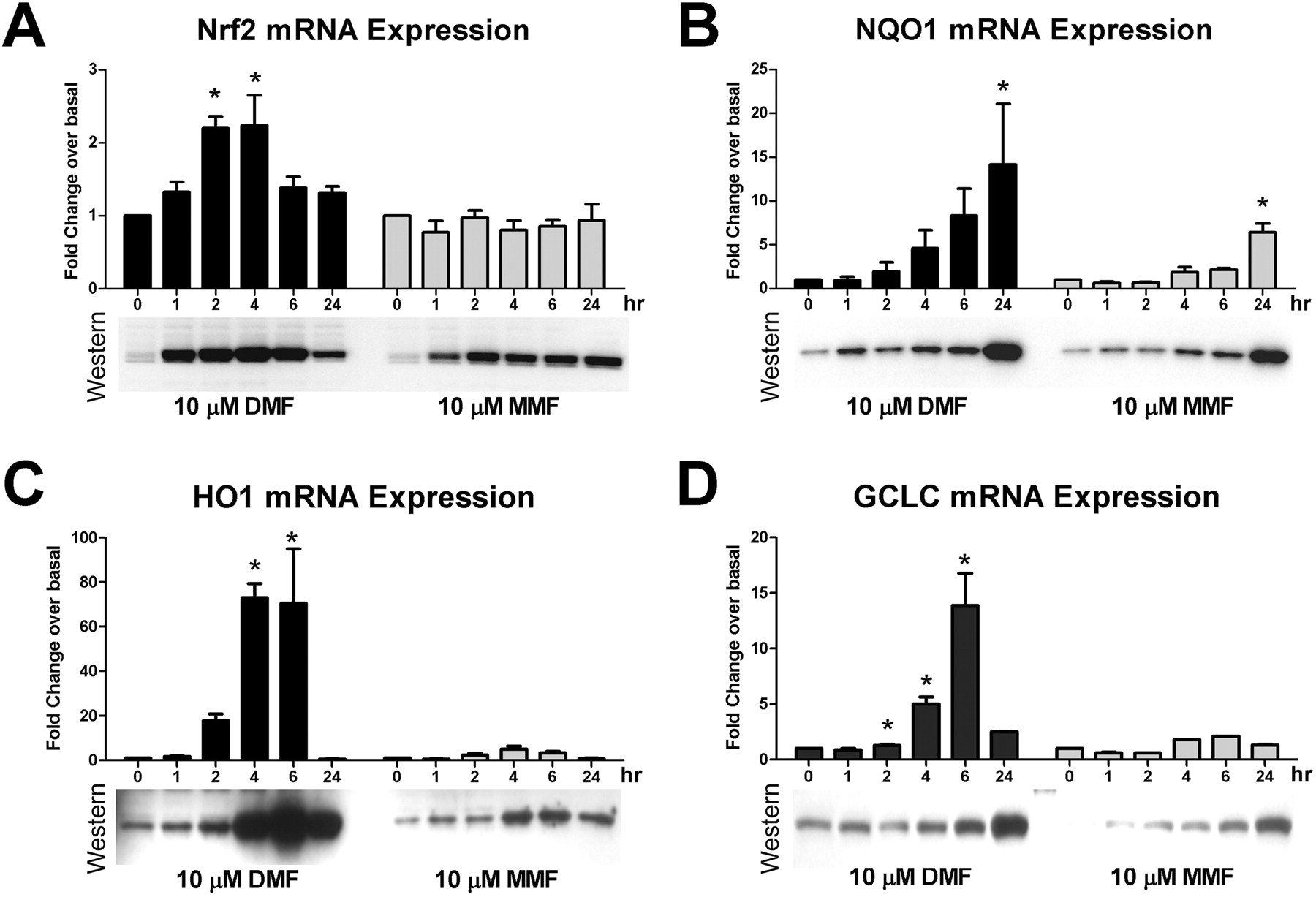
DMF and MMF induced up-regulation of canonical Nrf2 pathway antioxidant genes and proteins. Human astrocytes were treated with DMSO or 10 μM DMF or MMF, and replicate cultures were harvested at 1, 2, 4, 6, or 24 h after compound addition. Duplicate cultures were harvested at each time point for RNA extraction and subsequent qPCR and to generate a cell extract for Western immunoblotting. Relative mRNA (bar graphs) and protein levels [Western immunoblots of Nrf2 (A), NQO1 (B), HO1 (C), and GCLC (D)] were assessed. For qPCR data, the average fold change relative to normalized DMSO control was plotted. The error bar represents S.D. (n = 4 for all assays). *, p < 0.05 based on one-way ANOVA with Dunnett's post-test for multiple-sample comparison against the DMSO 0-h time point control.
MMF Treatment Increases Cellular Redox Potential.
To further explore the molecular changes induced by DMF and MMF treatment, primary cultures of human astrocytes were used to assess the functional consequences of stimulating the Nrf2 pathway. These and most subsequent experiments describe the use of MMF; although DMF seemed more potent in inducing the Nrf2 response in vitro (Fig. 3A), preclinical animal and human clinical data have demonstrated that DMF is quantitatively converted to MMF very rapidly after oral dosing (Biogen Idec, unpublished data). This indicates systemically that cells and tissues are only exposed to MMF after an oral DMF dose, making MMF the more relevant fumarate species for in vitro use. After overnight treatment with MMF, astrocytes were incubated with resazurin, an oxidized substrate that changes fluorescent properties when reduced to resorufin by metabolically active cells. This should thus provide a relative assessment of cellular redox potential based on the ability to reduce oxidized substrates, because the substrate was supplied in excess. Increasing concentrations of MMF significantly increased the levels of resorufin, with a maximal 35% increase over baseline at 30 μM MMF (Fig. 4A). GSH, a primary cellular biomolecule for detoxifying free radicals, was dose-dependently increased by MMF (maximal increase 18% at 30 μM) when treated cells were incubated with the thiol-containing molecule monochlorobimane (Fig. 4B). MMF also seemed to promote modest, but significant, increases (approximately 5%) in cellular ATP content, relative to DMSO controls (Fig. 4C). These increases in ATP levels may be caused by enhanced production of ATP, as would be expected to occur in response to changes in the mitochondrial membrane potential, which drives the electron transport chain and ATP biosynthesis. Supporting this hypothesis, MMF treatment significantly increased the mitochondrial membrane potential in a concentration-dependent fashion, as shown by increased TMRE/MitoFluor Green ratios (maximal increase 2.3-fold at 10 and 30 μM; Fig. 4D). TMRE accumulates in the mitochondria based on the membrane potential of this organelle, driving accumulation of the anionic probe. Taken together, these data suggest DMF and MMF treatment “primes” cells and increases their capacity for dealing with oxidative stress by increasing the intrinsic pathways that result in enhanced biosynthesis of detoxification molecules such as GSH.
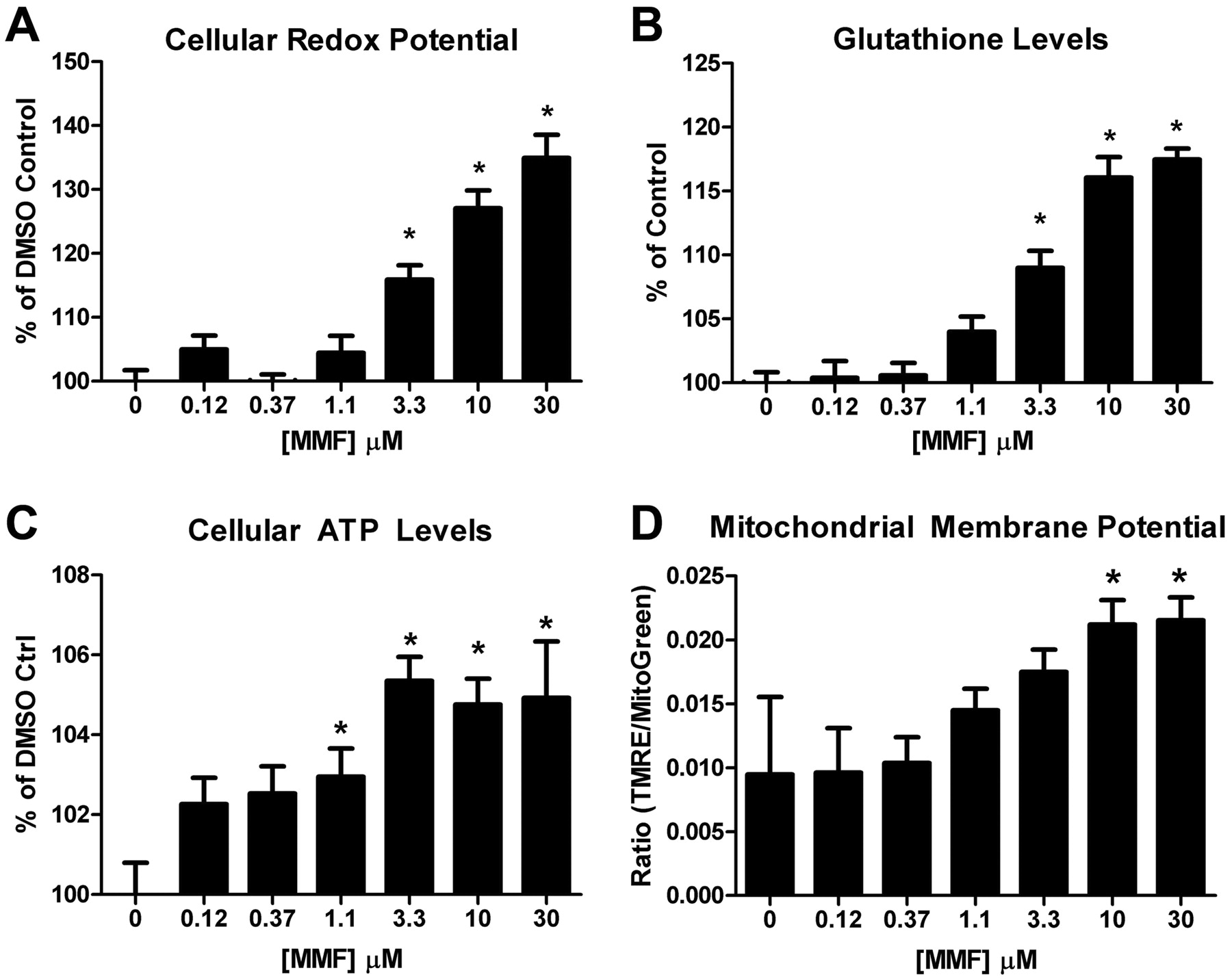
MMF treatment enhanced cellular redox potential by increasing GSH and ATP levels. Human astrocytes were treated with a titration of MMF and then assessed for cellular redox potential (A), GSH levels (B), total ATP levels (C), and mitochondrial membrane potential (D). Results in A to C are presented as a percentage relative to DMSO control-treated cells. In D, mitochondrial membrane potential is represented as the average ratio of TMRE intensity to MitoFluor Green intensity to normalize for total cellular mitochondrial content. Error bars represent S.D. (minimum n = 8). *, p < 0.05 based on one-way ANOVA analysis and Dunnett's multiple comparison post-test against DMSO controls.
DMF and MMF Enhance Viability after Oxidative Stress.
To begin characterizing the functional effects of DMF and MMF treatment, primary cultures of human astrocytes were subjected to oxidative stress by using H2O2 and assessed for changes in intracellular calcium accumulation. Changes in these levels were assessed by using Calcium 4 dye fluorescence recorded over a 120-min period in cells treated with a titration of MMF, then challenged with H2O2 (Fig. 5A). DMSO control cells challenged with H2O2 showed an approximate 50% increase in calcium over unchallenged controls, whereas increasing concentrations of MMF progressively reduced H2O2-induced increases back to baseline levels (Fig. 5B).

DMF and MMF promoted direct cytoprotection after oxidative challenge in astrocytes. The functional effects of MMF treatment were assessed in human astrocytes that were treated with a titration of MMF and then challenged with H2O2. A, real-time analysis of intracellular calcium as visualized by Calcium-4 fluorescence (RFU) in treated astrocytes followed for 120 min (recording once every minute) after a 50 μM H2O2 challenge. Different colored traces represent different MMF concentrations as indicated. Arrow denotes time of H2O2 addition. B, quantification of change in RFU based on difference of maximum and minimum signals over recording period. Graph represents average ΔRFU for each indicated condition. Error bars represent S.D. Each condition was done in quadruplicate. Red dashed line represents maximal H2O2-induced calcium accumulation; green dashed line represents basal calcium accumulation in the absence of H2O2 challenge. C and D, quantification of calcein AM fluorescence intensity (RFU) in LIVE/DEAD-labeled cells pretreated with a titration of DMF (C) or MMF (D) and then challenged with 0, 50, or 100 μM H2O2 followed by a 20-h recovery before analysis. E and F, replicate plates as in C and D fixed and stained with Hoechst dye. Graphs represent average cell nuclei counts in 10 fields per condition. Error bars represent S.D. in C to F. G to J, live imaging of cells from C to F: LIVE calcein AM (green) and DEAD ethidium homodimer (red) labeling. Presented images have identical histogram lookup tables for display comparison. G, negative control (Neg Ctrl) cells preincubated with 0.1% saponin to permeabilize plasma membranes. H, positive control cells treated with DMSO. I, control DMSO-treated and 50 μM H2O2-challenged cells. J, astrocytes pretreated with 11 μM MMF and challenged with 50 μM H2O2.
Because abnormal intracellular calcium accumulation is known to trigger apoptotic cascades and destabilize mitochondrial energy metabolism, the effects of DMF and MMF treatment and H2O2 challenge on cellular viability were assessed. To identify potential confounding effects of DMF and MMF cytotoxicity, both compounds were assessed and in both LIVE/DEAD and nuclei counting assays under conditions of no oxidative stress (0 μM H2O2). These results suggested that in these cells DMF was cytotoxic at concentrations of 33 and 100 μM (Fig. 5, C and E green bars), whereas MMF treatment (Fig. 5, D and F, green bars) resulted in less toxicity (only at 100 μM). Addition of 50 μM H2O2 to DMSO-treated cells in the absence of DMF or MMF resulted in approximately 66% reductions in fluorescence intensity in the LIVE/DEAD assay and 75% reductions in nuclei counts (Fig. 5, C–F). Nontoxic concentrations of DMF (Fig. 5, C and E) and MMF (Fig. 5, D and F) improved cellular viability in both assay formats. Consistent with Nrf2 pathway activation data (Figs. 2, B-D and 3), DMF seemed to be more potent than MMF in protecting astrocytes from oxidative challenge; however, the maximal protective response was similar for both compounds (Fig. 5, C–F). The cytoprotective effects conferred by DMF or MMF treatment were overcome by 100 μM H2O2 challenge, at which only minor trends in protection were observed (Fig. 5, C–F). To confirm the fluorescence intensity-based quantitative results (Fig. 5, C-F), imaging assessments from samples used in the LIVE/DEAD assay were performed. Saponin-treated negative controls (Fig. 5G) clearly showed a lack of live cells, whereas DMSO-positive controls showed a uniform confluent layer of viable cells (Fig. 5H). Challenging the DMSO-treated cells with 50 μM H2O2 resulted in a dramatic increase in dead cells and a change in morphology of the viable cells to a more rounded structure that is characteristic of cells undergoing apoptotic or other toxicity-related death (Fig. 5I). Pretreatment of the astrocytes with MMF resulted in the preservation of viability and morphology (Fig. 5J). However, MMF did not prevent the loss of all cells as can be seen from the decreased density of viable cells (compare Fig. 5, J and H). These data, using multiple different types of assay formats, suggested that both DMF and MMF were able to promote cytoprotective responses in cells such that they were better able to withstand oxidative stress and thus had improved viability after a toxic challenge.
To extend these data and determine whether the direct cytoprotective responses of DMF and MMF were consistent in other CNS cell types, cortical cultures from embryonic rat pups were used in a similar H2O2 challenge paradigm. Fixed cultures were immunostained for the neuronal marker protein βIII-tubulin, and nuclei were labeled with DAPI. Control cultures showed healthy neural cells, with all neurons extending both highly branched and unbranched neurites (Fig. 6A). Challenging with 10 μM H2O2 caused an 80% loss in neurons (Fig. 6, B and D). However, cells that were pretreated with MMF exhibited significant protection from the oxidative insult with a maximal 55% increase in viability at 10 μM MMF (Fig. 6, C and D). These data confirmed that MMF can act on multiple CNS-resident cell types and confer cytoprotective responses against oxidative stress.

MMF promotes cytoprotection of rat cortical neurons after oxidative challenge. A to C, representative images and quantitation from control rat primary cortical neuronal cultures from embryonic day 18 pups grown for 7 days in vitro (A), replicate cortical culture incubated with 10 μM H2O2 for 2 h then recovered for 20 h (B), and replicate cortical culture treated with 0, 3.3, 10, or 30 μM MMF for 20 h before 10 μM H2O2 challenge and 20-h recovery (C). All cultures were immunostained with βIII-tubulin (green) and DAPI (blue). Presented images have identical histogram lookup tables for display comparison. D, quantification of neuronal counts from five fields from each condition. Graph represents the mean percentage of each condition relative to non-H2O2 challenged controls. Error bars represent S.D. *, p < 0.05 based on one-way ANOVA analysis and Dunnett's multiple comparison post-test against “0 μM + H2O2” DMSO controls.
MMF-Induced Cytoprotection Is Nrf2-Dependent.
The role of Nrf2 in mediating DMF- or MMF-dependent cytoprotection was assessed by reducing Nrf2 mRNA and protein by using Nrf2-specific siRNA. Transfection of human astrocytes with control siRNA resulted in a small decrease in Nrf2 levels after induction with 10 μM DMF (Fig. 7, A and B). In contrast, cultures transfected with Nrf2 siRNA had lower basal levels of Nrf2 and did not seem to have increased Nrf2 protein after DMF treatment (Fig. 7, A and B). These data indicated that the Nrf2 siRNA was capable of reducing total Nrf2 protein and prevented the DMF-dependent increase in Nrf2. To determine the effects of reducing Nrf2 protein, astrocytes were transfected with either control or Nrf2 siRNA, treated with DMSO as a control or a titration of MMF for 20 h, and subjected to 50 μM H2O2 challenge. This analysis demonstrated that in control siRNA conditions MMF was able to confer cytoprotective responses on H2O2-challenged astrocytes in a concentration-dependent fashion (Fig. 7C), similar to previous data in untransfected cells (Fig. 5, D and J). However, in cells transfected with Nrf2 siRNA and challenged with 50 μM H2O2 MMF had no cytoprotective effects at any concentration tested (Fig. 7C), suggesting Nrf2 protein was required for MMF-dependent cytoprotection against oxidative stress.
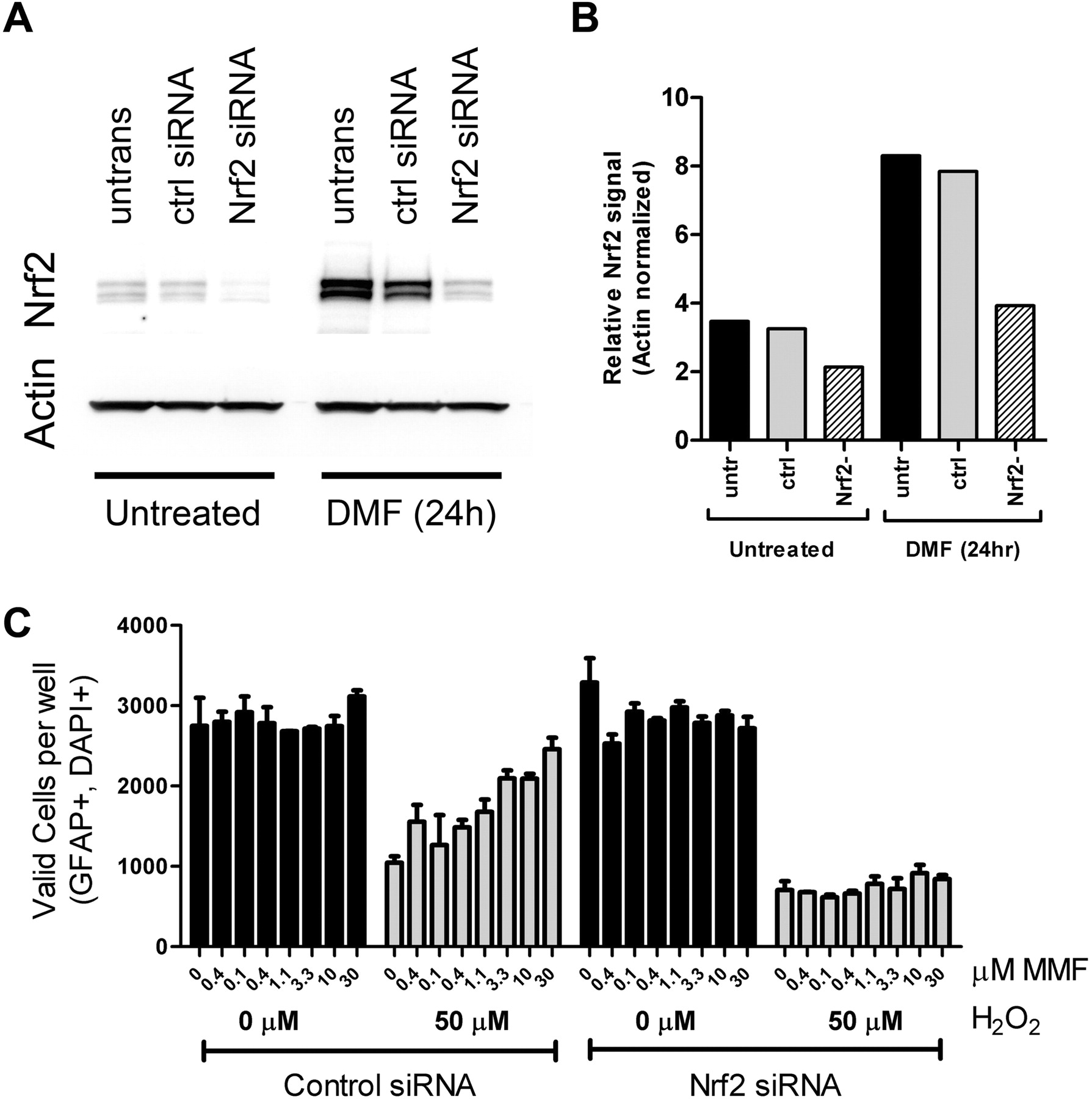
Astrocyte cytoprotection conferred by MMF depended on Nrf2. Human astrocytes that were untransfected or transfected with either control or Nrf2-specific siRNA were analyzed. A, replicate control and transfected astrocytes were separated into two different groups: “untreated” cells, which were transfected with indicated the siRNA and harvested 20 h after transfection; and “DMF 24h” cells, which were transfected, incubated for 20 h, and treated with 30 μM DMF for 20 h before preparation of cellular extracts. Western immunoblots of cell extracts were probed with antibodies to Nrf2 or actin. B, quantitation of Western immunoblots in A. C, astrocyte viability. Cells were transfected with control or Nrf2 siRNA, treated with a titration of MMF, and challenged with 0 μM H2O2 (black bars) or 50 μM H2O2 (gray bars). Valid cell counts were determined in cells that were positive for glial fibrillary acidic protein (GFAP) immunostaining and DAPI staining. The graph represents average cell counts per well in quadruplicate wells; error bars indicate S.D.
Discussion
Many studies have indicated that targeting the Nrf2 pathway presents a potentially potent mechanism to combat the oxidative stress associated with several different neurodegenerative diseases (Calkins et al., 2009). Targeting the Nrf2 pathway with small-molecule activators presents an attractive opportunity because the target is an intrinsic cellular pathway that is designed to be dynamically modulated. The need for this type of therapy in MS is clear, because current evidence indicates free radicals play a large role in MS pathogenesis (van Horssen et al., 2011), with damage to protein, lipid, and DNA in the tissues and cells proximal to lesion areas and in circulation (Vladimirova et al., 1998; Ferretti et al., 2005; van Horssen et al., 2008). It is noteworthy that these studies have shown that some antioxidant response proteins, including Nrf2, are elevated in MS lesions (van Horssen et al., 2008, 2010); however, the degree of intrinsic antioxidant induction is apparently insufficient to ultimately prevent disease progression. Our observations have shown that DMF and MMF can activate the Nrf2 pathway in multiple CNS-resident cell types both in vitro and in vivo, stabilizing levels of Nrf2 protein, resulting in the accumulation of active transcription factor and translocation into the nucleus, thereby conferring cytoprotective responses against oxidative stress. Profiling of known Nrf2 target genes, such as NQO1, suggests that astrocytes, OPCs, and perhaps neurons are capable of responding to MMF and DMF treatment, leading to induction of a prototypical Nrf2-dependent antioxidant response. Detailed profiling is currently underway to characterize CNS cell type- and brain region-specific transcriptional responses to DMF and MMF treatment.
In addition to characterization of Nrf2 pathway activation in CNS cells, a potential downstream functional mechanism by which DMF or MMF may promote resistance to oxidative stress has been identified. The studies described here demonstrated that MMF treatment can increase cellular GSH levels in astrocytes (Fig. 4B), which is consistent with the demonstrated up-regulation of GCLC (Fig. 3D), a rate-limiting enzyme in GSH biosynthesis. GSH is the main cellular antioxidant against ROS, thus increased levels of this thiol-containing tripeptide would promote increased ability to detoxify and survive oxidative stress. The MMF-dependent increases observed at 20 h after treatment are not necessarily inconsistent with previous findings, which demonstrated no acute changes to GSH levels over 60 min of MMF treatment (Schmidt and Dringen, 2010; Thiessen et al., 2010). The protein profiling analysis demonstrated that at least some enzymatic components involved in GSH biosynthesis, such as GCLC, were not up-regulated until several hours after treatment (Fig. 3D), indicating that 60 min may be an insufficient time to observe these changes. Recent in vivo evidence also supports a role for activation of the Nrf2 pathway in increasing CNS GSH levels. These data suggested that enhanced astrocyte GSH biosynthesis and secretion led to increased extracellular cysteine as a product of GSH metabolism, which was then transported into neurons via up-regulation of the excitatory amino acid transporter 3, and ultimately drove increased neuronal GSH due to increased levels of biosynthetic substrate (Escartin et al., 2011). Elucidation of this pathway suggests a potential role for cell type-specific neuroprotective responses and is consistent with the astrocyte data presented here. It is clear that increasing cellular GSH levels in the CNS could have a meaningful therapeutic impact on MS, because reduced brain GSH levels have been reported in patients with MS, as measured by magnetic resonance spectroscopy (Srinivasan et al., 2010; Choi et al., 2011). Therapies in clinical trials for MS, such as BG-12, which is an oral therapeutic containing DMF as the active ingredient (which is quantitatively converted to MMF after oral DMF delivery), could increase cellular GSH levels (Fig. 4B) and may offer an opportunity to restore redox balance in the CNS of patients. This could have a meaningful therapeutic impact by potentially offering greater protection against the neurodegenerative effects of disease-related increases in oxidative stress.
Mitochondrial dysfunction may also play a major role in the pathogenesis of neurodegenerative diseases, particularly in MS, where reduced mitochondrial function in the CNS has been observed (Dutta et al., 2006; Mahad et al., 2009), despite increased mitochondrial content (Blokhin et al., 2008; Witte et al., 2009). This seemingly paradoxical situation possibly reflects a compensatory mechanism for reduced energy output from individual mitochondrion. Our data obtained in astrocytes may indicate that MMF could potentially alleviate some of this mitochondrial dysfunction, because modest, but significant, increases in cellular ATP content (Fig. 4C) and mitochondrial membrane polarization (Fig. 4D) were observed after treatment. It is also important to note that mitochondria have a critical role in buffering intracellular calcium, and interestingly, MMF was able to prevent the H2O2-induced abnormal accumulation of intracellular calcium (Fig. 5, A and B). However, it was not clear if this was caused by improved mitochondrial buffering capacity via enhancement of the electrochemical gradient and subsequent increased calcium ionic flux or direct detoxification of the oxidative challenge via MMF-induced Nrf2 antioxidant response. The mechanism by which MMF may increase mitochondrial energy metabolism and membrane potential remains unclear.
Our results, using multiple types of assay formats, indicate that both DMF and MMF are able to promote cytoprotective responses in cells by activating the Nrf2 pathway, enabling them to better withstand oxidative stress. The attenuation of H2O2-induced calcium accumulation, along with potential mitigation of other cellular events related to toxic oxidative challenge, ultimately resulted in MMF- or DMF-dependent increase in viability in astrocytes and neurons. Nrf2 seemed to be required for these effects. The oxidative injury and challenge paradigms explored here are highly relevant to the mechanistic damage that occurs in MS, and collectively these preclinical studies using DMF and its primary metabolite MMF provide a compelling rationale for the use of BG-12 as a therapeutic agent in the treatment of MS.
Authorship Contributions
Participated in research design: Scannevin, Chollate, Jung, Shackett, Patel, Ryan, Lukashev, and Rhodes.
Conducted experiments: Scannevin, Chollate, Jung, Shackett, Patel, Bista, Zeng, and Ryan.
Contributed new reagents or analytic tools: Scannevin, Bista, Zeng, Ryan, and Yamamoto.
Performed data analysis: Scannevin, Chollate, Jung, Shackett, Patel, Bista, Zeng, Ryan, and Lukashev.
Wrote or contributed to the writing of the manuscript: Scannevin, Ryan, Lukashev, and Rhodes.
Acknowledgments
We thank Mark Hughes, Simon Whiteley, and Heather Sun for reviewing this manuscript and providing editing assistance.
Footnotes
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- MS
- multiple sclerosis
- CNS
- central nervous system
- Nrf2
- nuclear factor (erythroid-derived 2)-like 2
- DMF
- dimethyl fumarate
- MMF
- monomethyl fumarate
- qPCR
- quantitative polymerase chain reaction
- NQO1
- nicotinamide adenine dinucleotide phosphate dehydrogenase (quinone 1)
- Akr1b8
- aldo-keto reductase family 1, member B8
- HO1
- heme oxygenase 1
- GCLC
- glutamate-cysteine ligase catalytic subunit
- DMSO
- dimethyl sulfoxide
- GSH
- glutathione
- OPC
- oligodendrocyte precursor cell
- hOPC
- human OPC
- rOPC
- rat OPC
- hNeur
- human neuron
- DAPI
- 4′,6-diamidino-2-phenylindole
- TMRE
- tetramethyl rhodamine ester
- RFU
- relative fluorescence units
- siRNA
- small interfering RNA
- HDAC
- histone deacetylase
- ROS
- reactive oxygen species
- ANOVA
- analysis of variance
- WT
- wild type
- KO
- knockout
- AM
- acetoxymethyl ester.

- Received November 21, 2011.
- Accepted January 19, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}