


Abstract
Prostaglandin D2 (PGD2) is one of a family of biologically active lipids derived from arachidonic acid via the action of COX-1 and COX-2. PGD2 is released from mast cells and binds primarily to two G protein-coupled receptors, namely DP1 and DP2, the latter also known as chemoattractant receptor-homologous molecule expressed on Th2 cells. DP2 is predominantly expressed on eosinophils, Th2 cells, and basophils, but it is also expressed to a lesser extent on monocytes, mast cells, and epithelial cells. Interaction of PGD2 and its active metabolites with DP2 results in cellular chemotaxis, degranulation, up-regulation of adhesion molecules, and cytokine production. Chronic obstructive pulmonary disease (COPD) is a chronic progressive inflammatory disease characterized by elevated lung neutrophils, macrophages, and CD8+ T lymphocytes and mucus hypersecretion. Cigarette smoke contributes to the etiology of COPD and was used here as a provoking agent in a murine model of COPD. In an acute model, {2′-[(cyclopropanecarbonyl-ethyl-amino)-methyl]-6-methoxy-4′-trifluoro-methyl-biphenyl-3-yl}-acetic acid, sodium salt (AM156) and (5-{2-[(benzoyloxycarbonyl-ethyl-amino)-methyl]-4-trifluoromethyl-phenyl}-pyridin-3-yl)-acetic acid, sodium salt) (AM206), potent DP2 receptor antagonists, dose-dependently inhibited influx of neutrophils and lymphocytes to smoke-exposed airways. In a subchronic model, AM156 and AM206 inhibited neutrophil and lymphocyte trafficking to the airways. Furthermore, AM156 and AM206 treatment inhibited mucus cell metaplasia and prevented the thickening of the airway epithelial layer induced by cigarette smoke. These data suggest that DP2 receptor antagonism may represent a novel therapy for COPD or other conditions characterized by neutrophil influx, mucus hypersecretion, and airway remodeling.
Chronic obstructive pulmonary disease (COPD) is a complex disease in which airflow obstruction is not fully reversible. It is a chronic, progressive inflammatory condition of the small airways and lung parenchyma that is characterized by elevated pulmonary macrophages, neutrophils, and CD8+ T cells. Proinflammatory mediators, including IL-8, MCP-1, and IL-1β, are elevated in bronchoalveolar lavage fluid (BALF) and sputum samples obtained from patients with COPD (Keatings et al., 1996; Yamamoto et al., 1997; Hill et al., 1999; Soler et al., 1999). Chronic inflammation contributes to the decline in pulmonary function associated with chronic bronchitis, mucus hypersecretion, and emphysema via the release of proinflammatory mediators, reactive oxygen species, and tissue degradation enzymes (Corrigan and Kay, 1991; Saetta, 1999; Shapiro, 1999, 2002; Cosio and Majo, 2002; Stockley, 2002; Tetley, 2002). At present, there are no therapies that treat the underlying mechanisms of COPD. Corticosteroids, which are effective in other inflammatory conditions such as asthma are effective only against COPD exacerbations. Thus, a large unmet medical need exists for novel therapies aimed at ameliorating the progression of COPD.
Prostaglandin D2 (PGD2) is one of a family of biologically active lipids derived from arachidonic acid via the action of COX-1 and COX-2. Mast cells are the primary source of PGD2, but antigen-presenting cells and Th2 cells also contribute to the production of PGD2 (Urade et al., 1989, 1990; Tanaka et al., 2000). PGD2 binds primarily to two distinct G protein-coupled receptors, namely DP1 and DP2; DP2 is also known as chemoattractant receptor-homologous molecule expressed on Th2 cells. PGD2 is rapidly degraded after release and gives rise to several metabolites that are selective and potent agonists of the DP2 receptor (Hirai et al., 2001; Heinemann et al., 2003; Almishri et al., 2005). This suggests that PGD2 release may cause sustained activation of DP2.
At least one preclinical study has demonstrated an increase in PGD2 in the BALF after exposure to cigarette smoke (Hong and Lee, 1996). Furthermore, the concentrations of PGD2 in BALF recovered from COPD patients inversely correlated with forced expiratory volume in 1 s and peak expiratory flow, suggesting that PGD2 plays a role in the worsening of the disease (Csanky et al., 2009). However, the role for PGD2 in COPD remains to be further defined.
DP2 is predominantly expressed on eosinophils, Th2 cells, and basophils and is thought to play a key role in Th2-driven conditions, including allergic rhinitis and asthma (Pettipher, 2008). Relatively little attention has been given to DP2 in non-Th2-driven pathophysiology, including, for example, neutrophilia associated with COPD. This may be because, under normal conditions, human neutrophils do not express DP2 (Sawyer et al., 2002). However, DP2 expression has been found to be up-regulated on pulmonary neutrophils in patients with cystic fibrosis (Tirouvanziam et al., 2008), suggesting that, in some pathological conditions, DP2 is expressed on neutrophils as well. In rodents, however, DP2 is expressed on neutrophils under basal conditions and does not require activation of expression (Takeshita et al., 2004). Experiments in the Brown Norway rat reveal a role for DP2 in neutrophil migration in vivo. Injection of the selective DP2 agonist 13,14-dihydro-15-keto prostaglandin D2 (DK-PGD2) resulted in mobilization of leukocytes, including neutrophils, from the bone marrow into the circulating blood (Shichijo et al., 2003). Furthermore, in a murine model of contact hypersensitivity, a selective antagonist of DP2 inhibited recruitment of neutrophils to the skin (Boehme, 2009). Thus, rodent models of neutrophil trafficking may be useful in examining the PGD2-DP2 axis.
Exposure of animals to cigarette smoke induces pulmonary inflammation that is useful for the pharmacological evaluation of novel therapeutics for COPD. In acute models (<4 days) inflammation is predominantly neutrophilic (Leclerc et al., 2006; Morris et al., 2008; Doukas et al., 2009), whereas in subchronic exposure models (<30 days), the inflammatory phenotype is more complex and includes mucus hypersecretion and epithelial hyperplasia (Medicherla et al., 2008).
The goal of the studies described herein was to examine the efficacy of AM156 and AM206, novel DP2 receptor antagonists, in murine models of cigarette smoke exposure. First, we profiled our antagonists by use of in vitro radioligand membrane binding and whole-blood eosinophil shape-change assays. Next, we used a guinea pig pharmacodynamic assay to establish that these molecules could block in vivo effects of the selective DP2 agonist DK-PGD2. Last, we profiled AM156 and AM206 in acute and subchronic models of cigarette smoke exposure to determine the effect of DP2 antagonism on cellular influx, COPD-relevant inflammatory mediators, mucus hypersecretion, and airway remodeling.
Materials and Methods
Drugs
AM156 (Fig. 1A) and AM206 (Fig. 1B) are novel DP2 receptor antagonists developed by Amira Pharmaceuticals 099901 (Hutchinson et al., 2009). Representative examples of reference DP2 antagonists {4-chloro-6-dimethylamino-2-[4-(4-trifluoromethyl-benzoylamino)-benzyl]-pyrimidin-5-yl}-acetic acid, mono trifluoroacetate salt (Fig. 1C) and (5-fluoro-2-methyl-3-quinolin-2-ylmethyl-indol-1-yl)-acetic acid, sodium salt (Fig. 1D) outlined in patents from Actimis Pharmaceuticals, Inc. (San Diego, CA) (Ly et al., 2007) and Oxagen (Abingdon, Oxfordshire, UK) (Lovell, 2007), respectively, were synthesized in-house. DK-PGD2 was purchased from Cayman Chemical (Ann Arbor, MI). Rolipram (4-[3-(cyclopentyloxy)-4-methoxyphenyl]-2-pyrrolidinone) and dexamethasone ((11β,16α)-9-fluoro-11,17,21-trihydroxy-16-methylpregna-1,4-diene-3,20-dione) were purchased from Sigma-Aldrich (St. Louis, MO).

Chemical structures of AM156 (A), AM206 (B), and representative reference DP2 antagonists from Actimis (Ly et al., 2007) (C) and Oxagen (Lovell, 2007) (D).
In Vitro Potency Screening and Counterscreening
Membrane-Binding Assays for DP2, DP1, IP, and TP.
Membranes were isolated from human embryo kidney-293 cells stably transfected with human, mouse, or guinea pig DP2 receptor or human IP receptor. For the TP receptor and DP1 receptor counterscreens, membranes were prepared from enriched human platelets (Biological Specialty Corporation, Colmar, PA). For all experiments, DMSO or DP2 antagonists (diluted in DMSO) were added to membranes (4–17.5 μg of DP2/293, 10 μg of IP/293, 20 μg of human platelets), and tritiated ligand was added [(1 nM [3H]PGD2 (PerkinElmer Life and Analytical Sciences, Waltham, MA); 5 nM [3H]iloprost (GE Healthcare, Little Chalfont, Buckinghamshire, UK); 10 nM [3H]SQ 29,548 (PerkinElmer Life and Analytical Sciences); 2 nM [3H]BW A868C (American Radiolabeled Chemicals, Inc., St. Louis, MO)]. DP2 binding experiments were performed in the presence or absence of 0.2% human, mouse, or guinea pig serum albumin (Sigma-Aldrich). Reactions were incubated in 96-well deep-well plates for 60 min at room temperature. Membranes were harvested onto glass filter binding plates (UniFilter GF/C; PerkinElmer Life and Analytical Sciences) and washed three times with cold buffer containing 50 mM HEPES, pH 7.4, 0.5 M NaCl with use of a 96-tip cell harvester (Brandel, Gaithersburg, MD). Plates were dried for 60 min, and 50 μl of Microscint 20 (PerkinElmer Life and Analytical Sciences) was added to each well. Plates were sealed with TopSeal (PerkinElmer Life and Analytical Sciences), and the counts per minute were evaluated by use of a Packard TopCount NXT microplate scintillation counter (PerkinElmer Life and Analytical Sciences). The percentage inhibition of 3H ligand binding relative to the vehicle controls was graphed and the inhibitory concentration for 50% inhibition (IC50) was generated by nonlinear regression analysis of the data by use of MDL Assay Explorer (Elsevier MDL, San Ramon, CA).
Human Whole-Blood Eosinophil Shape Change.
Human blood was obtained from consenting adult volunteers by venipuncture and collected into EDTA tubes. Aliquots of fresh blood (100 μl) were incubated for 15 min at 37°C with 2 μl of DP2 antagonist diluted in 50% DMSO/water or with 50% DMSO/water as a vehicle control. PGD2 (50 nM final concentration) or vehicle was added, and incubation was continued for 5 min at 37°C. The reactions were placed on ice, and 250 μl of an ice-cold 1:4 dilution of Cytofix (BD Biosciences, San Jose, CA) in PBS was immediately added. Ammonium chloride lysing solution (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM disodium EDTA, pH 7.4) was added to lyse the red blood cells. The lysis was continued at room temperature for 10 min, and the tubes were centrifuged at 393g (5 min, 4°C). The supernatant was aspirated, and the cells were washed once with 3 ml of cold PBS and centrifuged at 393g (5 min, 4°C). The cells were resuspended in 200 μl of ice-cold 1:4 diluted Cytofix in PBS, and eosinophil shape change was analyzed on a FACScalibur (BD Biosciences) by analyzing forward light scatter of the autofluorescent cells. The percentage change of the PGD2-stimulated, vehicle-treated samples was set at 100%, and the percentage change of the nonstimulated, vehicle-treated samples was set at 0%. The percentage inhibition of eosinophil shape change by DP2 antagonist relative to the vehicle controls was graphed, and the IC50 was generated by nonlinear regression analysis of the data by use of MDL Assay Explorer (Elsevier MDL).
COX-1, COX-2, and Cerep Counterscreens.
Methods are outlined in supplemental data.
In Vivo
Animals.
Female BALB/c mice (Harlan, 8–10 weeks at study initiation) were used for pharmacokinetic and smoke studies. Male Hartley guinea pigs (200–300 g at study initiation; Charles River Laboratories, Inc., Wilmington, MA) were used for pharmacodynamic studies. Animals were housed in accordance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals. Animal protocols were approved by the Amira Pharmaceuticals Institutional Animal Care and Use Committee.
Pharmacokinetics.
The oral exposure of AM156 and AM206 was determined in fasted mice. Animals received an oral dose of AM156 or AM206 (10 mg/kg) in 0.5% methylcellulose (MC) vehicle and were then euthanized by CO2 inhalation at 1, 2, 4, 6, or 24 h after dose for AM156 and 1, 2, 4, 8, and 24 h after dose for AM206 (n = 2 animals per time point for each test compound). Blood (approximately 300 μl) was collected via cardiac puncture into EDTA-containing tubes and centrifuged at 1450g for 10 min. The plasma was removed and analyzed for AM156 or AM206 content by liquid chromatography-mass spectrometry.
Pharmacodynamics
Intravenous DK-PGD2 Challenge.
Methods were adapted from those detailed in Shichijo et al. (2003). Guinea pigs were primed with ovalbumin on day 0 by injection of 1 ml i.p. of a 100 μg/ml solution in Imject Alum (Pierce, Rockford, IL). They were then used in the DK-PGD2 procedure between days 14 and 21. Subjects were randomly assigned to receive either vehicle (0.5% MC, 4 ml/kg p.o.) or DP2 antagonist. Two hours after dosing, animals were anesthetized with ketamine/xylazine (70/10 mg/kg i.m.) and challenged with the selective DP2 agonist DK-PGD2 (1 mg/kg i.v.) or vehicle (10% methyl acetate in PBS, 1 ml/kg) via the dorsolateral penile vein. Thirty minutes after intravenous administration, blood was collected via the marginal ear vein into EDTA tubes for cell analysis and DP2 antagonist plasma concentration. After hypotonic lysis of red blood cells, leukocyte counts were determined by use of a hemacytometer. The number of cells per 0.1-mm3 field is presented.
Cigarette Smoke Exposure
General Exposure Procedures.
Mice were placed into a Plexiglas chamber (12 mice maximum per exposure group) and exposed to the smoke of seven cigarettes per day [3R4F (filters removed), University of Kentucky, Lexington, KY]. Smoke was generated by use of a peristaltic pump (Cole Parmer Masterflex L/S, Vernon Hills, IL) set to a delivery rate of 45 ml/min. Before entrance into the exposure chamber, smoke was diluted with air from an in-house air source. Mice were exposed to one cigarette every 15 min, for a total exposure time of 1.75 h. Ideally, air controls should be sham-treated by use of an identical procedure as used for the smoke-exposed mice minus the attachment of a cigarette to the apparatus. In our laboratory, however, we modified this procedure to accommodate space and equipment constraints. In this modified procedure, the filter tops were removed from the cages of air control groups and mice were exposed to room air for 1.75 h. In our experience, this modified procedure does not result in baseline values that differ from sham-exposed mice.
Acute Model.
Mice were exposed to smoke on days 0 and 1 and bronchoalveolar lavage (BAL) was performed on day 3, approximately 46 h after the final smoke exposure (Fig. 2A). AM156 (1 and 10 mg/kg) was prepared in 0.5% MC and dosed once daily. Rolipram (3 mg/kg q.d.) served as a positive control, whereas dexamethasone (0.3 mg/kg b.i.d.) served as a negative control. This dose of dexamethasone has been shown to inhibit lipopolysaccharide-induced pulmonary neutrophilia (Medicherla et al., 2008; Doukas et al., 2009). The vehicle for rolipram was 0.5% MC, whereas that for dexamethasone was 25% 2-hydroxypropyl-β-cyclodextrin (Sigma-Aldrich). AM156 and rolipram were administered orally 1 h before smoke. Dexamethasone was administered orally 1 h before smoke and again 6 h later. A previous experiment demonstrated no difference in smoke-induced responses between 0.5% MC and 25% 2-hydroxypropyl-β-cyclodextrin (data not shown), so for this experiment, only the vehicle (0.5% MC) for our compound of interest (AM156) was used.

A, study design for acute smoke exposure model. BALB/c females were exposed to the smoke of seven cigarettes on days 0 and 1, beginning at time 0 h. On these days, vehicle, AM156, and rolipram were dosed at −1 h. On day 2, vehicle and drug were dosed in the morning. Rolipram was dosed once daily. Dexamethasone was dosed twice a day. Bronchoalveolar lavage was performed on day 3. B, study design for variation of acute smoke exposure model. BALB/c females were exposed to the smoke of seven cigarettes on days 0, 1, and 2 beginning at time 0 h. AM156 and vehicle were dosed at −1 h. BAL was performed on day 3.
Efficacy of AM206 was explored in a variation of the acute smoke model. Here, mice were exposed to smoke on days 0 to 2, and BAL was performed on day 3 (Fig. 2B). AM206 (1 and 10 mg/kg) was prepared in 0.5% MC and dosed once daily. AM206 and vehicle were administered orally 1 h before smoke.
Subchronic Model.
Mice were exposed to cigarettes on days 0 to 1, 4 to 8, and 11 to 13 as outlined above in general exposure procedures (Fig. 3). For prophylactic treatment, mice were dosed 1 h before smoke with vehicle or AM156 for the duration of the study (days 0–13). For therapeutic treatment, mice were dosed with vehicle on days 0 to 4 and AM156 on days 5 to 13. BAL was performed approximately 22 h after the last smoke exposure. In a separate experiment, AM206 was dosed prophylactically (10 mg/kg) as described above for AM156.

Study design for subchronic smoke exposure model. BALB/c females were exposed to the smoke of seven cigarettes on days 0 to 1, 4 to 8, and 11 to 13 beginning at time 0 h. For prophylactic studies, vehicle, AM156, or AM206 was dosed on days 0 to 13. For therapeutic studies, vehicle was dosed on days 0 to 4, and vehicle or AM156 was dosed on days 5 to 13. On days of smoke exposure, treatments were administered 1 h before smoke. Bronchoalveolar lavage was performed on day 14.
Bronchoalveolar Lavage Procedure.
Mice were euthanized with isoflurane inhalation. Cardiac puncture was performed, and blood was collected into EDTA tubes for plasma analysis of drug concentration. The trachea was exposed and cannulated with a 20-gauge luer. BAL was performed by twice infusing and withdrawing 0.5 ml PBS. Cell suspensions were kept on ice until centrifugation (700g, 10 min). Supernatants were collected, measured in aliquots, and stored at −80°C until further analyses. Cell pellets were resuspended in 300 μl of 0.1% bovine serum albumin/PBS, and total leukocyte counts were determined on a Hemavet FS950 (Drew Scientific, Oxford, CT). A 100-μl aliquot of cell suspension was used for preparation of cytospin slides (Thermo-Shandon, Waltham, MA). Slides were stained with 3 Step Stain (Richard Allen Scientific, Kalamazoo, MI). Differential cell counts were performed on 300 cells using standard morphological features.
BALF Supernatant Analysis.
BALF from the subchronic AM156 experiment was analyzed for KC, MIP-2, IL-1β, IL-17, and MCP-1 in the BALF by ELISA (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions.
Mucin was analyzed by enzyme-linked lectin assay. In brief, 50 μl of BALF or mucin standard (bovine maxillary; Sigma-Aldrich) was added to a 96-well plate and dried overnight at 40°C. After washing, plates were blocked with 3% bovine serum albumin/PBS for 2 h. Plates were washed again, followed by incubation with UEA-1 lectin-horseradish peroxidase conjugate (Sigma-Aldrich) for 2 h. After washing, TMB substrate (BD Biosciences) was added, and color was allowed to develop for 30 min. The reaction was stopped with 1 M H2SO4, the resulting color change was read at 450 nm on a microplate reader (Molecular Devices, Sunnyvale, CA), and concentrations were determined relative to the standard curve.
Lung Tissue Homogenates.
In the subchronic experiments, lungs were collected from a subset of animals (n = 3/group), immediately frozen on dry ice, and stored at −80°C. Protease inhibitor (1.0 ml; Roche Diagnostics, Indianapolis, IN) was added to each lung upon removal from the freezer. Once thawed, lungs were homogenized with a Polytron tissue homogenizer (2200 rpm, 20 s; Glen Mills Inc., Clifton, NJ), sonicated with a Virsonic 100 (8 W, 10 s; VirTis, Gardiner, NY) and homogenized once more (2200 rpm, 10 s). Homogenates were centrifuged (900g, 15 min) and filtered through a 1.2-μm filter to remove debris. Protein concentrations of the resulting supernatants were determined by the Lowry method with use of a DC Protein kit (Bio-Rad, Hercules, CA) with bovine serum albumin (Pierce) to construct a standard curve. PGD2 was analyzed by enzyme immunoassay (Cayman Chemicals). Cytokines and chemokines were analyzed by ELISA (R&D Systems) according to the manufacturer's instructions. Results were normalized per milligram of protein.
Histology.
After collection of BALF, three lungs per group were fixated by inflation with 10% neutral-buffered formalin solution. Lungs were then excised and submerged in the formalin solution until tissue processing by an external histopathology laboratory (Pacific Pathology, San Diego, CA). Three 5-μm sections, 250 μm apart, were cut from paraffin-embedded lungs and mounted onto slides. Slides were then stained with periodic acid Schiff (PAS) to detect mucins.
Histological Quantitation.
Images were obtained with a Nikon Eclipse 50i microscope, and quantitative analysis was performed by use of NIS Elements image analysis software (Nikon Instruments, Melville, NY). The software was used to select the magenta-colored regions (PAS-positive cells). Images were obtained under 400× magnification, and the PAS-positive area as a percentage of each 400× field was calculated for two distinct airways per section level (6 total regions per lung). Images were obtained under 200× magnification, and epithelial thickness was determined by taking cross-sectional measurements from four representative areas per airway to determine the airway average. Two distinct airways were measured per lung section level, for a total of six airways per lung.
Statistical Analysis.
All values are expressed as the means ± S.E.M. Statistical analysis was performed by use of Student's t test for two-group analysis or one-way ANOVA for multiple group comparisons followed by either Tukey's or Newman-Keuls post hoc comparisons (GraphPad Prism, La Jolla, CA). P < 0.05 denotes statistical significance.
Results
In Vitro
Inhibition of [3H]PGD2 Binding to DP2 Expressing Membranes and PGD2-Mediated Eosinophil Shape Change in Whole Blood.
In [3H]PGD2 radioligand DP2 membrane-binding assays conducted in the presence of species-specific serum albumin, the IC50 values for AM156 were 24, 61, and 203 nM against human, mouse, and guinea pig DP2, respectively (Table 1). The IC50 values of AM206 for inhibition of [3H]PGD2 binding in the presence of species-specific albumin were 9.8, 206, and 62.5 nM against human, mouse, and guinea pig DP2, respectively (Table 1). AM156 inhibited PGD2-induced eosinophil shape change in human blood with an IC50 of 6.8 nM (Table 1), whereas the IC50 for AM206 in this assay was 15.5 nM. These data demonstrate that AM156 and AM206 bind to DP2 and inhibit DP2-mediated eosinophil shape change. Two representative reference DP2 antagonists (Oxagen and Actimis) displayed similar in vitro potencies (Table 1).
Species-specific DP2 binding and human whole blood eosinophil shape change potencies of AM156, AM206, and reference DP2 antagonists
Binding Selectivity and Enzyme Inhibition Counterscreens.
Both AM156 and AM206 are highly selective for human DP2 over human DP1, human TP, and human IP (Table 2). Additional selectivity data are presented in Supplemental Tables 1 and 2.
Inhibition of specific radioligand membrane binding to DP1, TP, and IP by AM156 and AM206
In Vivo
Pharmacokinetics.
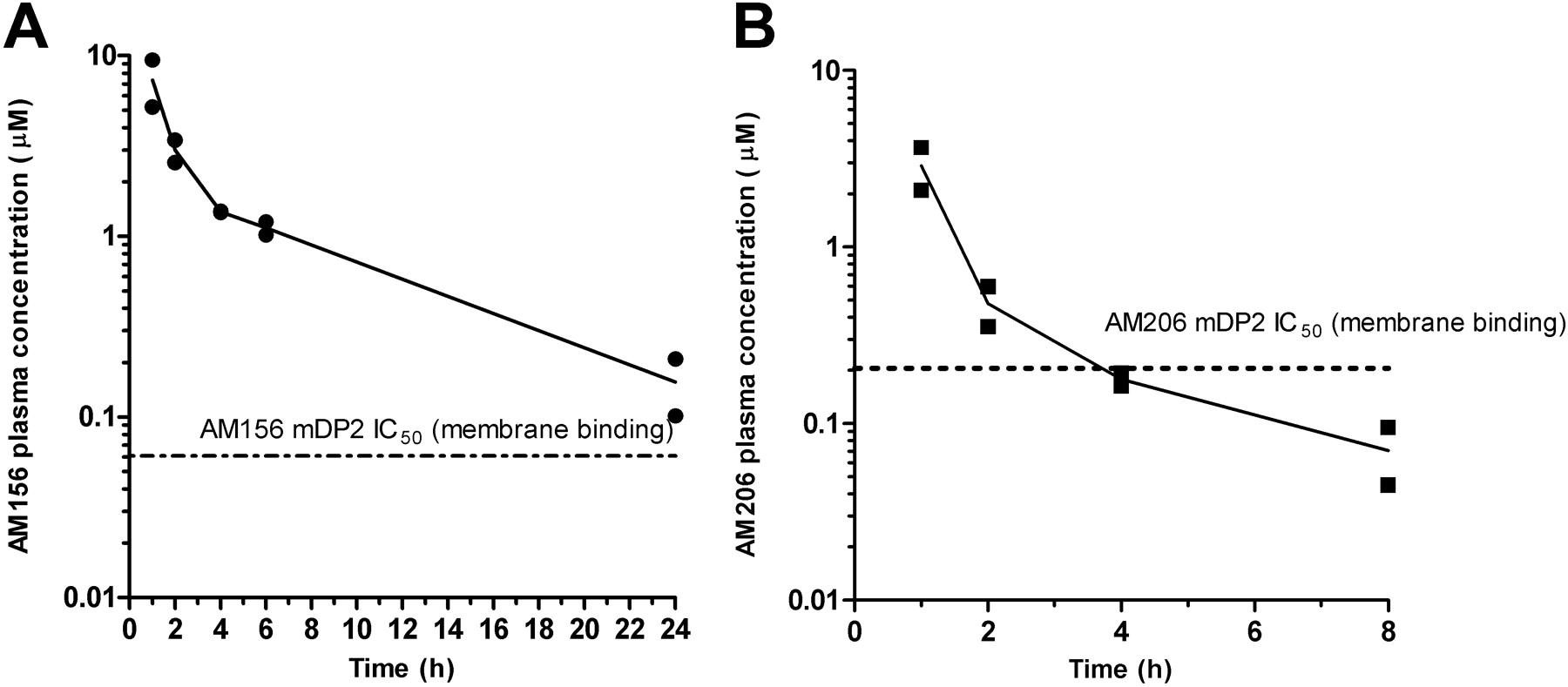
After oral administration of 10 mg/kg AM156, the peak plasma concentration of AM156 was 7.3 μM (1-h time point). AM156 concentrations then decreased to a trough value of 160 nM at 24 h (Fig. 4A). Oral administration of AM206 resulted in peak plasma concentrations of 2.9 μM (1-h time point) (Fig. 4B). At 8 h, AM206 concentrations were 70 nM. By 24 h, AM206 concentrations were below the limit of detection of the instrument (<10 nM).

Time-concentration profile for AM156 (A) and AM206 (B). Fasted mice were dosed with AM156 or AM206, and blood was sampled at 1, 2, 4, 6, and 24 h after oral dosing of AM156 (10 mg/kg) or at 1, 2, 4, 8, and 24 h after oral dosing of AM206 (10 mg/kg). Replicate plasma concentrations for each time point are graphed. The dashed line in each graph represents the in vitro IC50 for AM156 or AM206 against specific radioligand binding to mouse DP2 (AM156, 61 nM; AM206, 206 nM). AM206 plasma concentration was below detections limits (10 nM) at 24 h.
Pharmacodynamics.
Intravenous DK-PGD2 resulted in an increase in total leukocytes (Fig. 5). This increase was dose-dependently inhibited by AM156 and AM206 and by the reference DP2 antagonists from the patent literature (Lovell, 2007; Ly et al., 2007).

Pharmacodynamics of AM156, AM206, Oxagen, and Actimis DP2 antagonists. The DK-PGD2-induced increase in total leukocytes in the guinea pig is inhibited by AM156 (A), AM206 (B), Oxagen (Lovell, 2007) (C), and Actimis (Ly et al., 2007) (D).
Acute Cigarette Smoke Exposure Model.
In the AM156 experiment, acute cigarette smoke exposure caused an increase in total BALF leukocytes, which comprised macrophages, neutrophils, and lymphocytes (Fig. 6, A–D). Both total cells and macrophages were similarly affected by all treatment groups; dexamethasone produced a significant effect on macrophage influx (Fig. 6D). AM156 dose-dependently inhibited pulmonary neutrophilia (Fig. 6B). BALF neutrophils were also inhibited by rolipram but not by dexamethasone. There was a small but statistically significant increase in the number of lymphocytes recovered in the BALF (Fig. 6C), which was inhibited dose-dependently by AM156. Rolipram and dexamethasone were both effective against the lymphocyte influx. Based on these results, 10 mg/kg was selected for profiling in the subchronic smoke model.

A–D, inhibition of BALF total cells (A), neutrophils (B), lymphocytes (C), and macrophages (D) by AM156, rolipram, and dexamethasone (Dex) in the acute smoke exposure model. E–H, effect of AM206 on BALF total cells (E), neutrophils (F), lymphocytes (G), and macrophages (H). Values represent means ± S.E.M. *, P < 0.05; **, P < 0.01; ***, P < 0.001 versus smoke by ANOVA. #, P < 0.05; ##, P < 0.01 versus smoke by Student's t test.
In the variation of the acute smoke model, AM206 dose-dependently reduced the smoke-induced increase in BALF neutrophils (Fig. 6F). Lymphocytes were modestly increased by smoke and dose-dependently reduced by AM206 (Fig. 6G). Total BALF cells and macrophages were not affected by smoke in this model variation (Fig. 6, A and H).
Subchronic Cigarette Smoke Exposure Model.
Analysis of lung tissue homogenates revealed that lung PGD2 concentrations increased from 11.2 ± 0.7 ng/mg protein in air-exposed lungs to 34.2 ± 4.3 ng/mg protein in smoke-exposed lungs. Subchronic smoke exposure increased total BALF leukocytes (Fig. 7, A and E). Prophylactic treatment with AM156 significantly inhibited this increase in total cells. Therapeutic treatment with AM156 also reduced total cell numbers, but this did not reach statistical significance. AM206 caused a trend toward a reduction in total cells (Fig. 7E); however, this was not significant. Differential analysis of subtypes revealed that subchronic smoke exposure increased the numbers of neutrophils and lymphocytes (Fig. 7, B, C, F, and G). Only a modest increase in macrophages was observed (Fig. 7, D and H). AM156 reversed the smoke-induced effects on both neutrophils and lymphocytes after either prophylactic or therapeutic dosing regimens. No effect was seen on BALF macrophages. Likewise, AM206 reduced neutrophils and lymphocytes (Fig. 7, F and G). Trough plasma concentrations of AM156 were 114 and 169 nM, respectively, for prophylactic and therapeutic groups. Trough plasma concentrations of AM206 were below detection limits (<8 nM). These concentrations are consistent with those seen in the separate pharmacokinetic studies described above. These data suggest that it is not necessary to maintain plasma concentrations above the IC50 at trough.

Inhibition of BALF total cells (A, E), neutrophils (B, F), lymphocytes (C, G), and macrophages (D, H) by AM156 (A–D) and AM206 (E–H) in the subchronic smoke exposure model. Values represent means ± S.E.M. *, P < 0.05; **, P < 0.01 versus smoke by ANOVA. ##, P < 0.05 versus air by Student's t test; ###, P < 0.001 versus air by ANOVA.
BALF mucin was elevated after 10 days of smoke exposure (Fig. 8). Although not statistically significant, there was a trend toward reduction after treatment with AM156 and AM206. In the case of AM156, therapeutic dosing was as effective as prophylactic dosing. Histological analysis revealed PAS-positive cells in the airway epithelium, predominantly in the large airways, of smoke-exposed mice (Fig. 9, A and C). A reduction in PAS staining occurred after treatment with either AM156 (Fig. 9, A and C) or AM206 (Fig. 10A). Furthermore, epithelial thickening occurred in response to cigarette smoke exposure (Fig. 9, B and D). This was attenuated by both treatment with AM156 (Fig. 9, B and D) or AM206 (Fig. 10B). Moreover, therapeutic treatment with AM156 was nearly as effective as prophylactic treatment.

Inhibition of BALF mucin by AM156 (A) and AM206 (B). Values represent means ± S.E.M. ##, P < 0.05 versus air by ANOVA; ###, P < 0.01 versus air by ANOVA. Thera, therapeutic; Proph, prophylactic.

A and B, mucus cell metaplasia (A, 400× magnification) and epithelial thickening (B, 200× magnification) in PAS-stained lung sections from air controls, and smoke-exposed and AM156-treated mice. C, PAS staining was quantified by use of NIS Elements to determine the percentage of PAS-positive area per 400× field. D, Airway epithelial thickness was determined by taking cross-sectional measurements of representative airways. ###, P < 0.001 versus air by ANOVA; **, P < 0.01 versus smoke; and ***, P < 0.001 versus smoke by ANOVA.

Quantitation of mucus cell metaplasia (A) and epithelial thickening (B) in PAS-stained lung sections from air controls and smoke-exposed and AM206-treated mice. PAS-staining was quantified using NIS Elements to determine the percentage of PAS-positive area per 400x field (A). Airway epithelial thickness was determined by taking cross-sectional measurements of representative airways (B). ###, P < 0.001; ***, P < 0.001 versus smoke by ANOVA.
The AM156 experiment was further profiled via analysis of both BALF and lung homogenates. Cigarette smoke elevated KC, MIP-2, and MCP-1 in the BALF (Table 3). Neither KC nor MCP-1 was reduced by AM156. MIP-2 was modestly reduced with prophylactic treatment and unaltered by therapeutic with AM156. IL-1β and IL-17 were below the limits of quantitation for the assays (3 and 5 pg/ml, respectively).
Cytokines and chemokines in the BALF and lung homogenates
Cigarette smoke exposure resulted in elevated KC, MIP-2, MCP-1, and IL-1β in lung homogenates (Table 3). These concentrations were not altered by AM156. The lung concentration of IL-17 was not altered by smoke exposure; however, the basal concentration of IL-17 was reduced by DP2 antagonism.
Discussion
The results presented here are the first to demonstrate an effect of antagonism of the DP2 receptor on pulmonary inflammation, mucus cell metaplasia, and airway epithelial hyperplasia in a mouse model of COPD. DP2 antagonism inhibited cigarette smoke-induced influx of neutrophils and lymphocytes into the lungs. In the acute model, the degree of inhibition achieved was comparable with that of the well characterized phosphodiesterase 4 inhibitor rolipram. Dexamethasone failed to inhibit neutrophil influx, which is consistent with previous literature reports showing that rodent smoke models are steroid-resistant (Leclerc et al., 2006; Doukas et al., 2009).
The effects observed in the acute smoke model were further characterized in a subchronic smoke exposure model. In this setting, smoke exposure induced mucus cell metaplasia and airway epithelial thickening. The mucus hypersecretion, seen primarily in the large airways, could be detected both as released mucin in the BAL and histologically as a mucin-specific stain. Because lungs were lavaged before formalin fixation, histological analyses of small-airway inflammation, which would have been interesting, could not be performed accurately. Our results clearly demonstrate a beneficial effect of AM156 and AM206 in the subchronic cigarette smoke exposure model. As seen in the acute experimental model, the main anti-inflammatory effects were on inhibition of neutrophils and lymphocytes. In addition, there was a striking inhibition of both mucus cell metaplasia and airway epithelial hyperplasia. The longer duration of this model also allowed us to evaluate AM156 in both a prophylactic and therapeutic setting, the latter of which is more clinically relevant. Efficacy was achieved with both treatment paradigms. Because cigarette smoking is implicated in the etiology of COPD, DP2 antagonism may prove to be beneficial in the clinical setting, via prevention of airway inflammation, mucus hypersecretion, and airway remodeling.
The effects on neutrophil migration observed here are consistent with earlier reports in other murine models. In mice, DP2 is expressed on neutrophils, and intradermal injection of DK-PGD2, a selective DP2 agonist, increased staining of Ly-6G-positive neutrophils in the skin, demonstrating that activation of the receptor can have a direct effect on neutrophil recruitment (Takeshita et al., 2004). Furthermore, this was inhibited by ramatroban, a dual DP2/TP antagonist, but not by ridogrel, a selective TP antagonist. In contrast, the same authors failed to demonstrate in vitro chemotaxis of murine neutrophils in response to DK-PGD2, which perhaps suggests a role of the inflammatory milieu in the phenotypic response to DP2 agonists. In a murine model of contact hypersensitivity a selective antagonist of DP2 inhibited recruitment of neutrophils to the skin (Boehme et al., 2009). In vitro, cultured human neutrophils can up-regulate expression of DP2 (Carroll, 2006). Clinically, DP2 expression has been found to be up-regulated on sputum neutrophils in patients with cystic fibrosis (Tirouvanziam et al., 2008). These data suggest that in at least some pathological conditions, DP2 is expressed on human neutrophils. Therefore, it can be hypothesized that DP2 antagonism may provide a benefit in diseases such as COPD where neutrophils play a role in pathogenesis.
Chronic bronchitis is characterized by mucus hypersecretion and a pulmonary inflammation predominated by macrophages, neutrophils, and CD8+ T cells. Although each of these inflammatory cells plays an important role in the progression of the disease, neutrophils are the first cells to increase in the airways in response to cigarette smoke (D'Hulst A et al., 2005). Neutrophils, arriving in response to stimuli such as cigarette smoke, release mediators and enzymes that recruit additional inflammatory cells and break down alveolar tissue, leading to emphysema. Thus, inhibiting neutrophil recruitment with a DP2 antagonist may prevent the development of the chronic inflammatory environment, which contributes to disease progression.
Our results demonstrate that DP2 antagonism can inhibit the small, but significant, increase in BALF lymphocytes observed after cigarette smoke exposure. One mechanism by which DP2 antagonists prevent lymphocyte trafficking to the smoke-exposed lungs may be via direct action on circulating lymphocytes. In rats, the DP2 agonist DK-PGD2 stimulates the recruitment of lymphocytes from the bone marrow to the peripheral circulation, demonstrating a role for DP2 in lymphocyte trafficking (Shichijo et al., 2003). In vitro, Th2 lymphocytes chemotax in response to indomethacin, a DP2 agonist, and this was inhibited by an anti-DP2 antibody (Hirai et al., 2002). Thus, our results confirm that DP2 antagonism can inhibit lymphocyte trafficking in vivo.
Our data on mucus hypersecretion are consistent with previous findings with the dual DP2/TP antagonist ramatroban (Uller et al., 2007) and selective DP2 antagonists (Uller et al., 2007; Lukacs et al., 2008) in murine allergic settings. Although outside the scope of this manuscript, a possible mechanism by which DP2 antagonists affect mucus production may be via direct action on neutrophils to reduce elastase and other proteases.
The role of DP2 in epithelial cell proliferation and airway remodeling is an emerging concept indicated by this present report of DP2 antagonism inhibiting airway epithelial cell hyperplasia. While this manuscript was in preparation, similar results were reported with the selective DP2 antagonist AZ11805131 (Sargent et al., 2009). The epithelial cell acts as a first line of defense against harmful components in cigarette smoke, and DP2 is expressed on human bronchial epithelial cells (Chiba et al., 2006; Sargent et al., 2009). Intracellular expression of DP2 was demonstrated in NCI-H292 and normal human bronchial epithelial cells by both reverse transcription-polymerase chain reaction and flow cytometry (Chiba et al., 2007). The functional implications of DP2 expression on this cell type have yet to be fully elucidated; however, our data suggest that the efficacy of DP2 antagonists on cigarette smoke-induced pulmonary inflammation and mucus hypersecretion may be due to the direct effect of the DP2 antagonist on the epithelial cells, although, as stated above, a role for the DP2-neutrophil-epithelial cell axis cannot be ruled out.
We failed to demonstrate inhibition of KC, MIP-2, or IL-1β measured in either the BALF or lung homogenates, in agreement with those recently presented by Sargent et al. (2009). These results are in contrast to the inhibition of MIP-2, KC, and IL-1β levels in tissue homogenates by DP2 antagonism observed in an atopic dermatitis model (Boehme et al., 2009). We observed significant inhibition of neutrophil migration, even though the concentrations of the neutrophil chemoattractants KC and MIP-2 remained high in both BALF and lung tissue. This suggests that, in our model, the reduced number of neutrophils is not related to a DP2-dependent decrease in chemoattractants. Whereas the contrasting effects on cytokines and chemokines may simply reflect differences in the in vivo models used (smoke versus antigen or lung versus skin), we speculate that DP2 antagonists may be acting to down-regulate adhesion molecules on neutrophils, endothelial cells, or epithelial cells, thereby preventing adherence and subsequent chemotaxis of cells to the airways. Monneret et al. (2001) demonstrated that PGD2, probably acting via DP2, up-regulates expression of adhesion molecules on human eosinophils. Although this phenomenon has not been examined in murine granulocytes, it is possible that in the inflammatory milieu of cigarette smoke, DP2-dependent up-regulation of adhesion molecules on neutrophils or epithelial cells may occur. Thus, one mechanism by which DP2 antagonism could be inhibiting cell migration to the murine airways may be the down-regulation of cell surface adhesion molecules. Further exploration is needed to confirm this hypothesis.
In the naive lung, the resident alveolar macrophages are the predominant cell type. Mouse peritoneal macrophages can migrate toward the DP2 agonist, DK-PGD2 (Tajima et al., 2008). Our results in the acute smoke model support a role for DP2 in macrophage influx. However, neither macrophage influx nor local concentrations of MCP-1 were affected by AM156 in the subchronic model. Migration of peritoneal macrophages toward MCP-1 was unaltered in DP2-deficient mice (Tajima et al., 2008). Thus, the inability to reduce BAL macrophages with DP2 antagonism may be due to the role that MCP-1 may play in the recruitment of, and subsequent differentiation of, monocytes to macrophages in this subchronic smoke model.
It is noteworthy that we observed a reduction in basal lung IL-17 concentrations after treatment with a DP2 antagonist. In mice, Th17 cells are differentiated from Th0 cells in response to the production of IL-6 and transforming growth factor-β by dendritic cells (Romagnani, 2008). Elevations of IL-6 and transforming growth factor-β have been demonstrated in murine smoke models of COPD (Obot et al., 2004; Churg et al., 2006). The effect of DP2 antagonism on IL-17 confirms the modest reduction in IL-17 gene expression that Lukacs et al. (2008) observed in lungs from DP2 antagonist-treated mice relative to those from antigen-treated mice. IL-17 is implicated in promoting and sustaining neutrophilic inflammation; thus, the inhibitory effects of DP2 antagonism on pulmonary neutrophilia may, in part, be due to the reduction in basal lung IL-17 concentrations.
In summary, we report that DP2 antagonism with AM156 and AM206, potent and selective DP2 antagonists with favorable pharmacokinetic properties, inhibit cigarette smoke-induced pulmonary inflammation, mucus cell metaplasia, and airway epithelial hyperplasia. Furthermore, this is the first report that demonstrates the effectiveness of DP2 antagonism in this setting. Efficacy after therapeutic dosing of AM156 suggests that DP2 antagonists, such as AM156 or AM206, may provide novel and beneficial therapy for diseases such as COPD in which neutrophils and epithelial cells mediate disease progression.
Footnotes
This work was supported by Amira Pharmaceuticals.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.109.161919.
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- COPD
- chronic obstructive pulmonary disease
- AM156
- {2′-[(cyclopropanecarbonyl-ethyl-amino)-methyl]-6-methoxy-4′-trifluoro-methyl-biphenyl-3-yl}-acetic acid, sodium salt
- AM206
- (5-{2-[(benzoyloxycarbonyl-ethyl-amino)-methyl]-4-trifluoromethyl-phenyl}-pyridin-3-yl)-acetic acid, sodium salt)
- BAL
- bronchoalveolar lavage
- BALF
- bronchoalveolar lavage fluid
- COX
- cyclooxygenase
- PGD2
- prostaglandin D2
- DK-PGD2
- 13,14-dihydro-15-keto prostaglandin D2
- IL
- interleukin
- KC
- keratinocyte-derived chemokine
- PBS
- phosphate-buffered saline
- MCP-1
- monocyte chemoattractant protein-1
- MIP-2
- macrophage inflammatory protein-2
- DMSO
- dimethyl sulfoxide
- MC
- methylcellulose
- PAS
- periodic acid Schiff
- ANOVA
- analysis of variance
- AZ11805131
- 2-[2-(4-ethylsulfonyl-2-methylphenyl)-4-(trifluoromethyl)phenoxy]acetic acid.
- Received September 22, 2009.
- Accepted December 7, 2009.

- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}