


Abstract
Nonalcoholic steatohepatitis (NASH) comprises dysregulation of lipid metabolism and inflammation. Identification of the various genetic and environmental susceptibility factors for NASH may provide novel treatments to limit inflammation and fibrosis in patients. This study utilized a mouse model of hypercholesterolemia, low-density lipoprotein receptor knockout (LDLr-/-) mice fed a high-fat diet for 5 months, to test the hypothesis that farnesoid X receptor (FXR) deficiency contributed to NASH development. Either the high-fat diet or FXR deficiency increased serum alanine aminotransferase activity, whereas only FXR deficiency increased bile acid and alkaline phosphatase levels. FXR deficiency and high-fat feeding increased serum cholesterol and triglycerides. Although high fat led to macrosteatosis and hepatocyte ballooning in livers of mice regardless of genotype, no inflammatory infiltrate was observed in the livers of LDLr-/- mice. In contrast, in the livers of LDLr-/-/FXR-/- mice, foci of inflammatory cells were observed occasionally when fed the control diet and were greatly increased when fed the high-fat diet. Consistent with enhanced inflammatory cells, hepatic levels of tumor necrosis factor α and intercellular adhesion molecule-1 mRNA were increased by the high-fat diet in LDLr-/-/FXR-/- mice. In agreement with elevated levels of procollagen 1α1 and TGF-β mRNA, type 1 collagen protein levels were increased in livers of LDLr-/-/FXR-/- mice fed a high-fat diet. In conclusion, FXR deficiency induces pathologic manifestations required for NASH diagnosis in a mouse model of hypercholesterolemia, including macrosteatosis, hepatocyte ballooning, and inflammation, which suggest a combination of FXR deficiency and high-fat diet is a risk factor for NASH development, and activation of FXR may be a therapeutic intervention in the treatment of NASH.
Nonalcoholic fatty liver disease (NAFLD) is estimated to affect at least one quarter of the United States population and is the most common cause of abnormal liver function (Rector et al., 2008). NAFLD probably results from dysregulation of hepatic lipid metabolism as a component of the metabolic syndrome, which is collectively manifested as visceral obesity, dyslipidemia, atherosclerosis, and insulin resistance. Despite a benign prognosis for many cases, NAFLD can progress to non-alcoholic steatohepatitis (NASH), liver fibrosis, cirrhosis, and hepatocellular carcinoma, and a comprehensive understanding of the mechanism(s) of NAFLD-to-NASH transition remains elusive (Farrell and Larter, 2006).
Abnormality in lipid metabolism accompanied by chronic inflammation is a central pathway for the development of atherosclerosis and NASH (Loria et al., 2007; Schindhelm et al., 2007a,b). Indeed, a “two-hit” theory has been proposed, and the identification of secondary susceptibility factors related to lipid and cholesterol metabolism and inflammation that precipitate NASH in NAFLD patients is an important step in the treatment of this disease (James and Day, 1998). Susceptibility to NAFLD and NASH progression may include genetic variation, infection, environmental exposure, and dietary contribution, which may lead to chronic inflammation and/or oxidative stress.
Bile acids are critical regulators of lipid metabolism. However, dysregulation of bile acid homeostasis results in hepatic inflammation and injury. The central regulator and sensor for bile acids is the farnesoid X receptor (FXR), a ligand-activated nuclear receptor belonging to the nuclear receptor superfamily. Bile acids that can activate FXR include the primary and secondary bile acids: chenodeoxycholic acid, deoxycholic acid, lithocholic acid, and cholic acid, with decreasing potency (Parks et al., 1999; Wang et al., 1999). FXR is essential in regulating bile acid synthesis and transport. Mice with FXR deficiency have severe impairment of bile acid homeostasis (Sinal et al., 2000; Guo et al., 2003) and manifest systemic abnormalities, including altered lipid and cholesterol metabolism (Lambert et al., 2003; Ma et al., 2006), features known to be associated with the metabolic syndrome and NASH. In addition, FXR plays a pivotal role in regulating lipid metabolism (Urizar et al., 2000; Kast et al., 2001; Lambert et al., 2003). In addition, NASH has been reported to have a cholestatic presentation (Sorrentino et al., 2005). Indeed, bile acid levels were increased in the livers of some patients with NASH (Aranha et al., 2008) and in a murine steatohepatitis model induced by the methionine-choline-deficient (MCD) diet (Gyamfi et al., 2008).
Several risk factors associated with the development of atherosclerosis are interchangeable with those predicting NASH. Previous studies found that elevated plasma LDL levels in humans are closely associated with the risk of developing NASH and fibrosis (Musso et al., 2006; Singh et al., 2008). In addition, hepatic insulin resistance, a common outcome for NAFLD, is sufficient to cause atherosclerosis in mice (Biddinger et al., 2008). A previous report concludes that there is a universal correlation between NASH and insulin resistance (Chitturi et al., 2002). To this end, an emerging trend in NASH research is to utilize mouse models traditionally targeted for studies of atherosclerosis, including mice deficient in the LDL receptor (LDLr) or the ApoE gene (Yoshimatsu et al., 2004; Tous et al., 2005; Shiri-Sverdlov et al., 2006; Ma et al., 2008). The LDLr-/- mice have higher circulating LDL levels and develop atherosclerosis when fed a high-fat diet. LDLr-/- mice fed a high-fat/cholic acid diet manifest some features of NASH, including steatosis and inflammatory cell infiltration (Yoshimatsu et al., 2004), suggesting again that an increased bile acid level may be a risk factor for NASH development.
In the current study, we hypothesized that FXR deficiency induces the progression of NAFLD to NASH in metabolic syndrome-presenting, atherosclerosis-susceptible mice (i.e., LDLr-/- mice fed a high-fat diet). To test this hypothesis, LDLr-/- and LDLr-/-/FXR-/- mice were fed a control or high-fat diet for 5 months, and the requisite components of NASH, including hepatic steatosis, hepatocellular injury, inflammation, and fibrosis, were evaluated. The results showed that FXR deficiency led to the development of NASH in LDLr-/- mice fed a high-fat diet, indicating that FXR deficiency and/or altered bile acid homeostasis may increase the susceptibility to develop NASH in patients with metabolic syndrome.
Materials and Methods
Generation of LDLr-/-/FXR-/- Mice. Six-week-old LDLr-/- mice from the C57BL/6 genetic background were purchased from The Jackson Laboratory (Bar Harbor, ME). The generation of FXR-/- mice with a congenic C57BL/6 genetic background was described previously (Sinal et al., 2000; Guo et al., 2006). All mice were maintained and bred in the Laboratory of Animal Research at the University of Kansas Medical Center under a standard 12-h light/dark cycle, with access to chow and water ad libitum. All protocols and procedures have been approved by the Institutional Animal Care and Use Committee at the University of Kansas Medical Center. The LDLr-/-/FXR-/- mice were generated by cross-breeding LDLr-/- and FXR-/- mice. In detail, cross-breeding the LDLr-/- and FXR-/- mice generated an F1 generation with the LDLr+/- and FXR+/- genotype. This F1 generation was cross-bred between littermates to generate strains of LDL+/+/FXR+/+, LDLr-/-/FXR+/+, LDLr+/+/FXR-/-, and LDLr-/-/FXR-/- mice. The LDLr-/-/FXR-/- mice were normal in appearance, and the litter size ranged from three to five pups. For genotyping, tail DNA extraction and standard PCR amplification were performed using a Sigma RED Extract-N-Amp Tissue PCR Kit obtained from Sigma-Aldrich (St. Louis, MO).
The primers used for genotyping the FXR allele were described earlier (Sinal et al., 2000). The sequence of the primers used for genotyping the LDLr allele were obtained from the merchant and were as follows: primer 1, 5′-AGGTGAGATGACAGGAGATC-3′; primer 2, 5′-ACCCCAAGACGTGCTCCCAGGATGA-3′; and primer 3, 5′-CGCAGTGCTCCTCATCTGACTTGT. A 400-bp PCR product represents the wild-type allele and a 1.6-kb product for the knockout allele.
Diets and Sample Collection. At 3 to 6 weeks of age, male LDLr-/- and LDLr-/-/FXR-/- mice were divided into two groups (n = 10–14 mice per group) and fed either a control diet or a high-fat diet for 5 months. All diets were prepared by Bio-Serv (Frenchtown, NJ) and were based on a standard AIN-93G rodent diet (58.6% carbohydrate, 18.1% protein, 7.2% fat, 0.1% cholesterol, 5.1% fiber, 3.4% ash, and 10% moisture). The high-fat diet contains 0.15% cholesterol, 34% saturated fatty acids, 50% monounsaturated fatty acid, and 16% polyunsaturated fatty acid. The food consumption was monitored weekly, and the change in body weight was recorded monthly. Fasting serum was collected from the mice before and after the diet treatment. After 5 months of feeding, the mice were euthanized, and livers were removed, with one part snap-frozen in liquid nitrogen for gene expression analysis, one part frozen in the ornithine carbamyl transferase compound for hepatic steatosis analysis by Oil Red O staining and of hepatic collagen 1 protein expression by immunohistochemistry, and one part fixed in 10% phosphate-buffered saline-buffered formalin for histopathologic assessment.
Serum Parameters: ALT, ALP, Triglyceride, Cholesterol, Phospholipids, and Bile Acids. Serum was prepared from whole blood by centrifugation at 6000g using the Microtainer serum separator tubes (BD Biosciences, San Jose, CA). The kit for analyzing serum levels of ALT and ALP activity was obtained from Pointe Scientific (Canton, MI). Kits for analyzing triglycerides, cholesterol, phospholipids, and bile acids were obtained from Wako Bioproducts (Richmond, VA). All measurements were performed according to the manufacturer's instructions.
Histopathologic Analysis, Oil Red O Staining, and Collagen I Staining. Formalin fixed livers were cut into 5-μm sections and stained with hematoxylin and eosin for histopathology. The histopathologic alternations were examined, and the diagnosis of NAFLD and NASH was made by a pathologist (Dr. Ossama Tawfik, Department of Pathology, University of Kansas Medical Center). The Tissue-TEK OCT compound embedded frozen livers were sectioned at 8 μM and were stained with Oil Red O with a standard protocol. Staining and quantification for collagen I expression was described previously (Kim et al., 2006).
RNA Isolation, Northern Blotting, and Real-Time Quantitative PCR. Total RNA was isolated from the liver using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. The concentration of the total RNA was determined by spectrophotometry, and the integrity was confirmed by gel electrophoresis. Hepatic mRNA expression of the genes in lipid homeostasis was assessed by Northern blot analysis as described previously (Guo et al., 2003). Expression of the genes in inflammation and fibrosis was quantified by real-time quantitative PCR (Q-PCR) using the SYBR green chemistry. Primer sequences are listed in Table 1.
Primer sequences used for real-time quantitative PCR
Statistics. Unless otherwise stated, all values were expressed as the mean ± S.E. All data were analyzed by one-way analysis of variance followed by the Student-Newman-Keuls test. A P value less than 0.05 was considered significant.
Results
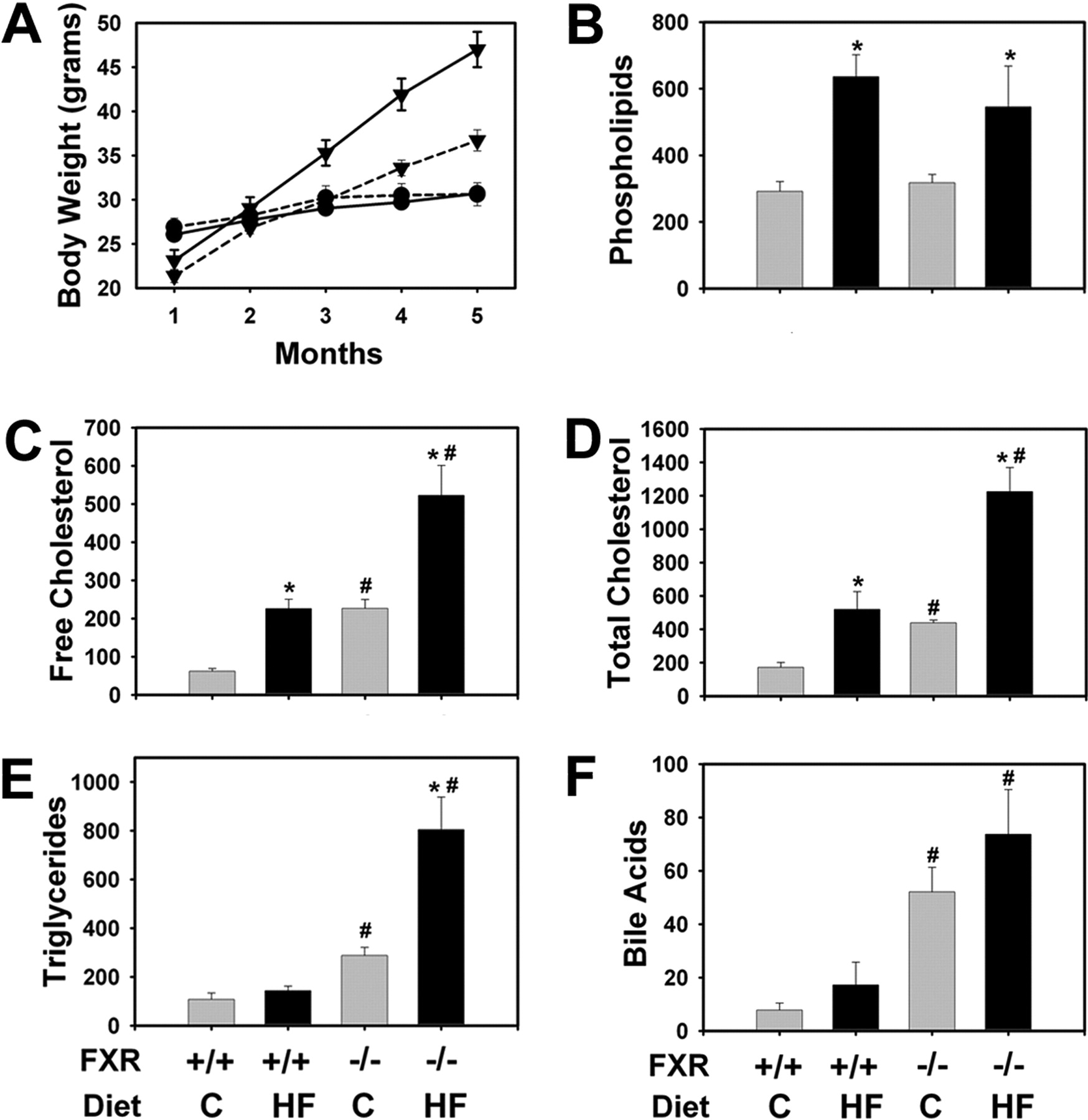
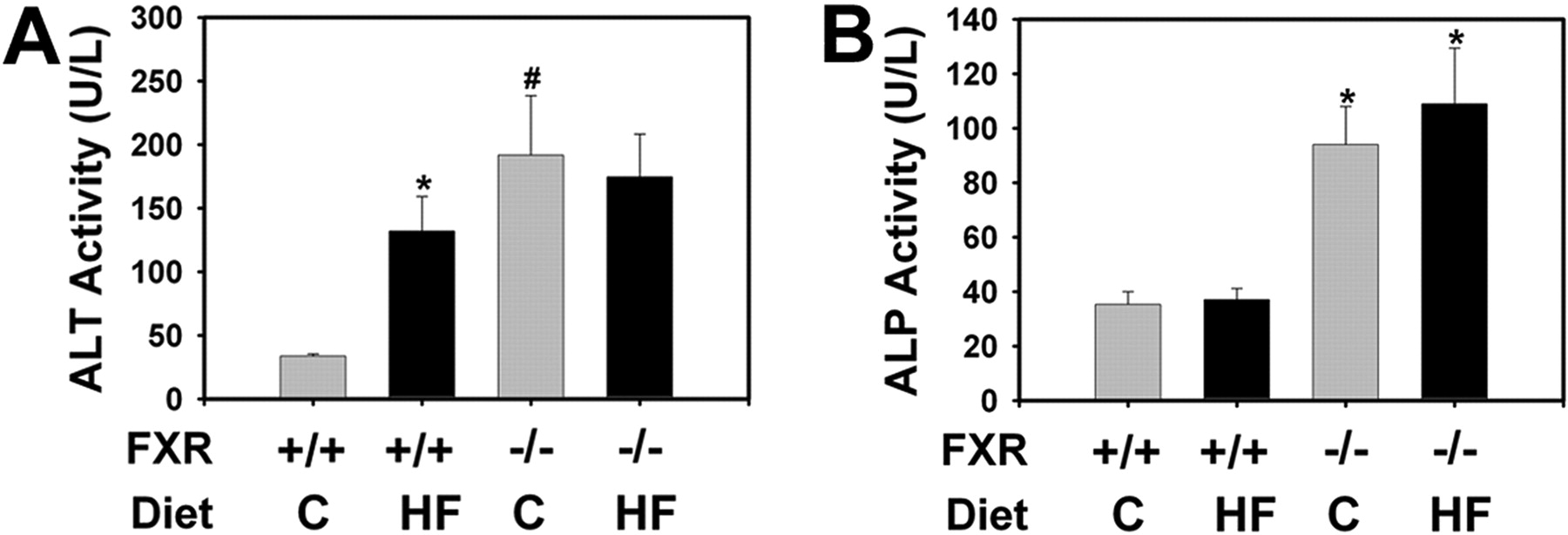
Serum Chemistry and Liver Histopathological Evaluation. The LDLr-/- and LDLr-/-/FXR-/- mice were fed a control or high-fat diet for 5 months. The LDLr-/- and LDLr-/-/FXR-/- mice gained a similar amount of weight when fed the control diet. Both strains gained more weight when fed the high-fat diet, with a faster rate in the LDLr-/- mice compared with LDLr-/-/FXR-/- mice (Fig. 1A). In agreement with the previous study (Ishibashi et al., 1994), the serum levels of cholesterol and phospholipids but not triglycerides were increased in LDLr-/- mice fed the high-fat diet for 5 months. Combined FXR deficiency increased the serum levels of cholesterol and triglycerides in mice fed the control diet, and the effect further increased in mice fed the high-fat diet (Fig. 1, B–E). FXR deficiency increased serum bile acids, and this was not affected by diet composition (Fig. 1F). Serum ALT levels were significantly increased in LDLr-/- mice fed the high-fat diet compared with LDLr-/- mice fed the control diet (Fig. 2A). Serum ALT levels were also markedly increased by FXR deficiency, and this increase was not altered by the high-fat diet. In contrast, serum ALP levels were selectively increased in the FXR-deficient mice and were not affected by high-fat feeding (Fig. 2B).

Effect of FXR deficiency on body weight and lipid levels in LDLr-/- mice fed a high-fat diet. The LDLr-/- mice and LDLr-/-/FXR-/- mice were fed either a control (C) diet or an HF diet for 5 months. A, body weight was determined in LDLr-/- mice (solid lines) and LDLr-/-/FXR-/- mice (dashed lines) fed control diet (circles) or high-fat diet (triangles). Serum levels of phospholipids (B), free (C) and total (D) cholesterol, triglycerides (E), and bile acids (F) were determined. Data are expressed as mean (milligrams per deciliter) ± S.E.M. n = 6 mice per group. *, significantly different from same genotype fed the control diet. #, significantly different from LDLr-/- mice fed the same diet. P < 0.05.
Liver histopathology revealed that the LDLr-/- mice fed the control diet had normal liver histology and normal lipid accumulation (Fig. 3, A and B). In contrast, the LDLr-/-/FXR-/- mice had mild macrosteatosis and scattered foci of inflammatory cell accumulation (Fig. 3, E and F). Despite manifesting more severe steatosis and hepatocyte ballooning, inflammatory cell accumulation was not detected in livers of LDLr-/- mice fed the high-fat diet (Fig. 3, C and D). In striking contrast, the LDLr-/-/FXR-/- mice fed the high-fat diet developed severe macrosteatosis, hepatocyte ballooning, and panlobular inflammation (Fig. 3, G and H). The generally required histopathologic observations for NASH diagnosis are macrosteatosis, lobular inflammation, and hepatocyte ballooning (Kleiner et al., 2005; Hübscher, 2006). These three features were all observed in the LDLr-/-/FXR-/- mice fed the high-fat diet.
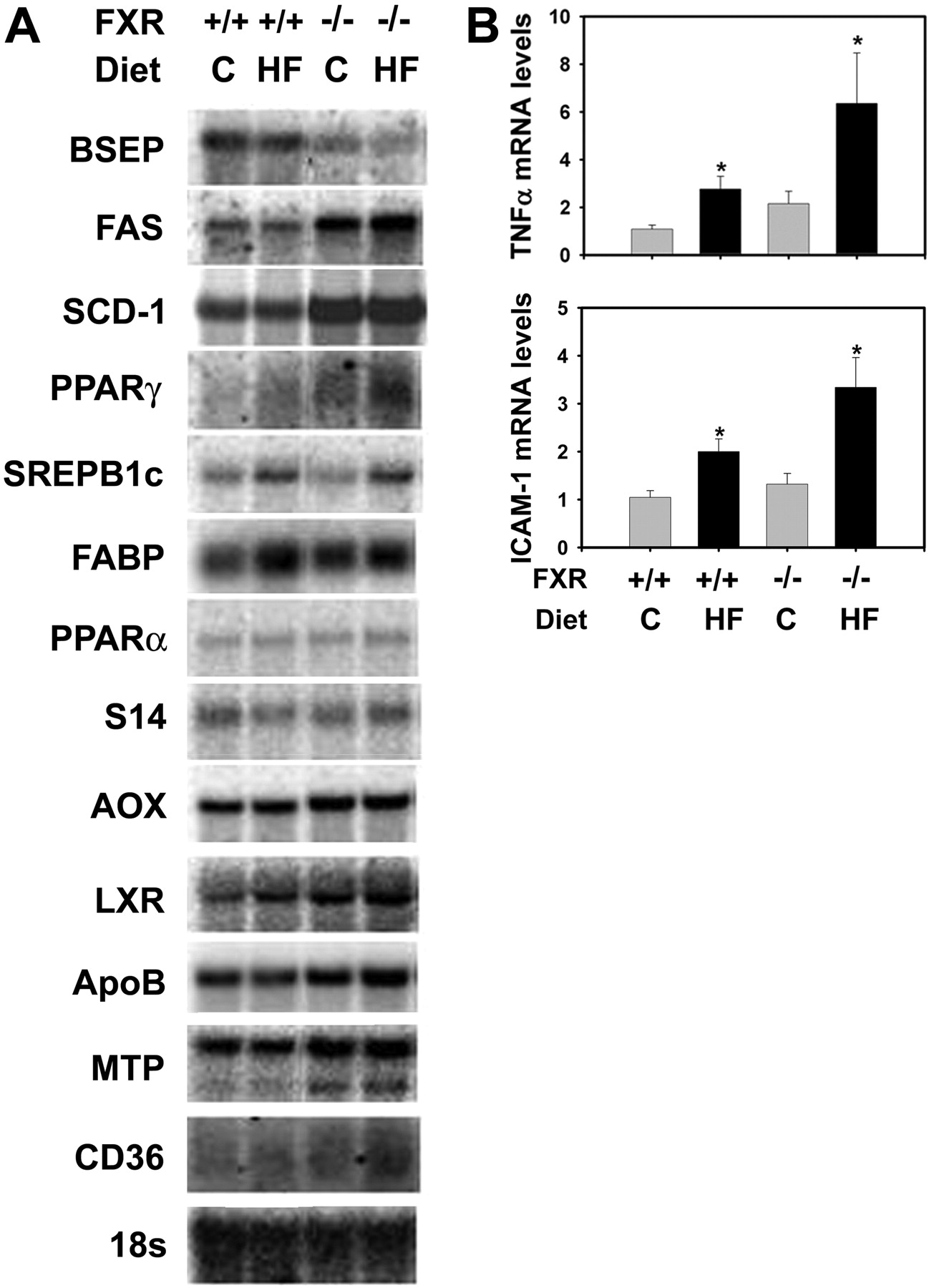
Expression of Genes Involved in Hepatic Lipid Homeostasis and Inflammation. Next, we determined the effect of FXR-deficiency and the high-fat diet on the expression of genes regulating hepatic lipid homeostasis at the mRNA level (Fig. 4A). In LDLr-/- mice fed control diet, a deficiency of FXR decreased hepatic mRNA levels of bile salt export pump and increased those of fatty acid synthase and stearyl-CoA desaturase-1. These data are in agreement with previous results indicating that FXR inhibits hepatic lipogenesis and promotes bile acid efflux into the bile (Sinal et al., 2000; Lambert et al., 2003; Watanabe et al., 2004). The high-fat diet increased the mRNA expression of sterol regulatory element binding protein 1c and fatty acid binding protein but decreased those of bile salt export pump in the liver of LDLr-/- mice. In livers of LDLr-/-/FXR-/- mice, these changes were further aggravated. In agreement with the observed inflammation in livers of LDLr-/-/FXR-/- mice fed the high-fat diet, the mRNA expression of the inflammatory genes, TNFα and ICAM-1, were markedly increased by the high-fat diet and FXR deficiency in an additive manner (Fig. 4B).

Effect of FXR deficiency on ALT and ALP activity in LDLr-/- mice fed a high-fat diet. The LDLr-/- mice and LDLr-/-/FXR-/- mice were fed either a control (C) diet or an HF diet for 5 months. Serum levels of ALT (A) and ALP (B) were determined. Data are expressed as mean (units per liter) ± S.E.M. n = 6 mice per group. *, significantly different from same genotype fed the control diet. #, significantly different from LDLr-/- mice fed the same diet. P < 0.05.

Effect of FXR deficiency on liver histopathology and steatosis in LDLr-/- mice fed a high-fat diet. The LDLr-/- mice and LDLr-/-/FXR-/- mice were fed either a control diet or a high-fat diet for 5 months. Liver histology in each group of mice was evaluated after hematoxylin and eosin staining (A, C, E, and G) of livers from each group. Lipid accumulation in livers from each group of mice was evaluated by Oil Red O staining (B, D, F, and H). Representative photomicrographs are shown from LDLr-/- mice fed the control diet (A and B), LDLr-/- mice fed the high-fat diet (C and D), LDLr-/-/FXR-/- mice fed the control diet (E and F), and LDLr-/-/FXR-/- mice fed the high-fat diet (G and H). Images are original magnifications ×20. The green circle in E indicates inflammatory cell infiltration.
Development of Liver Fibrosis. The guidelines of the American Association for the Study of Liver Diseases Single Topic Conference on NASH indicate that fibrosis is not necessary for the diagnosis of NASH, although it is often present. We evaluated the effect of the high-fat diet and FXR deficiency on the mRNA expression of fibrogenic genes in the liver (Fig. 5, A–D). FXR deficiency and the high-fat diet alone both increased liver procollagen mRNA expression in the LDLr-/- mice. Although this increase was apparently additive in LDLr-/-/FXR-/- mice fed the control diet, it did not achieve statistical significance. In LDLr-/- mice, the mRNA expression for α-smooth muscle actin (SMA) was increased slightly by the high-fat diet, although this did not achieve statistical significance. The α-SMA mRNA levels were increased significantly by FXR deficiency. The latter increase was not further affected by the high-fat diet. FXR deficiency significantly increased tissue inhibitor of metalloproteinase (TIMP)-1 expression (Fig. 5C). The mRNA expression of transforming growth factor (TGF) β was selectively increased in livers of LDLr-/-/FXR-/- mice fed the high-fat diet (Fig. 5D).

Effect of FXR-deficiency on expression of genes regulating lipid homeostasis and inflammation in LDLr-/- mice fed a high-fat diet. The LDLr-/- mice and LDLr-/-/FXR-/- mice were fed either a control (C) diet or an HF diet for 5 months. A, hepatic mRNA levels for genes in hepatic lipid homeostasis were determined by Northern blot analysis. B, hepatic levels of TNFα and ICAM-1 mRNA were determined by real-time Q-PCR. n = 6 mice per group. *, significantly different from same genotype fed control diet. #, significantly different from LDLr-/- mice fed the same diet. P < 0.05.
Minimal collagen staining was observed in livers of LDLr-/- mice fed with either the control (Fig. 5E; 1.0 ± 0.27) or high-fat (Fig. 5F; 1.46 ± 2.0) diet. Collagen levels were increased in livers of the FXR-/-/LDLr-/- mice fed the control diet (Fig. 5G; 2.96 ± 1.13). Collagen levels were greatest in livers of FXR-/-/LDLr-/- mice fed the high-fat diet and were localized primarily near vessels of periportal regions and the liver sinusoids (Fig. 5H; 4.30 ± 1.7).

Effect of FXR deficiency on fibrogenic gene expression and collagen deposition in LDLr-/- mice fed a high-fat diet. The LDLr-/- mice and LDLr-/-/FXR-/- mice were fed either a control (C) diet or an HF diet for 5 months. Hepatic levels of procollagen 1α1 (A), α-SMA (B), TIMP-1 (C), and TGF-β (D) mRNA were determined by real-time Q-PCR. n = 6 mice per group. *, significantly different from the same genotype fed the control diet. #, significantly different from LDLr-/- mice fed the same diet. P < 0.05. E to H, representative photomicrographs showing collagen 1 staining, in red color, in livers of LDLr-/- mice fed the control diet (E), LDLr-/- mice fed the high-fat diet (F), LDLr-/-/FXR-/- mice fed the control diet (G), and LDLr-/-/FXR-/- mice fed the high-fat diet (H).
Discussion
NAFLD results from imbalanced lipid homeostasis in the liver caused by increased fatty acid synthesis and influx and/or decreased fatty acid oxidation and VLDL excretion. The progression from NAFLD to NASH is of the greatest concern and is hypothesized to involve a “second hit” induced by oxidative stress and/or concurrent inflammation. The present study indicates that in mice, a combination of dietary and genetic factors influences the development of NASH. Specifically, the current study suggests that altered FXR status or bile acid signaling may be important susceptibility factors for the transition of NAFLD to NASH. FXR deficiency led to NASH development in LDLr-/- mice, which are susceptible to diet-induced hyperlipidemia, obesity, insulin resistance, and atherosclerosis, all features of metabolic syndrome.
Mice with deletion of the LDLr gene develop hyperlipidemia and atherosclerosis, especially after high-fat feeding (Ishibashi et al., 1994). In agreement with this result, this study showed that LDLr-/- mice became obese with high-fat diet feeding, levels of serum cholesterol were elevated, and these mice developed fatty plaques in the aorta, a major feature of atherosclerosis (data not shown). Moreover, these mice developed liver macrovesicular steatosis, a classic feature of NAFLD. LDLr-/- mice fed a high-fat diet, but not the control diet, developed liver steatosis, indicating that a combination of genetic deficiency and dietary risk factors contribute to steatosis development. To this end, the LDLr-/- mice fed the high-fat diet modeled several features consistent with metabolic syndrome and NAFLD. However, despite increased serum ALT activity levels and increased hepatic expression of inflammatory genes, inflammatory cell infiltration in the liver, a histologic necessity of NASH, was not detected in LDLr-/- mice fed the high-fat diet. This indicates that additional factors are required for the complete transition to NASH. One previous study showed that LDLr-/- mice develop NASH with high-fat diet feeding (Yoshimatsu et al., 2004). However, the high-fat diet used in that study contained 0.5% cholic acid, a primary bile acid that is known to cause hepatic toxicity. A recent study showed that high cholesterol content in the high-fat diet leads to NAFLD-to-NASH transition in ApoE-/- or LDLr-/- mice (Wouters et al., 2008). Additional studies showed that feeding rats for 3 weeks or intragastric feeding of C57BL/6 mice for 9 weeks with a high-fat diet led to severe NASH development (Lieber et al., 2004; Deng et al., 2005). Despite these reports, the LDLr-/- mice used in our study are from the congenic C57BL/6 background and were free of NASH, even after 5 months of feeding of the high-fat diet. However, it is possible that feeding the LDLr-/- mice a high-fat diet for a longer time (e.g., 8 months) would induce NASH. For example, wild-type mice fed a “western” diet for 8 months were reported recently to develop NASH (DeLeve et al., 2007).
Genetic susceptibility may be a potential stimulus for the more rapid progression of NASH. The present study clearly showed that FXR deficiency is a significant risk factor for the progression of NAFLD to NASH in mice. FXR deficiency alone induced modest but various features of NASH in mice fed the control diet, including steatosis and inflammatory cell infiltration. This observation is consistent with a previous report showing that FXR deficiency increased liver inflammation, especially after high-fat feeding for 12 weeks (Hanniman et al., 2005). Of importance, the histopathologic severity of steatosis, inflammation, and fibrosis was markedly increased by the combination of FXR deficiency and high-fat diet in the LDLr-/- mice. The baseline susceptibility of LDLr-/- mice to the metabolic syndrome probably reflects the early phase of NAFLD in patients. Taken together, our results indicate that superimposing FXR deficiency into an established model of metabolic syndrome (i.e., LDLr-/- mice) reproduces all key features of NASH. Future studies should include rescue experiments targeting the pathologic signaling pathways activated by FXR deficiency in the LDLr-/-FXR-/- model. Moreover, identification of animal models of NASH in which FXR is expressed, but dysfunctional, or where bile acid homeostasis is altered would be ideal for experiments determining whether genetic or pharmacologic activation of FXR “rescues” the mice from developing NASH.
FXR is essential in maintaining bile acid homeostasis (Sinal et al., 2000). Increased bile acid concentration in the liver has been shown to cause liver injury and inflammatory cell infiltration (Jaeschke et al., 2002; Kim et al., 2006). Thus, it is possible that a hepatic inflammatory response emerges in NASH as a consequence of increased bile acids because of dysfunctional FXR. Indeed, serum levels of bile acids are increased in some patients with metabolic syndrome and NASH (Aranha et al., 2008). Activation of FXR prevents overt bile acid toxicity and lowers serum lipid levels (Maloney et al., 2000; Liu et al., 2003). Thus, FXR activation may be beneficial in the prevention and treatment of cholestasis associated with NASH by lowering hepatic lipid and bile acid levels. To this end, FXR activation may limit hyperlipidemia and a second hit, such as bile acid-induced hepatic inflammation.
Bile acid-induced inflammation occurs in liver through multiple mechanisms, some of which are FXR-independent. For example, bile acids activate the membrane G-protein-coupled receptor TGR5 and epidermal growth factor receptor epidermal growth factor receptor on hepatic parenchymal and nonparenchymal cells (Rao et al., 2002; Keitel et al., 2007, 2008). This activates intracellular signaling pathways, including the extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and Akt signaling pathways (Rao et al., 2002; Qiao et al., 2003; Dent et al., 2005). Bile acid activation of ERK1/2 induces expression of the transcription factor early growth response factor-1 (Kim et al., 2006), which coordinates induction of several proinflammatory genes (McMahon and Monroe, 1996). Indeed, Egr-1 deficiency limited inflammation and cholestatic liver injury in bile duct-ligated mice, a model of obstructive cholestasis (Kim et al., 2006). It is interesting that we found that hepatic Egr-1 mRNA levels were significantly increased by FXR deficiency in the LDLr-/- mice (unpublished data). Taken together, the activation of inflammatory transcription factors like Egr-1 by elevated bile acids in the LDLr-/-FXR-/- mice may act as switch-promoting transition of simple steatosis to NASH.
The current study supports the use of mice susceptible to metabolic syndrome for the study of NAFLD and the transition to NASH. There is a close link between the epidemic of obesity and the widespread occurrence of NAFLD, a well recognized risk factor for NASH. One established animal model for the study of NASH development in rodents is through feeding the MCD diet (Weltman et al., 1996; Anstee and Goldin, 2006). The consumption of the MCD diet results in an acquired deficiency in mitochondrial β-oxidation. Although MCD diet-fed animals develop similar histopathological manifestation of liver steatosis, inflammation, and fibrosis, this diet fails to produce other features of the metabolic syndrome, such as obesity and insulin resistance (Weltman et al., 1996). These results indicate that the MCD diet-induced NASH model may not represent metabolic syndrome-associated NASH. The LDLr-/- mice manifest typical features of the metabolic syndrome, including obesity, especially upon feeding with the high-fat diet, increased serum lipids, increased circulating glucose and insulin resistance, and development of cardiovascular diseases, clinical pathologic features classically seen in patients with NAFLD and NASH. The LDLr-/-/FXR-/- mouse model may serve as a good model for the study of NAFLD-to-NASH transition. LDLr-/-/FXR-/- mice develop typical histological features for NASH in the liver, including macrovesicular steatosis, hepatocyte ballooning, and inflammatory cell infiltration.
Our model suggests that the combination of genetic and dietary risk factors constitutes the disease etiology for NASH development. It is known that mutation in LDLr or increase in LDL leads to hypercholesterolemia, a major risk factor in the development of several human diseases, including atherosclerosis, a component in metabolic syndrome (Goldstein and Brown, 1982). The LDLr knockout mouse is one of the most common mouse models in studying hypercholesterolemia-induced cardiovascular diseases. Although variation in the FXR gene is less studied, a recent study reported FXR gene polymorphism in humans with a population allele frequency ranging from 2.5 to 12% (Marzolini et al., 2007). It is interesting that in this study, FXR gene polymorphism has been shown to decrease FXR function. Thus, the current study indicates that increase in LDL and dysregulation of FXR-regulated pathway could contribute to NASH development.
In summary, the current study identified the deficiency of FXR as a significant risk factor in the development of NASH. Selective modulation of FXR in the liver may represent a promising therapeutic strategy in the future to prevent and treat NASH, especially for cases associated with bile acid abnormality.
Acknowledgments
We thank Noriko Esterly and Ruipeng Wang for excellent technical support.
Footnotes
-
This work was supported by National Institutes of Health [Grant P20 RR021940]; and by the American Heart Association [Grants 08351216, P20 RR015563, K12 HD052027, R01 DK031343].
-
B.K. and J.P.L. contributed equally to this work.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.108.144600.
-
ABBREVIATIONS: NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; FXR, farnesoid X receptor; MCD, methionine-choline-deficient; LDL, low-density lipoprotein; LDLr, LDL receptor; ALT, alanine aminotransferase; ALP, alkaline phosphatase; PCR, polymerase chain reaction; Q-PCR, quantitative PCR; TNF, tumor necrosis factor; ICAM, intercellular adhesion molecule; SMA, smooth muscle actin; TIMP, tissue inhibitor of metalloproteinase; TGF, transforming growth factor.
- Received August 7, 2008.
- Accepted October 22, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}