


Abstract
The prostaglandin D2 (PGD2) receptor type 2 (DP2) is a G protein-coupled receptor that has been shown to be involved in a variety of allergic diseases, including allergic rhinitis, asthma, and atopic dermatitis. In this study, we describe the preclinical pharmacological and pharmacokinetic properties of the small-molecule DP2 antagonist [2′-(3-benzyl-1-ethyl-ureidomethyl)-6-methoxy-4′-trifluoromethyl-biphenyl-3-yl]-acetic acid (AM211). We determine that AM211 has high affinity for human, mouse, rat, and guinea pig DP2 and it shows selectivity over other prostanoid receptors and enzymes. Antagonist activity of AM211 at the DP2 receptor was confirmed by inhibition of PGD2-stimulated guanosine 5′-O-[γ-thio]triphosphate binding to membranes expressing human DP2. A basophil activation assay and a whole-blood assay of eosinophil shape change were used to demonstrate the ability of AM211 to potently antagonize PGD2-stimulated functional responses in relevant human cells and in the context of a physiologically relevant environment. AM211 exhibits good oral bioavailability in rats and dogs and dose-dependently inhibits 13,14-dihydro-15-keto-PGD2-induced leukocytosis in a guinea pig pharmacodynamic assay. AM211 demonstrates efficacy in two animal models of allergic inflammation, including an ovalbumin-induced lung inflammation model in guinea pigs and an ovalbumin-induced mouse model of allergic rhinitis. AM211 represents a potent and selective antagonist of DP2 that may be used clinically to evaluate the role of DP2 in T helper 2-driven allergic inflammatory diseases.
Introduction
Prostaglandin D2 (PGD2) is one of a family of biologically active lipids derived from arachidonic acid via the action of the cyclooxygenase-1 and -2 enzymes (COX1 and COX2). PGD2 binds to three G protein-coupled receptors within the prostanoid family, namely PGD2 receptor type 1 (DP1), PGD2 receptor type 2 [DP2 or CRTH2, chemoattractant receptor-homologous molecule expressed on T helper 2 (Th2) cells], and the thromboxane A2 receptor (TP). Both DP1 and DP2 as well as TP have been suggested to be viable targets for the treatment of asthma and other allergic diseases (Pettipher et al., 2007).
DP2 is expressed on eosinophils, basophils, and Th2 lymphocytes and is also present in a number of human hematopoetic organs and tissues of the digestive system (Nagata et al., 1999a,b; Sawyer et al., 2002). DP2 is not expressed on human neutrophils in peripheral blood, but the receptor has been shown to be present on neutrophils in the sputum of patients with cystic fibrosis, whereas DP2 expression remains absent on their blood neutrophils (Tirouvanziam et al., 2008). By contrast, mouse and rat DP2 expression is highest in lung, brain, ovary, and spleen and, unlike human expression, is also observed on peripheral blood neutrophils (Abe et al., 1999; Shichijo et al., 2003). The DP2 knockout mouse is healthy and fertile, supporting the lack of obvious target-related safety concerns. Sequence variants of the human DP2 gene that result in increased messenger RNA stability have been associated with bronchial hyper-responsiveness and severe asthma (Huang et al., 2004).
Selective DP2 antagonists have shown efficacy in preclinical models of allergic rhinitis and asthma (Pettipher, 2008). One study using DP2 knockout mice demonstrated an attenuation of nasal eosinophilia, allergen-specific IgE production, and IL-4 production in a mouse model of allergic rhinitis (Nomiya et al., 2008). In addition, in a murine model mimicking the enhanced asthmatic response induced by viral infection, DP2-deficient mice were protected from double-stranded RNA-induced airway eosinophilia (Shiraishi et al., 2008). Selective agonist activation of mouse DP2 increased allergic lung and skin inflammation (Spik et al., 2005), and a small-molecule DP2 antagonist inhibited allergic airway and cutaneous inflammation (Pettipher, 2008; Boehme et al., 2009).
The development of a high-affinity DP2 antagonist for allergic inflammatory diseases offers the potential to control Th2 cytokine amplification of allergic inflammation and also potentially neutrophilic activation, which is characteristic of patients who are resistant to corticosteroid treatment. There are no marketed selective DP2 antagonists, but several compounds are reported to be in phase II clinical trials for allergic rhinitis, asthma, and chronic obstructive pulmonary disease (COPD) (Pettipher, 2008). Ramatroban [Baynas, BAY u3405 (+)-(3R)-3-(4-fluorobenzenesulfonamido)-1,2,3,4-tetra-hydrocarbazole-9-propionic acid], a nonselective dual TP/DP2 antagonist (five times more potent on TP than DP2), is clinically efficacious in allergic rhinitis and is marketed for this indication in Japan (Ohkubo and Gotoh, 2003). Some of the clinical efficacy of Ramatroban may be mediated by DP2 antagonism, because blood concentrations achieved at clinical doses are sufficient to block DP2 (Johnston et al., 1993; Sugimoto et al., 2003).
We have characterized AM211 [2′-(3-benzyl-1-ethyl-ureidomethyl)-6-methoxy-4′-trifluoromethyl-biphenyl-3-yl]-acetic acid and shown that it represents a novel, potent, and selective DP2 antagonist. We show that AM211 can decrease antigen-induced pulmonary inflammation in guinea pigs and antigen-induced nasal symptoms in a mouse model of allergic rhinitis. Based on its pharmacokinetic (PK) and pharmacodynamic (PD) profile, AM211 may hold promise as an oral agent for the treatment of persistent asthma and other allergic diseases associated with Th2-dependent inflammation.
Materials and Methods
Chemicals.
AM211 was synthesized at Amira Pharmaceutical's Department of Medicinal Chemistry (Fig. 1). For all in vitro studies, a stock concentration of 25 mM in dimethylsulfoxide (DMSO) was diluted to the appropriate final concentration in assay buffer. For the pharmacokinetic and in vivo studies, the compound was dosed by oral gavage as a solution in 0.5% methylcellulose unless otherwise specified.

Chemical structure of AM211.
[3H]PGD2 Radioligand Membrane Binding Assays.
HEK293/FlpIn cells (Invitrogen, Carlsbad, CA) stably expressing human, mouse, guinea pig, or rat DP2 were used for binding experiments. All stable cell lines were grown in Dulbecco's modified Eagle's medium (high glucose) + Glutamax (Invitrogen) supplemented with 10% fetal bovine serum, penicillin/streptomycin, sodium pyruvate, and 200 μg/ml hygromycin B (Invitrogen). Receptor binding was carried out with 4 to 20 μg of HEK293 membranes stably expressing DP2, 1 nM tritiated ligand ([ 3H]PGD2, PerkinElmer Life and Analytical Sciences, Waltham, MA) and varying concentrations of AM211 (0.01 nM–100 μM) in buffer containing 50 mM HEPES, 10 mM MnCl2, and 1 mM EDTA in the presence or absence of 0.2% human, mouse, guinea pig, or rat serum albumin. Nonspecific binding was determined using an excess (10 μM) of PGD2. Reactions were incubated for 60 min at room temperature, harvested onto glass filter binding plates (UniFilter GF/C; PerkinElmer Life and Analytical Sciences), and washed three times with cold buffer containing 50 mM HEPES, pH 7.4, and 0.5 M NaCl. Plates were dried, microscint was added, and cpm were recorded using a Packard TopCount NXT microplate scintillation counter (PerkinElmer Life and Analytical Sciences). The percentage of inhibition of [3H]PGD2 binding relative to the vehicle controls was graphed, and the IC50 was generated by nonlinear regression analysis.
[35S]GTPγS Membrane Binding Assay.
The [35S]GTPγS binding assay was carried out using 10 to 20 μg of CHO membranes from cells stably expressing human DP2, varying concentrations of AM211 (1 nM–30 μM), 50 nM [35S]GTPγS, 5 μM GDP, and 80 nM PGD2 in buffer containing 50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, and 0.2% human serum albumin, pH 7.35. Nonspecific binding was determined using an excess (10 μM) of GTPγS. Reactions were incubated for 60 min at 30°C, harvested onto glass filter binding plates (UniFilter GF/B), and washed three times with cold buffer containing 50 mM HEPES, pH 7.4, 0.1 M NaCl, and 5 mM MgCl2. Counts per minute were recorded using a Packard TopCount NXT microplate scintillation counter. The percentage of inhibition of [35S]GTPγS binding relative to the vehicle controls was graphed, and the IC50 was generated by nonlinear regression analysis. For agonist mode, varying concentrations of AM211 (10 nM–100 μM) or PGD2 (0.1 nM–3 μM) were incubated with 10 to 20 μg of CHO membranes from cells stably expressing human DP2, 50 nM [35S]GTPγS, and 5 μM GDP in buffer containing 50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, and 0.2% human serum albumin, pH 7.35. Agonist binding data were plotted as a percentage of the maximum PGD2-induced binding.
Human and Guinea Pig Whole-Blood Eosinophil Shape Change Assay.
Human blood was obtained by venipuncture from consenting adult volunteers, collected into EDTA vacutainer tubes, and used within 30 min of collection. Guinea pig blood was collected into EDTA vacutainer tubes from male Hartley guinea pigs via the marginal ear vein and used within 15 min of collection. Aliquots of fresh blood (100 μl) were incubated for 15 min at 37°C with varying concentrations of AM211 (0.1 nM–1 μM) (diluted in 50% DMSO/water) or with vehicle control. PGD2 (50 nM human blood or 500 nM guinea pig blood) was added, and the incubation was continued for 5 min. The reactions were placed on ice, and 250 μl of an ice-cold 1:4 dilution of Cytofix (BD Biosciences, San Jose, CA) in PBS was immediately added. The reactions were then transferred to 12 × 75-mm polystyrene tubes, and 3 ml of an ammonium chloride lysing solution (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM disodium EDTA, pH 7.4) was added to lyse the red blood cells. The lysis was continued at room temperature for 10 min, and the tubes were centrifuged at 1300 rpm for 5 min at 4°C. The cells were washed once with 3 ml of cold PBS, centrifuged, and resuspended in 200 μl of ice-cold 1:4 diluted Cytofix. Eosinophil shape change (ESC) was analyzed on a FACScalibur (BD Biosciences) by isolating the eosinophils from the other cells based on their intrinsic autofluorescence and then analyzing the percentage of total eosinophils with an increased forward scatter value. Maximum (100%) shape change was determined in the presence of 50 nM PGD2 (human blood) or 500 nM PGD2 (guinea pig blood). Minimum (0%) shape change was determined in the presence of PBS. The percentage of inhibition of eosinophil shape change by AM211 relative to the vehicle controls was graphed, and the IC50 was generated by nonlinear regression analysis.
Human Basophil CD203c Expression.
Blood was collected from consenting volunteers into citrate tubes and used within 2 h of draw. After sedimentation of red blood cells with dextran sulfate, a hypotonic lysis of remaining red blood cells was performed and the leukocytes were washed in Hanks' balanced salt solution and then resuspended in GAFS buffer (PBS containing 0.9 mM CaCl2, 0.5 mM MgCl2, 10 mM HEPES, 10 mM glucose, and 0.2% human serum albumin) at 4 × 106 cells/ml. Leukocytes (0.5 ml) were aliquoted into 1.2-ml polypropylene tubes and incubated for 10 min at 37°C with varying concentrations of AM211 (0.01 nM–300 nM) or vehicle (PBS/5% DMSO). PGD2 (100 nM final concentration) was added, and incubation was continued for 10 min at 37°C. The reactions were placed on ice, and 600 μl of ice-cold stain buffer (BD Pharmingen, San Diego, CA) was immediately added. The reactions were then transferred to FACS tubes, and the cells were pelleted. Cells were resuspended in 100 μl of stain buffer and 20 μl of an anti-human CD203c antibody (Beckman Coulter, Fullerton, CA) conjugated to R-phycoerythrin was added. Cells were incubated with the antibody for 30 min on ice before washing twice with stain buffer. The cells were resuspended in 400 μl of ice-cold 1:4 diluted Cytofix (BD Biosciences) and analyzed using a FACScalibur. Basophils were identified based on positive CD203c staining and low side scatter, and the percentage of basophils expressing high levels of CD203c (CD203chi) was determined. Maximum (100%) and minimum (0%) percentage of CD203chi cells was determined in the presence of 100 nM PGD2 and PBS, respectively. The percentage of inhibition of CD203chi cells by AM211 relative to the vehicle controls was graphed, and the IC50 was generated by nonlinear regression analysis.
Human Prostaglandin Receptor Binding Assays and COX Whole-Blood Assays.
A detailed description of the assay conditions for the COX-1 and COX-2 whole-blood assays can be found in Lorrain et al. (2009). For prostacyclin receptor (IP) binding experiments, AM211 was added to 10 μg of HEK293 membranes stably expressing human IP and 5 nM tritiated ligand ([ 3H]iloprost; GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) in buffer containing 10 mM HEPES, 10 mM MnCl2, pH 7.35. Nonspecific binding was determined using an excess (10 μM) of Iloprost (Cayman Chemical, Ann Arbor, MI). For DP1 binding experiments, AM211 was added to 20 μg of human platelet membranes and 2 nM tritiated ligand ([3H]3-[(2-cyclohexyl-2-hydroxyethyl)amino]-2,5-dioxo-1-(phenylmethyl)-4-imidazolidineheptanoic acid (BW A868C); PerkinElmer Life and Analytical Sciences) in buffer containing 50 mM HEPES, 10 mM MnCl2, and 1 mM EDTA pH 7.35. Nonspecific binding was determined using an excess (10 μM) of BW A868C (Cayman Chemical). For TP binding experiments, AM211 was added to 20 μg of human platelet membranes and 10 nM tritiated ligand ([ 3H]SQ 29,548 ([1S-[1α,2α(Z),3α,4α]]-7-[3-[[2-[(phenylamino)carbonyl]hydrazino]methyl]-7-oxabicyclo[2.2.1]hept-2-yl]-5-heptenoic acid) (PerkinElmer Life and Analytical Sciences) in buffer containing 50 mM HEPES, 10 mM MnCl2 and 1 mM EDTA pH 7.35. Nonspecific binding was determined using an excess (10 μM) of SQ 29,548 (Cayman Chemical). For prostaglandin F2α receptor (FP) binding, AM211 was added to 35 μg of HEK membranes stably expressing human FP and 1.5 nM tritiated ligand ([3H]PGF2α; PerkinElmer Life and Analytical Sciences) in buffer containing 10 mM 2-(N-morpholino)ethanesulfonic acid/KOH, 10 mM MnCl2, and 0.4 mM EDTA pH 7.35. Nonspecific binding was determined using an excess (5 μM) of PGF2α (Cayman Chemical). In each case, reactions were incubated for 60 min at room temperature. Membranes were harvested onto glass filter binding plates (UniFilter GF/C) and washed three times with cold buffer containing 50 mM HEPES, 0.5 M NaCl, pH 7.4 (DP1 and TP binding), 15 mM HEPES, 0.01% bovine serum albumin, pH 7.4 (IP binding), or 10 mM 2-(N-morpholino)ethanesulfonic acid/KOH, 0.01% bovine serum albumin (FP binding) using a Brandel 96-tip cell harvester (Brandel Inc., Gaithersburg, MD.). Plates were counted using a Packard TopCount NXT microplate scintillation counter (PerkinElmer Life and Analytical Sciences). The total binding (TB) was defined as the average cpm from the vehicle-treated samples and was set to 100%, and the nonspecific binding was defined as the average cpm from vehicle-treated samples in the presence of excess cold compound and this value was set to 0%. The percentage of inhibition of radioligand binding relative to the vehicle controls was calculated as follows: ((cpm TB − cpm sample)/(cpm TB − cpm nonspecific binding)) × 100.
Peroxisome Proliferator-Activated Receptor Assays.
The ability of AM211 to function as an agonist or antagonist at the human peroxisome proliferator-activated receptor (PPAR) α, δ, and γ was evaluated using commercially available LanthaScreen Coactivator Assay kits from Invitrogen. In brief, these kits evaluated the ability of AM211 to inhibit (antagonist mode) or enhance (agonist mode) binding of a PPAR coactivator peptide to a PPAR α, δ, and γ ligand-binding domain-glutathione transferase fusion protein. When AM211 was screened in antagonist mode, the following agonist molecules were used to stimulate binding of the coactivator peptide: 10 nM 2-methyl-2-[[4-[2-[[(cyclohexylamino)carbonyl](4-cyclohexylbutyl)amino]ethyl]phenyl]thio]-2-methylpropanoic acid (GW7647) for PPARα, 20 nM [4-[[[2-[3-fluoro-4-(trifluoromethyl)phenyl]-4-methyl-5-thiazolyl]methyl]thio]-2-methylphenoxy] acetic acid (GW0742) for PPARδ, and 100 nM rosiglitazone for PPARγ.
Animals.
Male Sprague-Dawley rats (Charles River Breeding Laboratories, Portage, MI) weighing 250 to 300 g with surgically implanted jugular vein catheters were used for pharmacokinetic experiments. Male Hartley guinea pigs (Charles River Breeding Laboratories; 200–300 g at study initiation) were used for intravenous DK-PGD2 and ovalbumin (OVA) studies. Female BALB/c mice (Harlan, Indianapolis, IN) weighing 19 to 22 g were used in the allergic rhinitis procedure. All animals were given free access to standard rodent/guinea pig chow and water and were allowed to acclimate for 1 week before study initiation. Animals were maintained on a 12-h light/12-h dark schedule with lights on at 6:00 AM. All experiments were conducted during the light phase of the light/dark cycle. All procedures were approved by the local Institutional Animal Care and Use Committee and carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Pharmacokinetics of AM211.
AM211 (2 mg/kg) was administered to fasted rats (n = 2) or dogs (n = 3) intravenously as a solution in 0.9% saline via a bolus injection into the jugular vein or orally at 5 to 10 mg/kg as a solution in 0.5% methylcellulose via an oral gavage to the stomach. Blood samples (approximately 300 μl) were taken from each animal via the jugular vein at various times up to 24 h postdose in sodium EDTA tubes. Plasma samples, prepared by centrifugation of whole blood, were stored frozen before analysis. To prepare the standard curve, known amounts of AM211 were added to thawed rat, dog, or guinea pig plasma (depending on the species being used) to yield a concentration range from 0.8 to 4000 ng/ml. Plasma samples were precipitated using acetonitrile (1:4, v:v) containing the internal standard buspirone. Ten microliters of the analyte mixture was injected using a LEAP PAL autosampler (LEAP Technologies, Carrboro, NC). The calibration curves were constructed by plotting the peak-area ratio of analyzed peaks against known concentrations. The lower limits of quantitation were 1 ng/ml when diluted in rat plasma, 1 to 4 ng/ml when diluted in dog plasma, and 20 to 40 ng/ml when diluted in guinea pig plasma. The data were subjected to linear regression analysis with 1/x2 weighting. Analyses were performed using an Agilent Zorbax SB-C8 column (2.1 × 50 mm; 5 μm) (Agilent Technologies, Santa Clara, CA) linked to a Shimadzu LC-10AD VP with SCL-10A VP system controller (Shimadzu, Kyoto, Japan). Tandem mass spectrometric detection was carried out on a PE Sciex API3200 (PerkinElmerSciex Instruments, Boston, MA) in the positive ion mode by multiple reaction monitoring. The mobile phases contained 10 mM ammonium acetate in water with 0.05% formic acid (solvent A) and 10 mM ammonium acetate in 50% acetonitrile/50% methanol with 0.05% formic acid (solvent B). The flow rate was maintained at 1 ml/min, and the total run time was 2.5 min. Analytes were separated using a linear gradient as follows: 1) mobile phase was held for 0.5 min at 5% B; 2) B was increased from 5 to 95% over the next 0.2 min; 3) B was held constant for 1.3 min at 95%, and 4) B was returned to the initial gradient conditions.
The pharmacokinetic parameters of AM211 were calculated by a noncompartmental analysis using WinNonlin (Pharsight, Mountain View, CA). The dose-adjusted area under the curve (AUC) was calculated by dividing the AUC by the dose. The oral bioavailablility was calculated by dividing the dose-adjusted AUC of the oral arm by the dose-adjusted AUC of the intravenous arm. Maximum plasma concentrations (Cmax) and their time of occurrence (Tmax) were obtained directly from the measured data.
Guinea Pig Intravenous DK-PGD2 Challenge.
Methods were adapted from those detailed in Shichijo et al. (2003). Guinea pigs were primed with ovalbumin on day 1 by intraperitoneal injection of 1 ml of a 100 μg/ml solution in Imject Alum (Thermo Fisher Scientific, Waltham, MA). They were then used in the DK-PGD2 procedure between days 15 and 22. Animals were randomly assigned to receive either vehicle (0.5% methylcellulose) or AM211 orally. Either 2 or 18 h after dosing of AM211, animals were anesthetized with ketamine/xylazine (70/10 mg/kg i.m.) and injected intravenously with 1 mg/kg of the selective DP2 agonist DK-PGD2 or vehicle (10% methyl acetate in PBS, 1 ml/kg) via the dorsolateral penile vein. Thirty minutes after intravenous DK-PGD2 administration, blood was collected via the marginal ear vein into EDTA tubes for cell analysis and AM211 plasma concentrations. After hypotonic lysis of red blood cells, leukocyte counts were determined using a hemacytometer. The number of cells per 0.1-mm3 field is presented. Maximum (100%) leukocytosis was determined in the presence of intravenous DK-PGD2 with a vehicle pretreatment, and basal (0%) leukocytosis was determined in the presence of PBS with a vehicle pretreatment. The percentage of inhibition of leukocytosis by AM211 relative to the vehicle controls was graphed, and the IC50 was calculated from the Prism (GraphPad Software, Inc., San Diego, CA) plot.
Measurement of Pulmonary Inflammation in Ovalbumin-Challenged Guinea Pigs.
Methods were adapted from those detailed in Muise et al. (2002). On day 1, all animals received a priming dose of ovalbumin by intraperitoneal injection [0.5 ml of a 100 μg/ml solution in Imject Alum (Thermo Fisher Scientific)]. An additional 0.5 ml of the ovalbumin solution was injected subcutaneously in the proximity of the cervical lymph nodes. On day 21, animals were randomly assigned to receive either vehicle (0.5% methyl cellulose) or AM211 (3 or 30 mg/kg) orally. One hour after dosing, animals were challenge with aerosolized ovalbumin (0.03% for 1 min) in an enclosed chamber. To enhance the DP2 component of the antigen challenge, guinea pigs received an intraperitoneal injection of DK-PGD2 (200 μg) 2 h after the aerosol ovalbumin. A second oral dose of vehicle or AM211 was given 6 h after antigen challenge. Twenty-four hours after challenge guinea pigs received an overdose of sodium pentobarbital (100 mg/kg s.c.), their trachea was exposed and cannulated, and bronchoalveolar lavage fluid (BALF) was collected (5 × 5 ml of PBS solution) and placed on ice. More than 90% of the fluid was recovered from each animal. Samples were centrifuged, and pellets were resuspended in 3 ml of PBS. Samples were loaded into a Thermo Fisher Scientific cytofunnel and spun for 5 min. Slides were allowed to dry and then stained using a diff-quick staining procedure and cover-slipped. Cells were visualized at a magnification of 40× using a Lab-Pro (Sunnyvale, CA) light microscope equipped with a digital camera and DigiPro software (LABO America, Inc., Fremont, CA). Four digital images were captured and later counted for cell totals and differential and expressed as the totals from the four images. At least 100 cells were counted for each subject.
Mouse Allergic Rhinitis.
The procedures for inducing allergic rhinitis symptoms were adapted from those detailed in Nakaya et al. (2006). In brief, mice were immunized by an intraperitoneal injection of 10 μg of OVA complexed with Imject Alum in a volume of 0.2 ml on days 1 and 8. Seven days later (day 15) mice were challenged intranasally with 20 μl of a 10 mg/ml solution of OVA. The challenge period occurred daily from days 15 to 19. Mice (seven/group) were randomly assigned to receive either compound (AM211, 10 mg/kg) or vehicle (methyl cellulose, 10 ml/kg) and treated by oral gavage 1 h before each OVA challenge. The number of sneezes were counted for 8 min immediately after the OVA challenge on days 15, 17, and 19 by an independent observer who was blinded to the treatment groups.
Data Analysis.
Results are expressed as means ± S.E.M. Where appropriate, data were statistically analyzed using Prism (GraphPad Software Inc.). A one-tailed t test, two-tailed t test, and one-factor or two-factor analysis of variance (ANOVA) were conducted where appropriate and as indicated. In the case of ANOVA, Dunnett's, Tukey's, or Newman-Keuls post-test were conducted as indicated. A P value less than 0.05 was considered to be statistically significant.
Results
Determination of AM211 Potency and Selectivity.
The potency and selectivity of AM211 for DP2 and other receptors in the prostaglandin family was determined using radioligand membrane binding with the relevant radioligand and cell membranes expressing the various receptors. AM211 displayed potent, concentration-dependent competition of [3H]PGD2 binding to cell membranes containing human, mouse, rat, or guinea pig DP2 (Fig. 2). The average IC50 values for the inhibition by AM211 of [3H]PGD2 binding to DP2 are presented in Table 1. [3H]PGD2 radioligand membrane binding experiments were also performed in the presence of species-specific serum albumin to determine whether the presence of an abundant blood protein would have a significant effect on the potency of AM211. In the absence of serum albumin, AM211 inhibited radiolabeled PGD2 binding to human, mouse, guinea pig, and rat DP2 with IC50 values of 4.9, 7.8, 4.9, and 10.4 nM, respectively. In the presence of 0.2% serum albumin, AM211 inhibited radiolabeled PGD2 binding to human, mouse, guinea pig, and rat DP2 with IC50 values of 12.2, 20.1, 22.9, and 34.2 nM, respectively. Therefore, there was only a small shift in IC50 in the presence of serum albumin, suggesting that AM211 would maintain high affinity in the context of the abundant blood protein, serum albumin. AM211 displayed high selectivity for DP2 versus other receptors in the prostanoid family because IC50 values for the inhibition of radioligand binding to human TP, IP, DP1, and FP were more than 100 μM (data not shown). AM211 was also tested for inhibition of the COX-1 and COX-2 enzymes and antagonist or agonist activity at the PPAR family of nuclear receptors and displayed no activity at these proteins at concentrations up to 100 μM (data not shown).

Effect of AM211 on the binding of [3H]PGD2 to membranes stably expressing DP2. Concentration-response curves of AM211 for the inhibition of [3H]PGD2 binding to membranes expressing human, mouse, guinea pig, or rat DP2 are shown. Membranes from HEK293 cells recombinantly expressing DP2 were incubated with varying concentrations of AM211 and 1 nM [3H]PGD2. Data represent the mean ± S.E.M. of three to six independent experiments.
IC50 values for AM211 in the in vitro assays
All IC50 values are the mean ± S.D. with n values indicated in parentheses.
Determination of Antagonist Activity and Lack of Agonist Activity Using a [35S]GTPγS Membrane Binding Assay.
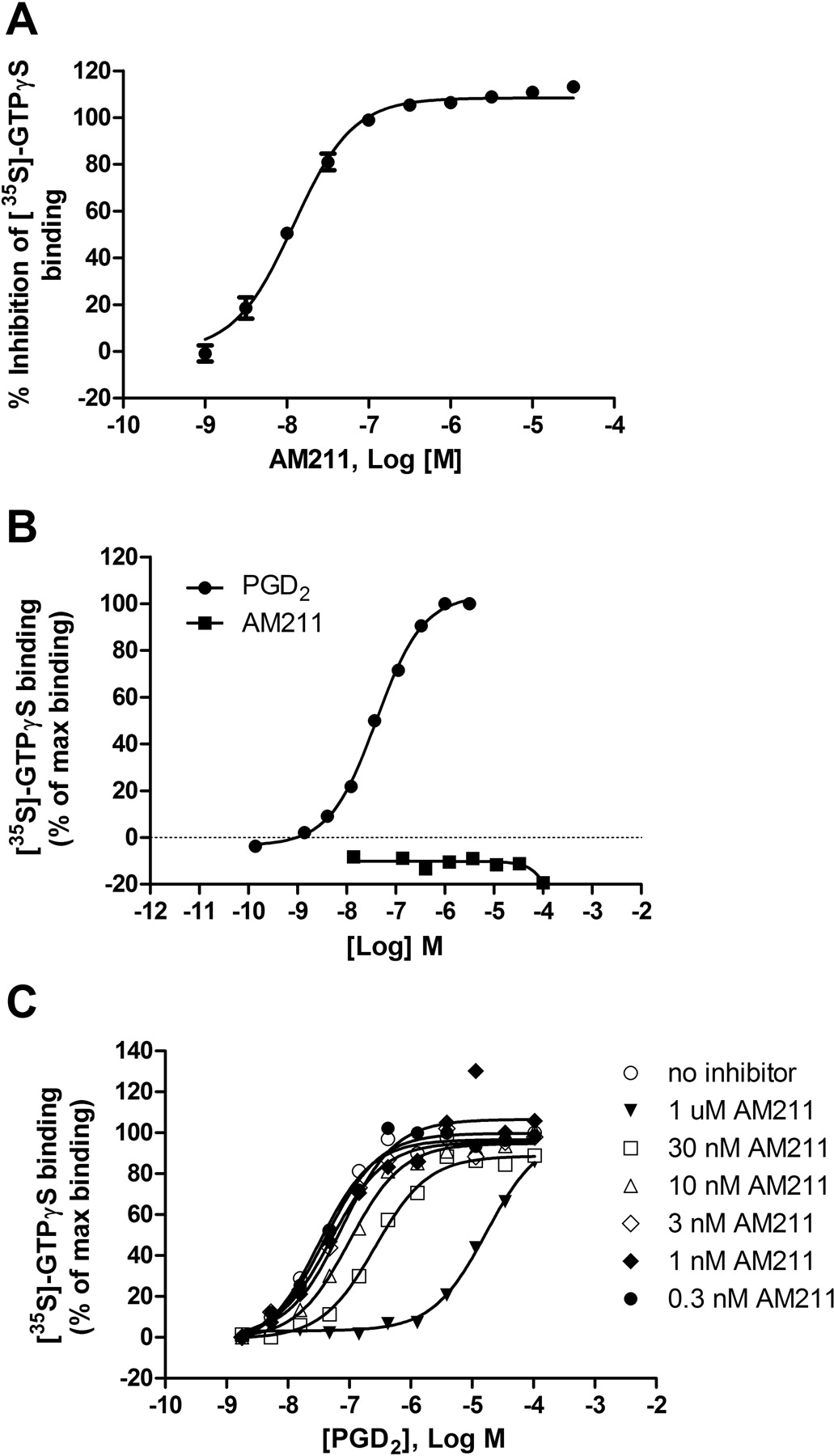
To determine that AM211 acted as a functional antagonist at DP2, it was evaluated for inhibition of ligand-stimulated GTP binding using a [35S]GTPγS membrane binding assay. AM211 showed concentration-dependent inhibition of PGD2-stimulated [35S]GTPγS binding to membranes containing human DP2 (Fig. 3A). The average IC50 value for antagonism of PGD2-induced [35S]GTPγS binding was 13.3 nM (Table 1). AM211 was also tested for agonist activity at DP2 by incubating increasing concentrations of AM211 with membranes expressing human DP2 in the absence of the endogenous ligand. AM211 showed no agonist activity at DP2 at concentrations up to 100 μM, whereas PGD2 showed concentration-dependent stimulation of [35S]GTPγS binding with an EC50 value of 44 nM (Fig. 3B). To characterize the nature of the antagonism more fully, a [35S]GTPγS binding assay was performed that evaluated concentration-response curves of PGD2 in the absence and presence of various concentrations of AM211. AM211 caused a rightward shift of the PGD2 concentration-response curves without altering the maximal PGD2 response (Fig. 3C). These data are consistent with competitive and reversible antagonism.

Effect of AM211 on the binding of [35S]GTPγS to membranes stably expressing human DP2. A, determination of antagonist activity. Concentration response curve of AM211 for the inhibition of [35S]GTPγS binding to membranes expressing human DP2 is shown. Membranes from CHO cells recombinantly expressing human DP2 were incubated with varying concentrations of AM211 and 80 nM PGD2. Data represent the mean ± S.E.M. of six independent experiments. B, evaluation of agonist activity at human DP2. Membranes from CHO cells recombinantly expressing human DP2 were incubated with various concentrations of AM211 (10 nM–100 μM) and [35S]GTPγS in the absence of PGD2. As a control, membranes from CHO cells recombinantly expressing human DP2 were incubated with various concentrations of PGD2 (0.1 nM–3 μM) and [35S]GTPγS. Plotted is a representative curve of the [35S]GTPγS binding as a percentage of the maximum PGD2-stimulated binding from one of two independent experiments. PGD2 stimulates binding of [35S]GTPγS to human DP2 with an EC50 of approximately 44 nM, C, concentration response curves of PGD2 in the presence of the indicated concentrations of AM211. Plotted is the [35S]GTPγS binding as a percentage of the maximum PGD2-stimulated binding. Shown are representative curves from one of two independent experiments.
Determination of Antagonist Potency of AM211 in Blocking PGD2-Induced Up-Regulation of Basophil CD203c Expression.
Incubation of basophils with PGD2 results in increased surface expression of the basophil activation marker CD203c (Monneret et al., 2005). Basophils can be identified from other leukocytes in the blood based on their expression of CD203c and low side scatter (SSC) value as indicated by the R1 gate in Fig. 4A. As shown in Fig. 4B, vehicle-treated basophils have low expression of CD203c (CD203clo) as determined by the low percentage (4.7%) of total basophils in the CD203chi (R2) gate (left). However, basophils stimulated with PGD2 up-regulate expression of CD203c as demonstrated by the increase in percentage of total basophils in the CD203chi (R2) gate after treatment with 100 nM PGD2 (38.7%) (Fig. 4B, center). We evaluated the ability of AM211 to inhibit this functional response because basophils represent a relevant human cell type that endogenously expresses DP2. As shown in Fig. 4B, right, 10 nM AM211 decreased the percentage of total basophils that were CD203chi to the same level as the vehicle control (R2 = 3.4%), demonstrating 100% inhibition of the PGD2-induced up-regulation of basophil CD203c expression (Fig. 4B). When the concentration of AM211 was varied, we observed potent, concentration-dependent antagonism of PGD2-induced CD203c expression on human basophils with an average IC50 value of 2.5 nM (Fig. 4C; Table 1).
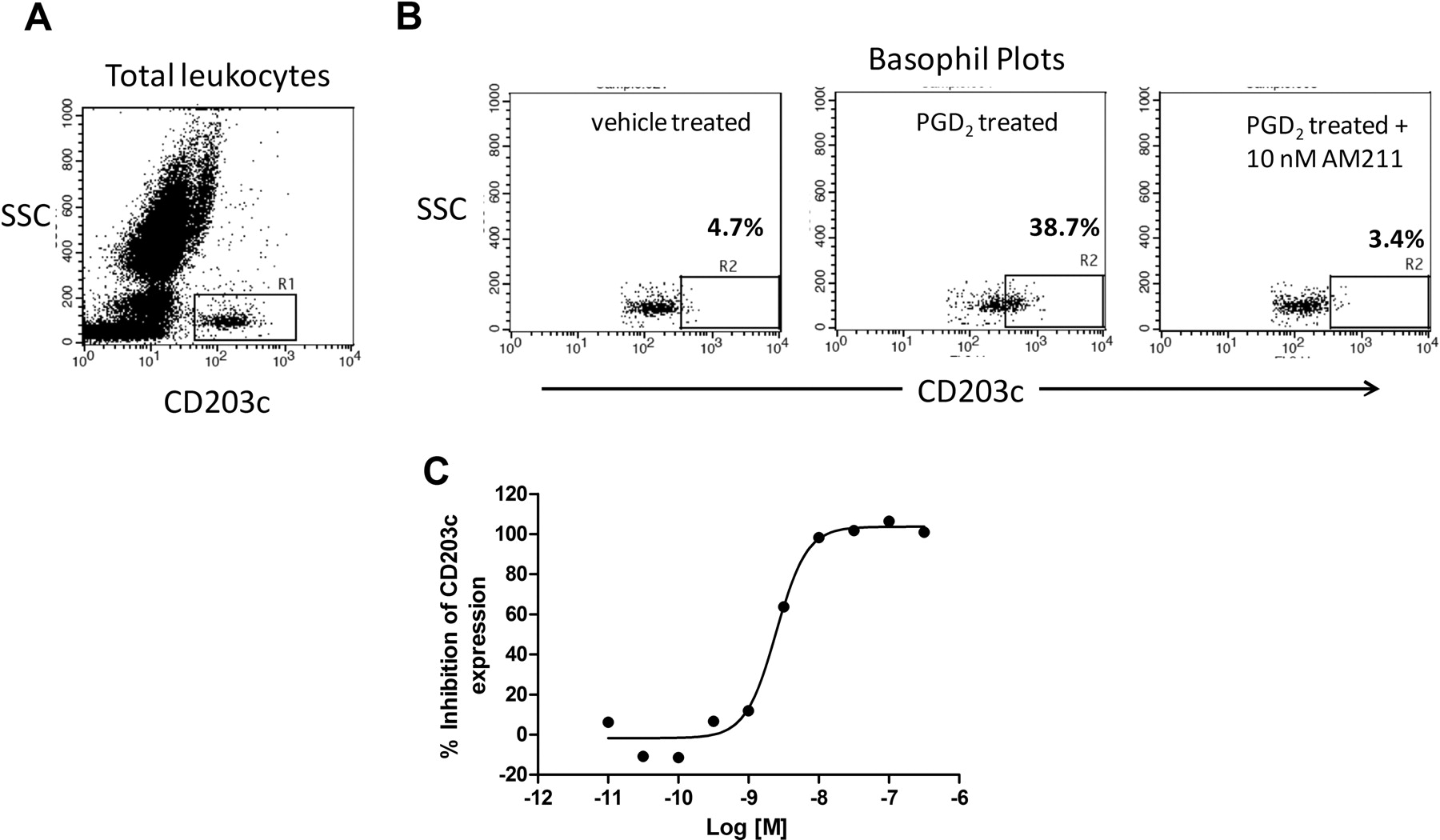
Effect of AM211 on human basophil CD203c expression. Isolated human leukocytes were stimulated with 100 nM PGD2 in the absence or presence of varying concentrations of AM211 and then stained with an antibody against CD203c conjugated to R-phycoerythrin. A, representative FACS plot of SSC versus CD203c expression on unstimulated total human leukocytes. R1 gate indicates the basophil population of cells (CD203+SSClo). B, SSC versus CD203c expression on the human basophil population after treatment with vehicle (left), 100 nM PGD2 (center), or 100 nM PGD2 in the presence of 10 nM AM211 (right). R2 gate indicates the population of basophils that express high levels of CD203c (CD203chi). C, concentration response curve for the inhibition by AM211 of PGD2-induced CD203c expression on human basophils. Shown is the mean percentage of inhibition for two independent experiments.
Determination of Antagonist Potency of AM211 in Blocking PGD2-Induced Eosinophil Shape Change in Human and Guinea Pig Whole Blood.
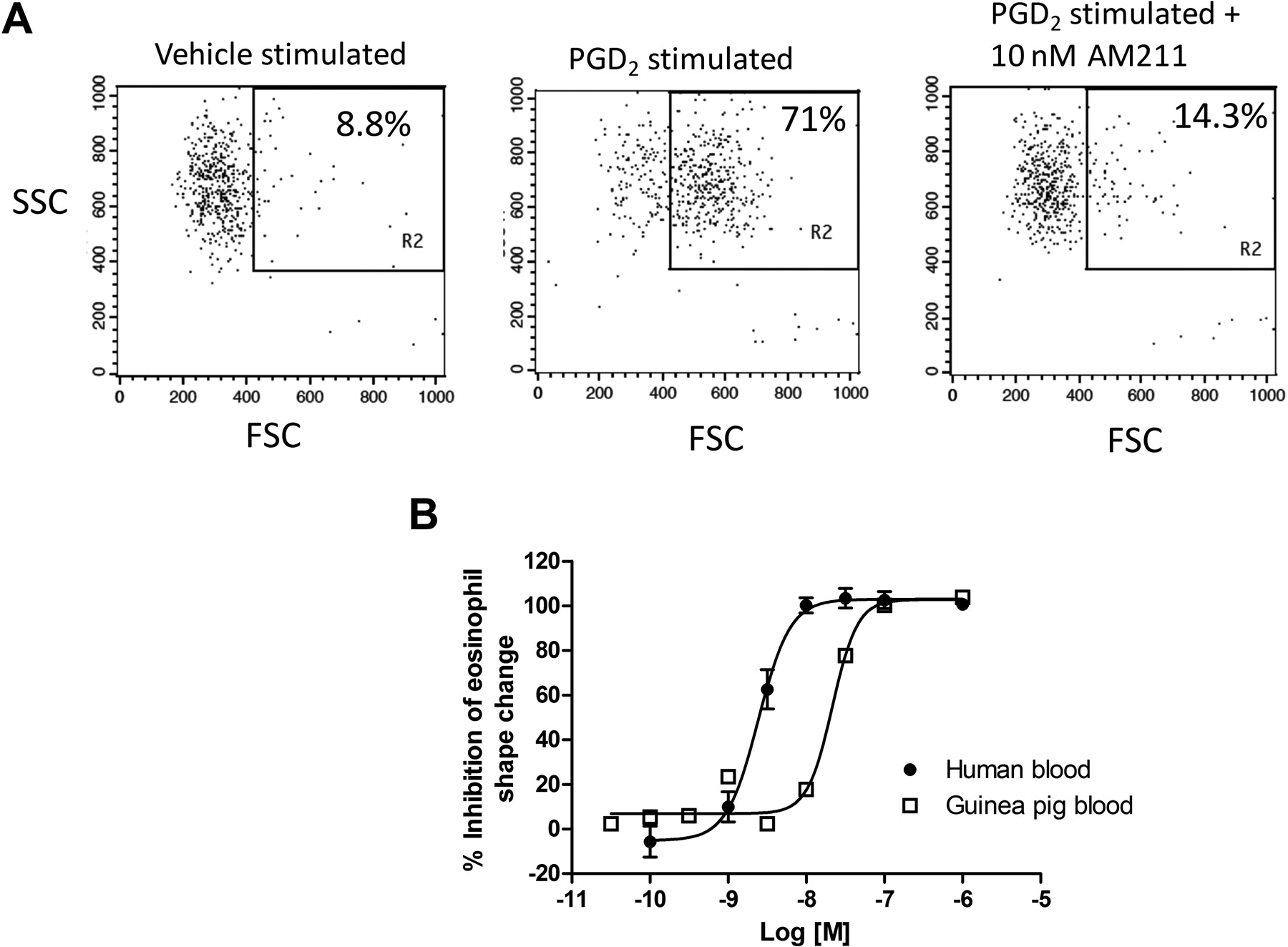
Incubation of blood with PGD2 activates DP2 expressed on eosinophils and initiates eosinophil degranulation and changes in eosinophil morphology (Stubbs et al., 2002; Schratl et al., 2007). The change in cell morphology, or shape, can be analyzed by flow cytometry and is evident by an increase in the forward light scatter (FSC) value (Stubbs et al., 2002; Schratl et al., 2007). We evaluated the ability of AM211 to inhibit PGD2-induced eosinophil shape change in human whole blood because these cells represent another important target cell in vivo and whole blood represents a physiologically relevant environment. As shown in Fig. 5A, left, vehicle stimulated blood results in only a small percentage of total eosinophils that fell into the FSChi shape change gate (8.8%). However, when human blood was stimulated with 50 nM PGD2, the percentage of total eosinophils that changed shape and appeared FSChi increased (71%) (Fig. 5A, middle). When blood was incubated with PGD2 in the presence of 10 nM AM211, the extent of eosinophil shape change was decreased to approximately basal levels (14.3%) (Fig. 5A, right). When the concentration of AM211 was varied, we observed concentration-dependent antagonism of PGD2-induced eosinophil shape change (Fig. 5B). The average IC50 value for antagonism of PGD2-induced eosinophil shape change was 2.7 nM (Table 1). The eosinophil shape change functional response to DP2 ligands has been difficult to generate in mice and rats, so evaluating AM211 potency in this assay was not possible in these rodent species. Therefore, we determined the potency of AM211 in inhibiting PGD2-induced eosinophil shape change in guinea pigs because it represents one of our efficacy species. The EC50 of PGD2 in inducing eosinophil shape change in guinea pig whole blood was shifted 5- to 10-fold relative to the EC50 in human whole blood (data not shown). Therefore, we treated the guinea pig blood with a 10-fold higher concentration of PGD2 relative to the human blood samples (50 versus 500 nM). When AM211 was evaluated for inhibition of eosinophil shape change in guinea pig blood, we observed a slight decrease in potency. The average IC50 value of AM211 for inhibition of eosinophil shape change in guinea pig blood was 21.5 nM (Fig. 5B; Table 1). These data demonstrate potent inhibition by AM211 of eosinophil shape change in a relevant cell type and a physiologically relevant environment and provide the most accurate estimate of the in vivo potency of AM211.

Effect of AM211 on eosinophil shape change in whole blood. Human or guinea pig whole blood was treated with 50 nM (human blood) or 500 nM (guinea pig blood) PGD2 in the absence or presence of varying concentrations of AM211, and eosinophil shape change was evaluated based on the changes in FSC value of the autofluorescent cells (eosinophils). A, representative FACS plots showing SSC versus FSC analysis of eosinophils after treatment of human blood with vehicle (left), 50 nM PGD2 (center), or 50 nM PGD2 in the presence of 10 nM AM211 (right). B, concentration response curves for the inhibition by AM211 of PGD2-induced eosinophil shape change in human and guinea pig whole blood. Shown are the mean ± S.E.M. from seven independent experiments using human blood and the mean from two independent experiments using guinea pig blood.
Pharmacokinetic Profiles of AM211 in Animals.
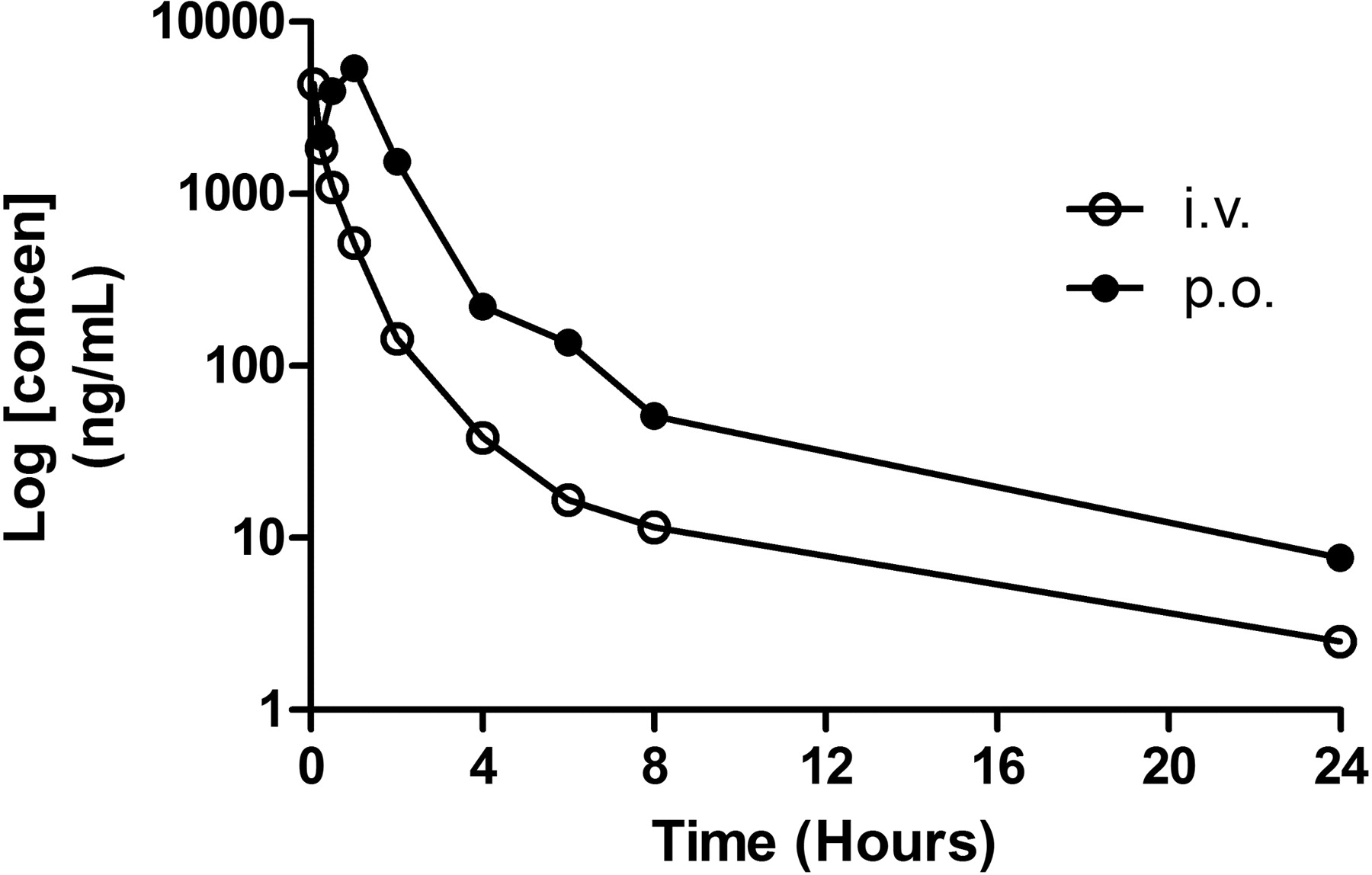
The pharmacokinetics of AM211 was assessed in Sprague-Dawley rats, beagle dogs, and guinea pigs over a 24-h period after oral and/or intravenous dosing. After intravenous administration of AM211, plasma clearance values (ClPl) in rats and dogs were 14 and 2.4 ml/min/kg, respectively, which suggests moderate to low clearance with respect to liver blood flow (Fig. 6; Table 2). Half-life values (t½) for rat and dog after intravenous dosing were 2.4 and 3.1 h, respectively (Fig. 6; Table 2). After oral administration in rat and dog, AM211 was rapidly absorbed with a time to maximum concentration (Tmax) of 1 and 0.3 h and a maximum concentration (Cmax) of 5.4 μg/ml (∼11 μM) and 13.5 μg/ml (∼27 μM), respectively (Fig. 6; Table 2). AM211 showed excellent bioavailability (%F = 48–78%) after oral dosing in both rat and dog. The guinea pig PK was not as good as rat and dog PK; however, based on the PK profile and the potency in guinea pig, it was determined that twice-daily dosing of AM211 at 30 mg/kg in guinea pig should provide adequate exposure for sustained coverage of the receptor in vivo.

Pharmacokinetic profile of AM211. Plasma concentrations of AM211 were analyzed at various times after a single intravenous (i.v.) dose of 2 mg/kg or an oral (p.o.) dose of 10 mg/kg in Sprague-Dawley rats. Plotted is the mean plasma concentration from two different rats.
Pharmacokinetic parameters for AM211 in various species after either intravenous or oral administration
Effects of AM211 on Guinea Pig Intravenous DK-PGD2-Induced Blood Leukocytosis.
In vivo, activation of DP2 by the selective agonist, DK-PGD2, leads to a dose- and time-dependent increase in the number of leukocytes in the peripheral blood and this leukocytosis is inhibited by DP2 receptor antagonists (Shichijo et al., 2003). Therefore, this assay represents a simple and robust way to evaluate the pharmacodynamic response of DP2 antagonists in vivo and provides information on dose selection for the longer and more difficult efficacy studies. AM211 was evaluated in this guinea pig pharmacodynamic model at both an early time point of 2-h post-AM211 dose as well as a late time point of 18-h post-AM211 dose to evaluate inhibitory activity at time points near peak (Cmax) and trough (Cmin) blood concentrations. In the 2-h postdose study, intravenous DK-PGD2 increased peripheral blood leukocytes by 2-fold from a basal level of ∼12 ± 1 cells/ml (×108) in PBS-challenged animals to ∼25 ± 2 cells/ml (×108) in DK-PGD2-challenged, vehicle-pretreated animals (P < 0.01; Fig. 7A). Oral dosing of AM211 2 h before the intravenous DK-PGD2 administration caused a dose-dependent decrease in the number of peripheral blood leukocytes. After a 30 mg/kg dose of AM211, the number of peripheral blood leukocytes decreased to ∼14 ± 1 cells/ml (×108), which represents ∼80% inhibition of the DK-PGD2-induced leukocytosis relative to the PBS control (∼12 ± 1 cells/ml × 108) (Fig. 7, A and C). Significant inhibition of 59, 62, and 80% was achieved in the 1, 10, and 30 mg/kg dose groups (P < 0.05). Based on these results, the ED50 for AM211 is calculated to be ∼0.85 mg/kg (Fig. 7C). The concentration of AM211 in plasma recovered 2.5 h after an oral dose was 22 ± 3, 61 ± 29, 222 ± 98, and 727 ± 313 nM in the 0.1, 1, 10, and 30 mg/kg dose groups, respectively. Using these plasma concentrations we estimate that the plasma EC50 for AM211 in the guinea pig leukocytosis assay is ∼55 nM.

Effect of AM211 on DK-PGD2-induced leukocytosis in guinea pigs. A and B, total cells/ml (×108) recovered in blood 30 min after injection of DK-PGD2. Animals were orally dosed with vehicle (Veh) or 0.1, 1, 10, or 30 mg/kg AM211 either 2 h (A) or 18 h (B) before the injection of DK-PGD2. PBS animals did not receive DK-PGD2. C, graph of the percentage of inhibition of leukocytosis versus dose after either a 2- or 18-h pretreatment with AM211. All values are the means ± S.E.M. from four to eight animals (for the 2-h group n = 4, 5, 5, 6, 4, and 4, respectively for PBS, Veh, 0.1, 1, 10, and 30 mg/kg; for the 18-h group n = 4, 7, 8, 8, 8, and 4, respectively for PBS, Veh, 1, 10, 10, and 100). ##, P < 0.01; ###, P < 0.001 versus PBS control group. *, P < 0.05; **, P < 0.01 versus vehicle response, Neuman-Keuls post hoc after one-factor ANOVA.
In the 18-h guinea pig leukocytosis study, intravenous DK-PGD2 increased peripheral blood leukocytes from 12 ± 1 cells/ml (×108) in PBS-challenged animals to 20 ± 2 cells/ml (×108) in DK-PGD2-challenged, vehicle-pretreated animals (P < 0.001; Fig. 7B). The leukocytosis response was dose-dependently reduced by oral AM211 administered 18 h before DK-PGD2, resulting in 20 ± 3, 18 ± 3, 16 ± 1, and 14 ± 13 cells/ml (×108) after 1, 10, 30, and 100 mg/kg, respectively (Fig. 7B). This correlates to 0, 29, 50, and 77% inhibition after AM211 doses of 1, 10, 30, and 100 mg/kg, respectively relative to the PBS control (12 ± 1 cells/ml × 108) (Fig. 7C). Significant inhibition was achieved in the 30 and 100 mg/kg dose groups (P < 0.05), and the ED50 was estimated at ∼30 mg/kg. The concentrations of AM211 in plasma recovered 18.5 h after oral AM211 were below the lower limit of quantitation of 20 ng/ml (∼40 nM) for all animals in the 0.1, 1, and 10 mg/kg dose groups. A single animal had quantifiable amounts in the 30 mg/kg dose group of 84 nM, whereas the remaining animals were below the lower limit of quantitation. All animals, with the exception of one, were quantifiable in the 100 mg/kg dose group and gave an average of 201 ± 164 nM. Because most values were below the lower limit of quantitation, we were unable to accurately calculate an EC50 associated with the ED50 of 30 mg/kg, but based on the available data, the EC50 can best be described as <40 nM.
Effects of AM211 on Ovalbumin-Induced Pulmonary Inflammation in Guinea Pigs.
To evaluate the ability of AM211 to inhibit pulmonary inflammation, OVA-primed guinea pigs were pretreated with either vehicle or AM211, then challenged with aerosolized OVA (Fig. 8A). To enhance the DP2 component of the antigen challenge, guinea pigs received an intraperitoneal injection of DK-PGD2 (200 μg) 2 h after the aerosolized OVA. Based on the pharmacodynamics of AM211 in the guinea pig leukocytosis study and the relatively poor pharmacokinetics of AM211 in guinea pig, a second oral dose of AM211 (or vehicle) was given 6 h after antigen challenge (Fig. 8A). Pulmonary inflammation was assessed by measuring cell counts in BALF 24 h after the OVA challenge. Total cells increased from 50 ± 10 per image (×4) in nonchallenged (control) animals to 184 ± 31 per image (×4) in vehicle-dosed, OVA-challenged animals. The increase in total cells resulted primarily from increases in eosinophil and neutrophil numbers with minimal increase in alveolar macrophages and lymphocytes (Fig. 8B). Oral treatment with AM211 produced a dose-dependent decrease in cellular influx, reaching statistical significance at a dose of 30 mg/kg. Total cells decreased to 85 ± 15 per image (×4) in animals treated with 30 mg/kg AM211, and this was associated with a significant decrease in eosinophils and neutrophils, demonstrating an inhibition of antigen-induced pulmonary inflammation (Fig. 8B).

Effect of AM211 on antigen-induced pulmonary inflammation in guinea pigs. Animals were sensitized to OVA on day 1 by intraperitoneal and subcutaneous injections. On day 21, animals were challenged with aerosolized OVA and DK-PGD2. A, day-21 time line indicating the dosing times for AM211 (3 or 30 mg/kg), OVA (0.03%), and DK-PGD2 (200 μg). B, graph of the number of cells/image (×4) in BALF collected 24 h after OVA challenge after dosing vehicle (n = 10), AM211 at 3 mg/kg [AM211(3); n = 8], or AM211 at 30 mg/kg [AM211(30); n = 8]. Control animals were dosed with vehicle followed by a PBS challenge (n = 4). Plotted are the means ± S.E.M. *, P < 0.05 versus vehicle dose group, two-factor ANOVA. #, P < 0.05 versus vehicle dose group, two-tailed t test.
Evaluation of AM211 in a Mouse Model of Allergic Rhinitis.
A mouse model of allergic rhinitis was used to determine the effects of AM211 on nasal symptoms. OVA-primed mice display a significant increase in sneezing behavior when challenged intranasally with OVA. Daily intranasal challenge with OVA produces a greater response (increased number of sneezes) over several consecutive days, which is probably a result of increased accumulation of inflammatory cells in the nasal mucosa (Nakaya et al., 2006). Therefore, OVA challenge was delivered daily across a 5-day challenge phase (days 15–19), and the numbers of sneezes in an 8-min period immediately after the challenge were recorded on days 15, 17, and 19 (Fig. 9A). Daily oral administration of 10 mg/kg AM211 1 h before each intranasal OVA challenge significantly reduced sneezing behavior on days 17 and 19 (Fig. 9B). When the number of sneezes during the 8-min counting period on days 15, 17, and 19 were averaged AM211 dosed at 10 mg/kg showed a statistically significant decrease in OVA-induced sneezing (Fig. 9C).

Effect of AM211 on nasal symptoms in a mouse model of allergic rhinitis. A, timeline indicating dosing times for the allergic rhinitis model. Mice were sensitized by intraperitoneal injections of OVA/alum on days 1 and 8 and then challenged with intranasal OVA on days 15 to 19. AM211 was dosed orally at 10 mg/kg 1 h before each OVA challenge on days 15 to 19. Sneezes in an 8-min period immediately after intranasal OVA were counted on days 15, 17, and 19 (as indicated by the thick hash marks). B, graph of the number of sneezes per 8-min period on days 15, 17, and 19 in vehicle- and AM211-treated mice. C, average number of sneezes across the three observation periods. All values are means ± S.E.M. of seven mice per group. #, P < 0.05 versus vehicle control group, one-tailed t test.
Discussion
In this study we describe the novel DP2 antagonist AM211. We show that AM211 displays high affinity for human, mouse, rat, and guinea pig DP2, is highly selective for DP2 over other prostanoid receptors, and acts as a functional antagonist. The nature of the antagonism was evaluated by performing concentration-response curves of PGD2 in the presence of various concentrations of AM211. AM211 resulted in a rightward shift in the PGD2 concentration-response curve without lowering the maximum binding. This type of “surmountable antagonism” was also observed in the human whole-blood eosinophil shape change assay (data not shown) and suggests that AM211 functions as a competitive antagonist at DP2.
The potency of AM211 for DP2 was evaluated in the absence and presence of serum albumin using a radioligand membrane binding assay. This represents a rapid way to screen for compounds that have a low protein shift and are, therefore, more likely to remain potent in whole blood. The very minor increase in IC50 in the presence of serum albumin suggested that AM211 would maintain high potency in blood. Indeed, the average IC50 in the ESC human blood assay was 2.7 nM. The human blood ESC assay is useful not only for rank-ordering compounds in vitro, but also as a pharmacodynamic measure of receptor antagonism in whole blood after oral dosing. As a result, it has become a useful assay for evaluating the PK/PD relationship of DP2 antagonists in phase I clinical trials.
DP2 is expressed on eosinophils, basophils, and Th2 cells and mediates proinflammatory responses of PGD2 that are important in asthma including degranulation of eosinophils and basophils and release of the Th2 cytokines IL-4, IL-5, and IL-13 from Th2 cells (Nagata et al., 1999a,b; Gervais et al., 2001; Sawyer et al., 2002; Xue et al., 2005). Our data demonstrate that AM211 has similar potencies in inhibiting functional responses in human eosinophils and basophils. These results demonstrate that AM211 antagonizes DP2 with equivalent potency on two distinct populations of human cells, both of which play important roles in allergic inflammatory diseases.
The potency observed in vitro for AM211 in the guinea pig ESC blood assay (IC50 = 21.5 nM) was comparable with the potency observed in vivo in the guinea pig leukocytosis model (EC50 = 55 nM). Originally described in brown Norway rats, DP2 activation by DK-PGD2 has been shown to increase peripheral blood leukocytes (Shichijo et al., 2003). This effect was reversed in animals pretreated with the dual DP2/thromboxane A2 receptor antagonist ramatroban. In the present study we used this pharmacodynamic response in guinea pigs to determine the in vivo ED50 and associated in vivo plasma EC50 for AM211. We first established that intravenous DK-PGD2 caused a significant increase in peripheral blood leukocytes in OVA-primed guinea pigs. We used OVA-primed guinea pigs based on earlier work showing that these guinea pigs have a greater response to DK-PGD2 than do naive animals. The reason for this effect is not known, but probably relates to an increased number of DP2-expressing Th2 cells after OVA priming. Oral administration of AM211 attenuated the DK-PGD2-induced increase in blood leukocytes in a dose-dependent manner, resulting in an ED50 of ∼0.85 mg/kg and an EC50 of ∼55 nM when given 2 h before the DK-PGD2. We observed a rightward shift in the ED50 when AM211 was dosed 18 h before the DK-PGD2 (ED50 of ∼30 mg/kg). An accurate EC50 could not be obtained from the 18-h study because of the bioanalytical limitation in quantitating the concentration of AM211 in the guinea pig plasma. However, based on the available data, the 18-h EC50 is best described as <40 nM. In radioligand binding experiments using membranes from cells stably expressing guinea pig DP2, AM211 had an IC50 of 23 nM in the presence of 0.2% guinea pig serum albumin. The IC50 of AM211 in the guinea pig whole-blood ESC assay was 22 nM. Together, these results demonstrate a good correlation between the in vitro and in vivo potencies of AM211 and suggest that potency in an in vitro binding assay or ESC assay may be a good predictor of the plasma concentration needed to achieve functional activity in an in vivo setting.
DP2 is thought to play a role in human allergic rhinitis. Indirect evidence is derived from the findings that the ligand PGD2 is present in the nasal lavage fluid of patients with allergic rhinitis in response to allergen challenge (Naclerio et al., 1983; Miadonna et al., 1999) and DP2 expression is increased in allergic human nasal mucosa relative to nonallergic mucosa (Shirasaki et al., 2009). In Japan, ramatroban (Bayans), a dual DP2/TP antagonist, is clinically used for the treatment of allergic rhinitis. We show here that AM211 is effective in a mouse model of allergic rhinitis as demonstrated by the attenuation of sneezing elicited by an intranasal antigen challenge. Others have shown that DP2-deficient mice are protected against cedar pollen-induced allergic rhinitis (Nomiya et al., 2008), and treatment with ramatroban, although not selective for DP2, attenuated sneezes and nasal rubs (Nomiya et al., 2008). Furthermore, ramatroban attenuated nasal airway resistance and eosinophil infiltration in guinea pigs (Narita et al., 1996). In addition, we have reported efficacy with structurally distinct DP2-selective antagonists in a murine model of allergic rhinitis (Stearns et al., 2009; Stebbins et al., 2010b). These data suggest that selective DP2 antagonists may demonstrate efficacy in allergic rhinitis patients.
DP2 has been suggested to play a significant role in asthma. Single-nucleotide polymorphisms in DP2 are linked to asthma severity and associated with increased risk of atopy, including allergic rhinitis and wheeze (Hsu et al., 2002; Huang et al., 2004; Seibert et al., 2008). In our guinea pig model of allergic asthma, AM211 demonstrated significant inhibition of ovalbumin-induced inflammatory cell influx into bronchial alveolar lavage fluids. This effect was primarily a reflection of the inhibition of eosinophil influx. These results are consistent with previous reports in similar animal models (Uller et al., 2007; Pettipher, 2008). However, in addition to inhibiting eosinophil influx, we observed a significant reduction in neutrophil influx. In rodents, DP2 is expressed on neutrophils under basal conditions and injection of the selective DP2 agonist DK-PGD2 results in mobilization of neutrophils from the bone marrow into the circulating blood (Shichijo et al., 2003; Takeshita et al., 2004). Selective antagonists of DP2 have been shown to inhibit neutrophil recruitment to the skin and lung in rodent models of contact hypersensitivity and COPD (Boehme et al., 2009; Stebbins et al., 2010a). Although circulating human neutrophils do not express DP2, expression has been found on sputum neutrophils from patients with cystic fibrosis (Sawyer et al., 2002; Tirouvanziam et al., 2008). Therefore, it is reasonable to speculate that cell surface expression of DP2 on neutrophils may increase locally in disease settings and that blocking DP2 in humans will affect conditions associated with neutrophilia, such as COPD, severe asthma, and cystic fibrosis.
DP2 represents a promising target for the treatment of asthma and other allergic inflammatory diseases. The receptor is expressed on several cell types known to be involved in allergic inflammatory responses, and PGD2 release from activated mast cells during the early response to allergen challenge may provide an essential link between the early- and late-phase response through the recruitment and activation of Th2 cells and eosinophils. PGD2 has the remarkable property of being able to stimulate the release of IL-4, IL-5, and IL-13 from human Th2 cells in the absence of costimulation and this can be inhibited by DP2 antagonists (Pettipher, 2008). Once activated, the Th2 cells can enhance the production of IgE from plasma cells, which further increases the allergic inflammatory response. Therefore, DP2 antagonists represent highly promising therapeutics for allergic inflammatory disease through their inhibition of Th2 cell recruitment, activation, and survival.
Although several DP2 antagonists are in clinical development, detailed preclinical pharmacology of most of these compounds is lacking (Pettipher, 2008). However, the in vitro and in vivo pharmacology of one antagonist, MK-7246 ({(7R)-7-[[(4-fluorophenyl)sulfonyl](methyl)amino]-6,7,8,9-tetrahydropyrido[1,2-α]indol-10-yl}acetic acid), has been described (Gervais et al., 2011). MK-7246 displayed similar potencies to AM211 in the inhibition of [3H]PGD2 binding to DP2 from various species and in the inhibition of eosinophil shape change in human whole blood. Although the functional endpoints in the basophil assays were different (CD203c expression versus CD11b expression), the potencies in inhibiting the basophil responses were similar between MK-7246 and AM211. Gervais et al. established a correlation between PK and PD in cynomologus monkey and sheep, whereas we have demonstrated a correlation between PK and PD in guinea pig. It is noteworthy that MK-7246 displayed insurmountable antagonism of the DK-PGD2-induced decrease in forskolin-stimulated cAMP production, whereas AM211 functioned as a surmountable antagonist in two distinct functional assays. It is unclear whether this difference in the nature of the antagonism will translate to functional differences in vivo. Both compounds had favorable PK profiles in rat and dog with excellent oral bioavailability in both species and showed efficacy in preclinical animal models. Further profiling of these compounds in patients will require characterization of the safety and PK in phase I clinical studies.
In summary, AM211 is a potent and selective DP2 antagonist with favorable pharmacokinetic and pharmacodynamic properties in preclinical species and is effective in animal models of allergic inflammation. DP2 has been suggested to play a significant role in asthma, and AM211 represents a promising tool for the characterization of the role of DP2 in this and other allergic inflammatory diseases.
Authorship Contributions
Participated in research design: Bain, Lorrain, Stebbins, King, and Evans.
Conducted experiments: Bain, Lorrain, Stebbins, Broadhead, Santini, Prodanovich, Darlington, Lee, and Baccei.
Contributed new reagents or analytic tools: Stearns and Truong.
Performed data analysis: Bain, Lorrain, Stebbins, Broadhead, Santini, Prodanovich, Darlington, Lee, and Baccei.
Wrote or contributed to the writing of the manuscript: Bain, Lorrain, Hutchinson, Prasit, and Evans.
Footnotes
This work was supported by Amira Pharmaceuticals.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.111.180430.
-
ABBREVIATIONS:
- PGD2
- prostaglandin D2
- DP2
- PGD2 receptor type 2
- DP1
- PGD2 receptor type 1
- DK-PGD2
- 13,14-dihydro-15-keto-PGD2
- AM211
- [2′-(3-benzyl-1-ethyl-ureidomethyl)-6-methoxy-4′-trifluoromethyl-biphenyl-3-yl]-acetic acid
- Th2
- T helper 2
- COPD
- chronic obstructive pulmonary disease
- BALF
- bronchoalveolar lavage fluid
- TP
- thromboxane A2 receptor
- IP
- prostacyclin receptor
- FP
- prostaglandin F2α receptor
- PBS
- phosphate-buffered saline
- ESC
- eosinophil shape change
- PK
- pharmacokinetic
- PD
- pharmacodynamic
- GTPγS
- guanosine 5′-O-[γ-thio]triphosphate
- COX
- cyclooxygenase
- OVA
- ovalbumin
- DMSO
- dimethylsulfoxide
- ANOVA
- analysis of variance
- CHO
- Chinese hamster ovary
- HEK
- human embryonic kidney
- AUC
- area under the curve
- IL
- interleukin
- Veh
- vehicle
- TB
- total binding
- PPAR
- peroxisome proliferator-activated receptor
- FSC
- forward light scatter
- SSC
- side scatter
- FACS
- fluorescence-activated cell sorting
- BAY u3405
- (+)-(3R)-3-(4-fluorobenzenesulfonamido)-1,2,3,4-tetra-hydrocarbazole-9-propionic acid
- BW A868C
- 3-[(2-cyclohexyl-2-hydroxyethyl)amino]-2,5-dioxo-1-(phenylmethyl)-4-imidazolidineheptanoic acid
- SQ 29,548
- [1S-[1α,2α(Z),3α,4α]]-7-[3-[[2-[(phenylamino)carbonyl]hydrazino]methyl]-7-oxabicyclo[2.2.1]hept-2-yl]-5-heptenoic acid
- GW7647
- 2-methyl-2-[[4-[2-[[(cyclohexylamino)carbonyl](4-cyclohexylbutyl)amino]ethyl]phenyl]thio]-2-methylpropanoic acid
- GW0742
- [4-[[[2-[3-fluoro-4-(trifluoromethyl)phenyl]-4-methyl-5-thiazolyl]methyl]thio]-2-methylphenoxy] acetic acid
- MK-7246
- {(7R)-7-[[(4-fluorophenyl)sulfonyl](methyl)amino]-6,7,8,9-tetrahydropyrido[1,2-α]indol-10-yl}acetic acid.
- Received February 10, 2011.
- Accepted April 8, 2011.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}