


Abstract
NF-E2-related factor 2 (Nrf2) is a transcription factor that is activated by oxidative stress and electrophiles that regulates the expression of numerous detoxifying and antioxidant genes. Previous studies have shown that Nrf2 protects the liver from xenobiotic toxicity; however, whether Nrf2 plays a role in lipid homeostasis in liver is not known. Accordingly, wild-type and Nrf2-null mice were fed a high-fat diet (HFD) for up to 4 weeks. Hepatic gene expression and lipid profiles were analyzed for changes in fatty acid, triglyceride, and cholesterol status. It is interesting to note that HFD reduced the mRNA expression of Nrf2 and its target genes in wild-type mice. The mRNA expression of lipogenic and cholesterologenic transcriptional factors and their target genes, such as sterol regulatory element-binding proteins 1c and 2, fatty acid synthase, acetyl-CoA carboxylase 1, fatty acid elongase, 3-hydroxy-3-methylglutaryl coenzyme A synthase and reductase, and low-density lipoprotein receptor mRNA expression were higher in Nrf2-null mice compared with wild-type mice after feeding a HFD, suggesting that Nrf2 may suppress these pathways. Hepatic triglycerides and cholesterol levels were not different between genotypes, whereas concentrations of hepatic free fatty acid and malondialdehyde equivalents were higher in Nrf2-null mice compared with wild-type mice 4 weeks after HFD feeding. Overall, these results suggest that Nrf2 inhibits lipid accumulation and oxidative stress in mouse liver after feeding a HFD, probably by interfering with lipogenic and cholesterologenic pathways.
Fatty liver disease afflicts 20% of the U.S. population (U.S. Department of Health and Human Services, 2004). Approximately 15 to 20% of patients with fatty liver develop a severe form of hepatic disease known as nonalcoholic steatohepatitis (NASH) (McCullough, 2002). High prevalence of fatty liver and NASH warrants research into the signaling mechanisms responsible for hepatic lipid accumulation in individuals consuming diets with a high proportion of fat.
Hepatic lipid homeostasis is maintained by the collective balance of lipid synthesis, catabolism, and secretion. Circulating and tissue lipids, including fatty acids and cholesterol, are essential nutrients that regulate lipid metabolism genes by transcription factors, such as sterol regulatory element-binding proteins (SREBPs). SREBPs are key transcription factors that regulate fatty acid and cholesterol metabolism in liver (Horton et al., 2002). SREBP-1c is known to regulate the expression of various lipogenic genes, such as fatty acid synthase (FAS), acetyl-CoA carboxylase 1 (ACC1), fatty acid elongase (FAE), and stearoyl-CoA desaturase-1 (SCD-1) (Bené et al., 2001; Moon et al., 2001; Matsuzaka et al., 2002).
β- and ω-Oxidation, regulated by the nuclear receptor peroxisome proliferator-activated receptor (PPAR)α, are important steps in fatty acid catabolism. Target genes of PPARα include acyl-CoA oxidase (AOX), carnitine palmitoyltransferase-1 (Cpt-1), cytochrome P450 4a10 (Cyp4a10), and Cyp4a14 (Hashimoto et al., 1999; Johnson et al., 2002).
SREBP-2 is a transcription factor that regulates several genes involved in cholesterol synthesis and uptake, such as HMG-CoA synthase, HMG-CoA reductase, and low-density lipoprotein (LDL) receptor (Cuthbert and Lipsky, 1992). Liver X receptor (LXR)α also regulates genes for cholesterol catabolism and export, including cholesterol 7-α hydroxylase (Cyp7a1), Abca1, and Abcg5/8 (Repa et al., 2002). Abca1 exports high-density lipoprotein cholesterol from hepatocytes into blood (Neufeld et al., 2002). Abcg5 and Abcg8 aid in the secretion of plant sterols and cholesterol from hepatocytes into bile (Yu et al., 2002). Cyp7a1 and sterol 12-α hydroxylase (Cyp8b1) are hepatic enzymes in the neutral pathway for the conversion of cholesterol to bile acids (Eloranta and Kullak-Ublick, 2005). Microsomal triglyceride transfer protein (MTP) is known to play an integral role in the assembly and secretion of very low-density lipoprotein (VLDL), and impaired VLDL secretion leads to fatty liver (Lettéron et al., 2003).
Oxidative stress has been implicated in the transition from fatty liver to NASH (Sakaida and Okita, 2005). Recent research has identified Nrf2 as a key transcription factor for combating hepatic oxidative stress (Kobayashi et al., 2006). Nrf2 is sequestered in the cytoplasm under physiological conditions by Kelch-like ECH-associated protein 1 (Keap1). Upon oxidative or electrophilic stress, Nrf2 translocates to the nucleus, and it regulates the transcriptional activation of a battery of cytoprotective genes (Itoh et al., 2004). Nrf2 induces expression of antioxidant enzymes and phase II detoxifying proteins, such as glutathione transferase (Gst), NAD(P)H:quinone oxidoreductase 1 (Nqo1), and heme oxygenase-1 by binding to antioxidant responsive elements in the promoters of these genes (Ishii et al., 2002; Kobayashi et al., 2006). Numerous studies have shown that Nrf2 mitigates various pathologies, including carcinogenesis, chemical toxicity, respiratory distress, and inflammatory diseases (Chan and Kan, 1999; Chan et al., 2001; Braun et al., 2002; Iida et al., 2004). Nrf2 is moderately expressed in liver, a major organ for lipid homeostasis (Petrick and Klaassen, 2007). In liver, Nrf2 plays a protective role against acetaminophen hepatotoxicity by regulating expression of antioxidant genes and phase II drug-metabolizing enzymes (Chan et al., 2001; Okawa et al., 2006).
Although previous studies have demonstrated that Nrf2 reduces xenobiotic toxicity, it is not known whether Nrf2 interferes with high-fat diet (HFD)-induced hepatic lipotoxicity. In the present study, male C57BL/6 (wild-type) and Nrf2-null mice were fed a normal diet or a HFD for 1 or 4 weeks. This short-term study was designed to investigate the role of Nrf2 in preventing lipid accumulation, before the onset of fatty liver and/or steatohepatitis. Liver lipid extracts and serum, as well as hepatic mRNA, were analyzed for changes in fatty acid, triglyceride, and cholesterol status. We hypothesized that feeding a HFD would increase hepatic fatty acid, cholesterol, and malondialdehyde (MDA) equivalent levels in both genotypes by 4 weeks, with greater accumulation in Nrf2-null mice. These changes in the Nrf2-null mice should be reflected in gene expression patterns favoring synthesis and retention of lipids.
Materials and Methods
Materials. Serum and hepatic free-cholesterol, nonesterified fatty acids, and triglycerides were quantified using colorimetric assays from Wako Chemicals USA, Inc. (Richmond, VA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless noted.
Animals. Eight-week-old male C57BL/6 wild-type mice were obtained from Charles River Laboratories, Inc. (Wilmington, MA). Male Nrf2-null mice, generated on a mixed C57BL/6 and AKR background, were obtained from Dr. Jefferson Chan (University of California, Irvine, CA). They were backcrossed with C57BL/6 mice for seven generations, resulting in greater than 97% C57BL/6 background (congenic confirmed by The Jackson Laboratory, Bar Harbor, ME). All mice were housed in the same animal care facility controlling for temperature, humidity, and light. Mice (n = 5–10/group) were fed commercial diets containing either a high amount of fat [HFD; 35% (w/w) lard, 0.15% (w/w) cholesterol; F5194, BioServ, Inc., Frenchtown, NJ] or a normal amount of fat [normal diet (ND); 4.4% (w/w) fat; 8604, Harlan Teklad, Madison, WI] for 1 or 4 weeks. Lard contained 46% oleic acid (most of monounsaturated fatty acids), 23% palmitic acid, 9% stearic acid, and 1% myristic acid (most of saturated fatty acids), and 14% linoleic acid and 0.2% eicosanoic acid (most of n-6 polyunsaturated fatty acids). Individual body weights were recorded weekly. After 1 or 4 weeks, blood samples were collected, and livers were harvested and weighed. At both time points, food was removed the night before collecting tissues. Animals received humane care as outlined in the Institute of Laboratory Animal Resources (1996). Studies were approved by the University of Kansas Medical Center Institutional Animal Care and Use Committee.
RNA Isolation. Total RNA was isolated using TRIzol reagent according to the manufacturer's protocol (Invitrogen, Carlsbad, CA). The concentration of total RNA in each sample was quantified spectrophotometrically at 260 nm.
Branched DNA Signal Amplification Assay. The mRNA expression of mouse Nrf2, Gst mu 6 (Gstm6), Abcg5, Abcg8, and LXRα were quantified using the branched signal amplification assay with modifications (QuantiGene, High Volume branched DNA Signal Amplification kit; Panomics, Fremont, CA) (Hartley and Klaassen, 2000). Multiple oligonucleotide probe sets (containing capture, label, and blocker probes) (Supplemental Table 1) specific to mouse LXRα mRNA transcripts were designed using ProbeDesigner software version 1.0 (Bayer Corp., Diagnostics Division, Emeryville, CA). Other probe sets designed specifically for mice include Nrf2, Gstm6, Abcg5, and Abcg8, as described previously (Maher et al., 2006; Knight et al., 2007; Petrick and Klaassen, 2007).
Real-Time PCR. The mRNA level of mouse Nqo1, CD36, FAS, SCD1, Cyp4a10), liver fatty acid-binding protein (L-FABP), ACC1, FAE, LDL receptor, HMG-CoA synthase, HMG-CoA reductase, AOX, Cpt-1, Cyp4a10, Cyp4a14, Cyp7a1, Cyp8b1, MTP, SREBP-1c, SREBP-2, PPARα, PPARγ, PGC-1α, PGC-1β, and 18s rRNA were quantified by TaqMan real-time polymerase chain reaction (PCR) as described previously (Gyamfi et al., 2008). Primers and probes (Supplemental Table 2) were designed using Primer Express 2.0 (Applied Biosystems, Foster City, CA). The amplification reactions were carried out in an ABI Prism 7900 sequence detection system (Applied Biosystems). The amount of mRNA was calculated using the comparative CT method, which determines the amount of target normalized to an endogenous reference. Each gene was normalized to 18s rRNA.
Quantification of Serum and Hepatic Free-Cholesterol, Nonesterified Fatty Acids, and Triglycerides. Liver lipid content was extracted as described previously (Zhou et al., 2006). In brief, 100 mg of frozen liver tissue was homogenized in 1 ml of buffer containing 18 mM Tris, pH 7.5, 300 mM mannitol, 50 mM EGTA, and 0.1 mM phenylmethylsulfonyl fluoride. Five hundred microliters of homogenate was mixed with 4 ml of chloroform/methanol (2:1) and incubated overnight at room temperature with occasional shaking. The next day, 1 ml of H2O was added, vortexed, and centrifuged for 5 min at 3000g. The lower lipid phase was collected and concentrated by vacuum. The lipid pellets were dissolved in a mixture of 270 μlof isopropanol and 30 μl of Triton X-100. Serum and liver lipid samples were analyzed spectrophotometrically for free-cholesterol (505 nm), nonesterified fatty acids (550 nm), and triglycerides (600 nm) in accordance with the manufacturer's protocols.
TBARS Assay. Livers were assayed for thiobarbituric acid reactive substances (TBARS) using components of the TBARS kit (ZeptoMetrix, Buffalo, NY). Frozen samples (100 mg) were homogenized in 1 ml of ice-cold 1.15% KCl the day of assay. One hundred microliters of sample homogenates, as well as MDA standards, were incubated with SDS and 0.8% thiobarbituric acid (20% acetic acid, pH 3.5) in the presence of 0.8% butylated hydroxytoluene at 95°C for 1 h. After incubation, samples were cooled on ice, and then they were centrifuged at 3000 rpm for 15 min. Supernatants were read on the spectrophotometer at 532 nm. TBARS were expressed as micromoles of MDA equivalents per gram of liver.
Hematoxylin and Eosin and Oil-Red O Staining. Liver samples were fixed in 10% neutral formalin, and then they were embedded in paraffin. Sections were stained with hematoxylin and eosin for histological examination. Frozen liver sections were stained with Oil-Red O to examine the accumulation of fat in hepatocytes. In brief, the sections were fixed in 10% neutral formalin. After two changes of 60% isopropanol, Oil-Red O was added with agitation for 20 min, followed by washing in 60% isopropanol. The sections were then counterstained with hematoxylin and rinsed in distilled water.
Statistical Analysis. The software package STATISTICA release version 4.5 (StatSoft, Tulsa, OK) was used for statistical analysis. Statistical analysis included one-way analysis of variance, followed by Duncan's multiple range test. Differences were considered statistically significant at p < 0.05.
Results
Body Weight and Liver Weight/Body Weight Ratio in Wild-Type and Nrf2-Null Mice Fed a HFD. Both wild-type and Nrf2-null mice fed a HFD equally gained weight over the 4 weeks (data not shown). Liver weight/body weight ratio was decreased 22% by the HFD in wild-type mice 4 weeks after HFD feeding, but it was not changed by HFD in Nrf2-null mice.
Hepatic mRNA Expression of Nrf2 and Its Targets in Wild-Type and Nrf2-Null Mice Fed a HFD. It is interesting to note that feeding a HFD to wild-type mice for 4 weeks decreased mRNA expression of Nrf2 56% (Fig. 1A). A similar trend in reduced Nrf2 mRNA was observed at 1 week, although it was not statistically significant. The mRNA expression of Nrf2 target genes, including Nqo1 and Gstm6 was quantified. Analogous to previous reports, the basal mRNA expression of Nqo1 and Gstm6 is reduced in Nrf2-null mice, compared with wild-type mice (Chanas et al., 2002; Aleksunes et al., 2006). Similar to the reduced Nrf2 mRNA, the gene expression of Nqo1, was decreased 36 and 56% in wild-type mice fed a HFD for 1 and 4 weeks, respectively, but not in Nrf2-null mice (Fig. 1B). The mRNA expression of Gstm6, another target gene of Nrf2, was decreased 22% in wild-type mice fed a HFD 1 and 4 weeks, but it was not changed in Nrf2-null mice (Fig. 1B).
Serum and Hepatic Cholesterol and Nonesterified Fatty Acids in Wild-Type and Nrf2-Null Mice Fed a HFD. There were no differences in the basal serum and hepatic lipids between wild-type and Nrf2-null mice. Notable changes in hepatic and serum lipid profiles in wild-type and Nrf2-null mice were evident after feeding a HFD for 4 weeks. Compared with normal diet fed mice, 4 weeks of feeding a HFD increased serum free-cholesterol by 96% in wild-type, and 57% in Nrf2-null mice (Fig. 2A). In contrast, hepatic free-cholesterol was not altered after feeding a HFD, although there was a trend for increased hepatic cholesterol in both genotypes (Fig. 2B). Serum free fatty acids were increased above control levels in both genotypes after feeding a HFD (Fig. 2A). Hepatic free fatty acids were increased in both genotypes after feeding a HFD for 4 weeks, with higher levels observed in Nrf2-null mice (Fig. 2B). Serum triglycerides were not changed in any group after feeding a HFD (Fig. 2A). Hepatic triglycerides were similarly increased above control levels in both genotypes after feeding a HFD (Fig. 2B).

A, Nrf2 mRNA expression in wild-type mouse liver 1 and 4 weeks after feeding a HFD. Total liver RNA was isolated from wild-type mice fed a ND and a HFD, and it was analyzed as described under Materials and Methods. B, Nrf2 target mRNA expression in wild-type and Nrf2-null mouse liver fed a ND and a HFD. Total hepatic RNA was isolated from mice fed a ND and a HFD, and it was analyzed as described under Materials and Methods. Data are presented as mean ± S.E.M. (each group; n = 5–10 animals). Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. Asterisks (*) represent statistically significant difference (p < 0.05) compared with either wild-type or Nrf2-null mice fed a ND, respectively; daggers (†) represent a statistically significant difference (p < 0.05) from the matched wild-type control mice within the same diet group.
To determine whether the lipid in livers was evident histologically, hematoxylin and eosin and Oil-Red O staining were conducted. Histological examination revealed no basal differences in livers from wild-type and Nrf2-null mice fed a normal diet (data not shown). After feeding a HFD for 4 weeks, hematoxylin and eosin staining showed more microvesicular lipid accumulation in livers compared with the normal diet group, with no appreciable differences between genotypes. As expected, there was no histological evidence of steatohepatitis (infiltration of inflammatory cells and pericellular necrosis) in either genotype after feeding a HFD for 4 weeks. Liver sections from the mice fed a HFD for 4 weeks showed more Oil-Red O staining compared with the mice fed a normal diet in both genotypes (data not shown). There were no significant differences in Oil-Red O staining between genotypes after feeding a HFD, similar to hepatic triglyceride concentrations.
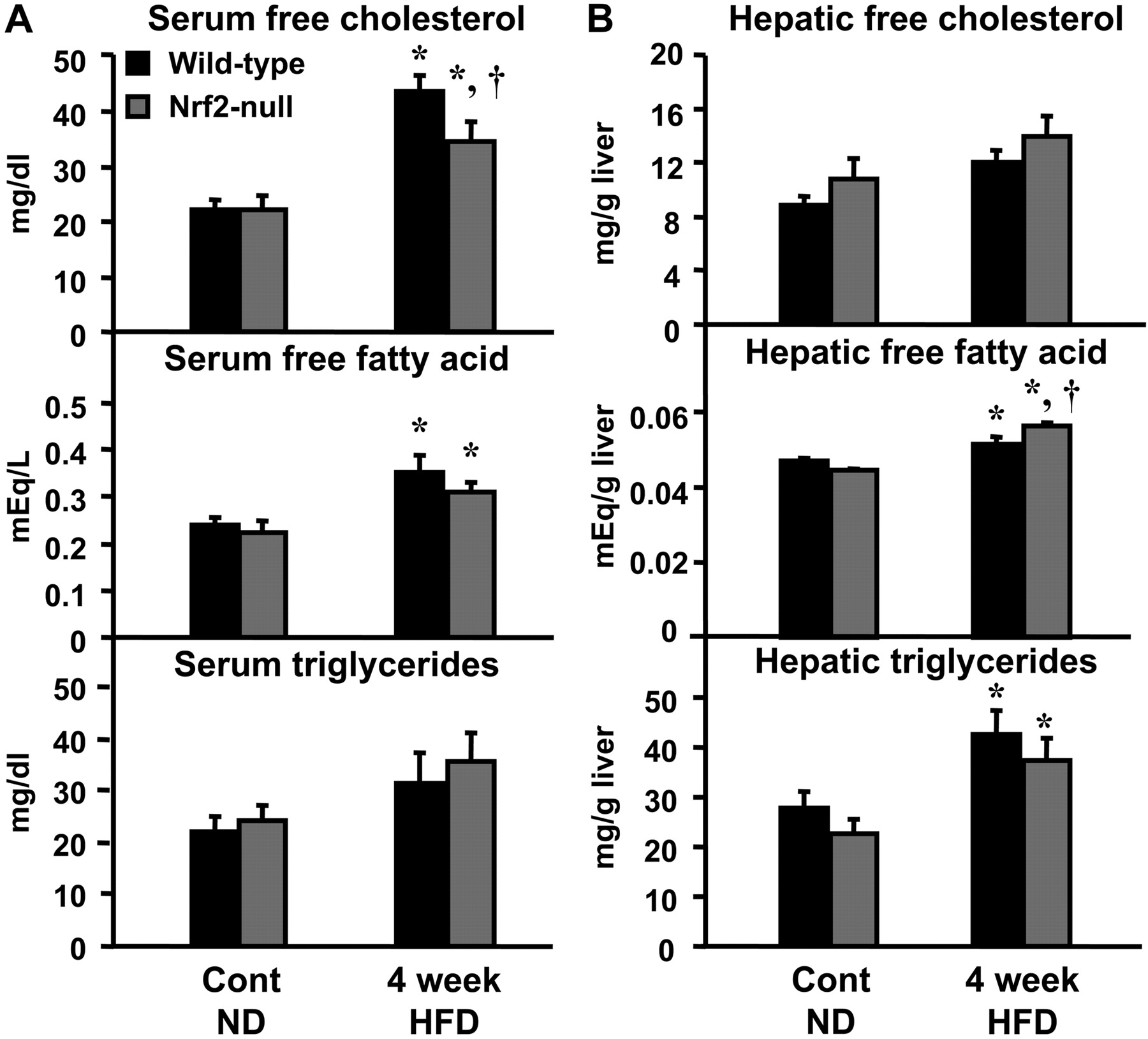
A, serum free-cholesterol, nonesterified fatty acids, and triglycerides in wild-type and Nrf2-null mice after HFD feeding. At 4 weeks after HFD feeding, free-cholesterol, nonesterified fatty acids, and triglycerides were quantified in serum. Data are expressed as mean ± S.E.M. (each group; n = 5–10 animals). B, hepatic free-cholesterol, nonesterified fatty acids, and triglycerides in wild-type and Nrf2-null mice after HFD feeding. At 4 weeks after HFD feeding, free-cholesterol, nonesterified fatty acids, and triglycerides were quantified in liver. Data are expressed as mean ± S.E.M. (each group; n = 5–10 animals). Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. Asterisks (*) represent statistically significant difference (p < 0.05) compared with either wild-type or Nrf2-null mice fed a ND, respectively; daggers (†) represent a statistically significant difference (p < 0.05) from the matched wild-type control mice within the same diet group.
Hepatic Malondialdehyde Levels in Wild-Type and Nrf2-Null Mice Fed a HFD. To evaluate the degree of lipid peroxidation, hepatic TBARS were quantified and reported as equivalents of MDA. Levels of MDA equivalents were unchanged in wild-type mice but were increased 36% in Nrf2-null mice compared with wild-type mice after feeding a HFD for 4 weeks (Fig. 3).
Based upon the aforementioned observations, subsequent experiments were designed to determine the influence of reduced (wild-type mice) and absent Nrf2 (Nrf2-null mice) on the regulation of genes involved in cholesterol and fatty acid metabolism and transport in response to a HFD. Altered expression of genes in these pathways may play a role in the observed differences in the patterns of serum and hepatic biomarkers in wild-type and Nrf2-null mice fed a HFD (Fig. 2).

Hepatic MDA equivalents in wild-type and Nrf2-null mice after HFD feeding. At 4 weeks after HFD feeding, TBARS were quantified in liver using an MDA standard curve, and data are expressed as mean MDA equivalents ± S.E.M. (each group, n = 5–10 animals). Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. Asterisks (*) represent statistically significant difference (p < 0.05) compared with either wild-type or Nrf2-null mice fed a ND, respectively; daggers (†) represent a statistically significant difference (p < 0.05) from the matched wild-type control mice within the same diet group.
Hepatic Expression of Cholesterol-Related Genes in Wild-Type and Nrf2-Null Mice Fed a HFD. Cholesterol levels are regulated by the balance of uptake, synthesis, catabolism, and export. SR-B1 is responsible for hepatic uptake of high-density lipoprotein cholesteryl esters. LDL receptor, HMG CoA synthase, and HMG CoA reductase are SREBP-2 target genes that are responsible for hepatic uptake and/or synthesis of cholesterol. There are no basal differences in the hepatic expression of SR-B1, LDL receptor, HMG CoA synthase, and reductase between genotypes. SR-B1 mRNA was increased 41% after feeding a HFD to wild-type mice for 1 week, and it was further up-regulated in Nrf2-null mice (Fig. 4). By 4 weeks of HFD feeding, expression of SR-B1 mRNA remained elevated only in Nrf2-null mice. Compared with vehicle-fed counterparts, LDL receptor mRNA expression was increased 80 and 83% in wild-type and Nrf2-null mice, respectively, after 1 week of a HFD diet, and it was further increased in Nrf2-null mice after feeding a HFD for 4 weeks (Fig. 4). mRNA expression of HMG CoA synthase and HMG CoA reductase was not changed in wild-type mice during the duration of the study, but it was elevated above control levels in Nrf2-null mice after feeding a HFD for 4 weeks by 164 and 291%, respectively (Fig. 4). Therefore, absent expression of Nrf2 in null mice is associated with enhanced levels of cholesterol synthesis and uptake genes in mice fed a HFD.
Genes responsible for the metabolism (Cyp7a1 and 8b1) and efflux (Abca1, Abcg5, and Abcg8) of cholesterol were also quantified. Compared with wild-type mice, the basal expression of Cyp8b1 was lower in Nrf2-null mice. There were no differences in the constitutive hepatic levels of Cyp7a1, Abca1, Abcg5, and Abcg8 between wild-type and Nrf2-null mice. Opposite regulation of the expression of Cyp7a1 and 8b1 genes was observed in mice fed a HFD (Fig. 5). Compared with control mice, Cyp7a1 mRNA was increased at least 150% in both genotypes after 4 weeks of a HFD diet. mRNA expression of Cyp8b1 was reduced 55 and 48% in wild-type mice fed a HFD for 1 and 4 weeks, respectively, and it was decreased 62% in Nrf2-null mice fed a HFD for 1 week. mRNA expression of Abca1, Abcg5, and Abcg8 was not changed in either genotype in response to HFD feeding for 1 or 4 weeks (data not shown). Data are summarized in Table 1.
Summary of high-fat diet-induced changes in fatty acid/cholesterol gene mRNA expression

Hepatic cholesterol uptake and synthesis mRNA expression in wild-type and Nrf2-null mice after HFD feeding. Total hepatic RNA was isolated from mice fed a ND and a HFD, and it was analyzed as described under Materials and Methods. Data are presented as mean ± S.E.M. (each group, n = 5–10 animals). Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. Asterisks (*) represent statistically significant difference (p < 0.05) compared with either wild-type or Nrf2-null mice fed a ND, respectively; daggers (†) represent a statistically significant difference (p < 0.05) from the matched wild-type control mice within the same diet group. Data are summarized in Table 1.
Hepatic Expression of Fatty Acid-Related Genes in Wild-Type and Nrf2-Null Mice Fed a HFD. The mRNA expression of the fatty acid transporters FAT/CD36 and L-FABP and the SREBP-1c target lipogenic enzymes FAS, ACC1, SCD1, and FAE were quantified. There were no basal differences in the expression of these genes between wild-type and Nrf2-null mice. mRNA levels of CD36 and SCD1 were not changed in either genotype after feeding a HFD for 1 or 4 weeks (data not shown). Compared with control mice, L-FABP mRNA was increased 159 and 115% in wild-type mice after consuming a HFD for 1 and 4 weeks, respectively (Fig. 6). L-FABP mRNA was also increased above control levels in Nrf2-null mice after a HFD for 1 or 4 weeks. Expression of L-FABP was higher in Nrf2-null mice compared with wild-type counterparts after 1 and 4 weeks of feeding a HFD. FAS mRNA was reduced 42 and 65% in wild-type mice after 1 and 4 weeks of a HFD, respectively, but it was unchanged from control levels in Nrf2-null mice. It is interesting to note that the pattern of gene regulation of FAS is similar to that of Nqo1, a target gene of Nrf2. Compared with normal diet fed genotype control mice, ACC1 mRNA was decreased in wild-type mice after 4 weeks on a HFD by 37%, but it was increased 49% in Nrf2-null mice after 1 week of a HFD. Gene expression of FAE increased above control levels only in Nrf2-null mice fed a HFD for 4 weeks. In general, expression of lipid synthesis and intracellular transport genes in mice consuming a HFD was higher in Nrf2-null mice compared with wild-type mice, consistent with the higher hepatic levels of fatty acids in the mutant mice (Fig. 2).

Hepatic cholesterol catabolism mRNA expression in wild-type and Nrf2-null mice after HFD feeding. Total hepatic RNA was isolated from mice fed a ND and a HFD, and it was analyzed as described under Materials and Methods. Data are presented as mean ± S.E.M. (each group, n = 5–10 animals). Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. Asterisks (*) represent statistically significant difference (p < 0.05) compared with either wild-type or Nrf2-null mice fed a ND, respectively; daggers (†) represent a statistically significant difference (p < 0.05) from the matched wild-type control mice within the same group. Data are summarized in Table 1.
Fatty acids are metabolized in mitochondria/peroxisomes and microsomes via β-oxidation and ω-oxidation, respectively. The mRNA expression of the fatty acid oxidation genes AOX, Cpt-1, Cyp4a10, and Cyp4a14 was determined. Nrf2-null mice exhibit higher basal expression of Cyp4a14 compared with wild-type mice. Constitutive levels of AOX, Cpt-1, and Cyp4a10 were similar between genotypes. AOX mRNA expression was unchanged by a HFD in both genotypes (data not shown). Compared with control mice, mRNA expression of Cpt-1 was decreased in wild-type mice after 1 and 4 weeks of a HFD by 35 and 30%, respectively, but it was unchanged in Nrf2-null mice (Fig. 7). Cyp4a10 mRNA decreased 66 and 53% from control levels in wild-type mice after 1 and 4 weeks of a HFD, respectively. Similar reductions in Cyp4a10 levels (70 and 41%) were observed in Nrf2-null mice after 1 and 4 weeks of feeding a HFD, respectively. Cyp4a14 mRNA decreased more markedly than Cyp4a10 in both genotypes after 1 and 4 weeks on a HFD. Although expression was lower than control mice, there were no differences in Cyp4a14 mRNA levels between genotypes at 1 and 4 weeks of HFD feeding. Reduced expression of fatty acid oxidation genes in response to a HFD is consistent with elevated serum and hepatic fatty acid levels observed in both genotypes (Fig. 2).
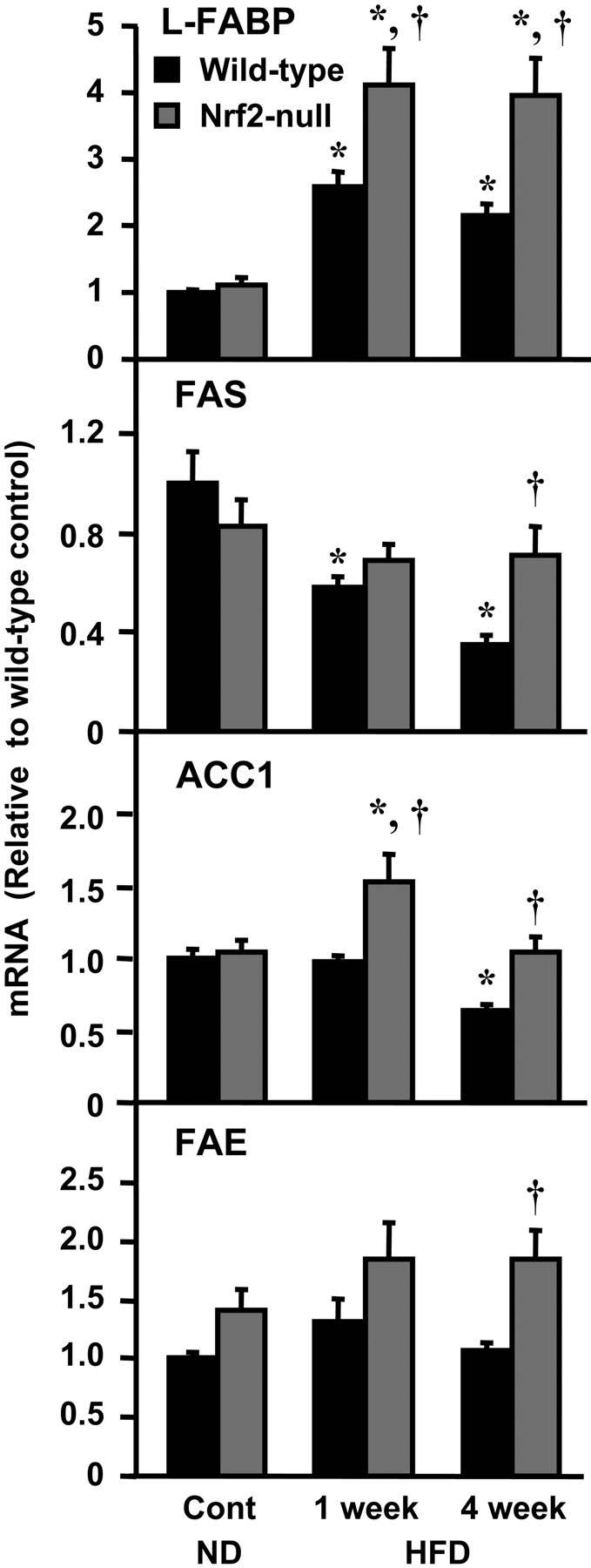
Hepatic fatty acid uptake and synthesis genes mRNA expression in wild-type and Nrf2-null mice after HFD feeding. Total hepatic RNA was isolated from mice fed a ND and a HFD, and it was analyzed as described under Materials and Methods. Data are presented as mean ± S.E.M. (each group, n = 5–10 animals). Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. Asterisks (*) represent statistically significant difference (p < 0.05) compared with either wild-type or Nrf2-null mice fed a ND, respectively; daggers (†) represent a statistically significant difference (p < 0.05) from the matched wild-type control mice within the same diet group. Data are summarized in Table 1.

Hepatic fatty acid oxidation genes and MTP mRNA expression in wild-type and Nrf2-null mice after HFD feeding. Total hepatic RNA was isolated from mice fed a ND and a HFD, and it was analyzed as described under Materials and Methods. Data are presented as mean ± S.E.M. (each group, n = 5–10 animals). Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. Asterisks (*) represent statistically significant difference (p < 0.05) compared with either wild-type or Nrf2-null mice fed a ND, respectively; daggers (†) represent a statistically significant difference (p < 0.05) from the matched wild-type control mice within the same diet group. Data are summarized in Table 1.
MTP is the rate-limiting step for the assembly and secretion of apolipoprotein B-containing lipoproteins. Basal expression of MTP was lower in livers from Nrf2-null mice, compared with wild-type mice. Relative to control mice, MTP mRNA was decreased 59 and 52% after 1 week of a HFD to wild-type and Nrf2-null mice, respectively (Fig. 7). Data are summarized in Table 1.
Hepatic Expression of Transcription Factor and Coregulator Genes in Wild-Type and Nrf2-Null Mice Fed a HFD. Similar transcriptional changes in cholesterol and fatty acid pathways were observed in mice with reduced (wild-type) and absent (Nrf2-null) Nrf2 expression. Nrf2 may directly regulate lipid homeostasis genes or indirectly by altering levels of lipid-related transcription factors and coregulators. Levels of SREBP-1c, SREBP-2, PPARα, PPARγ, PGC-1α and PGC-1 β mRNA were quantified. Only PPARγ demonstrated a basal difference in hepatic expression between wild-type and Nrf2-null mice. SREBP-1c mRNA increased 346 and 555% above control values in wild-type and Nrf2-null mice, respectively, when on a HFD for 1 week (Fig. 8). Compared with mice fed a normal diet, the HFD reduced SREBP-2 mRNA by 38% in wild-type mice at 1 week, which returned to control values by 4 weeks. After 4 weeks on the HFD, SREBP-2 mRNA increased 73% in Nrf2-null mice, compared with normal diet controls. PPARα mRNA similarly increased in both genotypes after 1 and 4 weeks on the HFD. Relative to control values, mRNA expression of PPARγ after 4 weeks on a HFD was increased 84% in wild-type and 74% in Nrf2-null mice. Compared with wild-type mice, HFD-induced expression of PPARγ was much lower in Nrf2-null mice. PGC-1α mRNA tended to decrease in wild-type mice fed a HFD for 1 or 4 weeks, but it was unchanged in Nrf2-null mice. Feeding a HFD did not change the mRNA expression of PGC-1β in wild-type mice. In contrast, PGC-1β mRNA was increased 70 and 143% above controls in Nrf2-null mice after feeding a HFD for 1 and 4 weeks, respectively (Fig. 8). LXRα mRNA levels were unchanged after feeding the HFD (data not shown).
Discussion
Nrf2 is activated in response to oxidative and electrophilic injury in an attempt to mitigate subsequent injury by upregulating expression of several detoxifying and antioxidant enzymes (Kobayashi et al., 2006). The absence of Nrf2 in genetically engineered mice increases susceptibility to toxicants, including acetaminophen, hyperoxia, benzo[a]pyrene, and dextran sulfate (Chan et al., 2001; Ramos-Gomez et al., 2001; Cho et al., 2002; Khor et al., 2006). Likewise, activation of Nrf2 either pharmacologically or genetically by deleting its sequestering partner Keap1 protects the liver from xenobiotic toxicity (Okawa et al., 2006; Farombi et al., 2007). Accumulating evidence suggests that Nrf2 may also have roles in lipid metabolism. For example, microarray analysis of hepatic genes in gallstone-susceptible (C57L/J) and gallstone-resistant (AKR/J) mice shows differential mRNA expression patterns, including many antioxidant, lipid peroxidation genes, and fatty acid metabolism-related genes. Gallstone-resistant mice have increased Nrf2 expression compared with gallstone-susceptible mice (Dyck et al., 2003). In addition, it has been shown that oxidized n-3 fatty acids react directly with Keap1, inducing Nrf2-directed gene expression (Gao et al., 2007). However, it is not known whether Nrf2 plays a role in lipid homeostasis in liver. To examine this, wild-type and Nrf2-null mice were fed a HFD for 1 and 4 weeks.
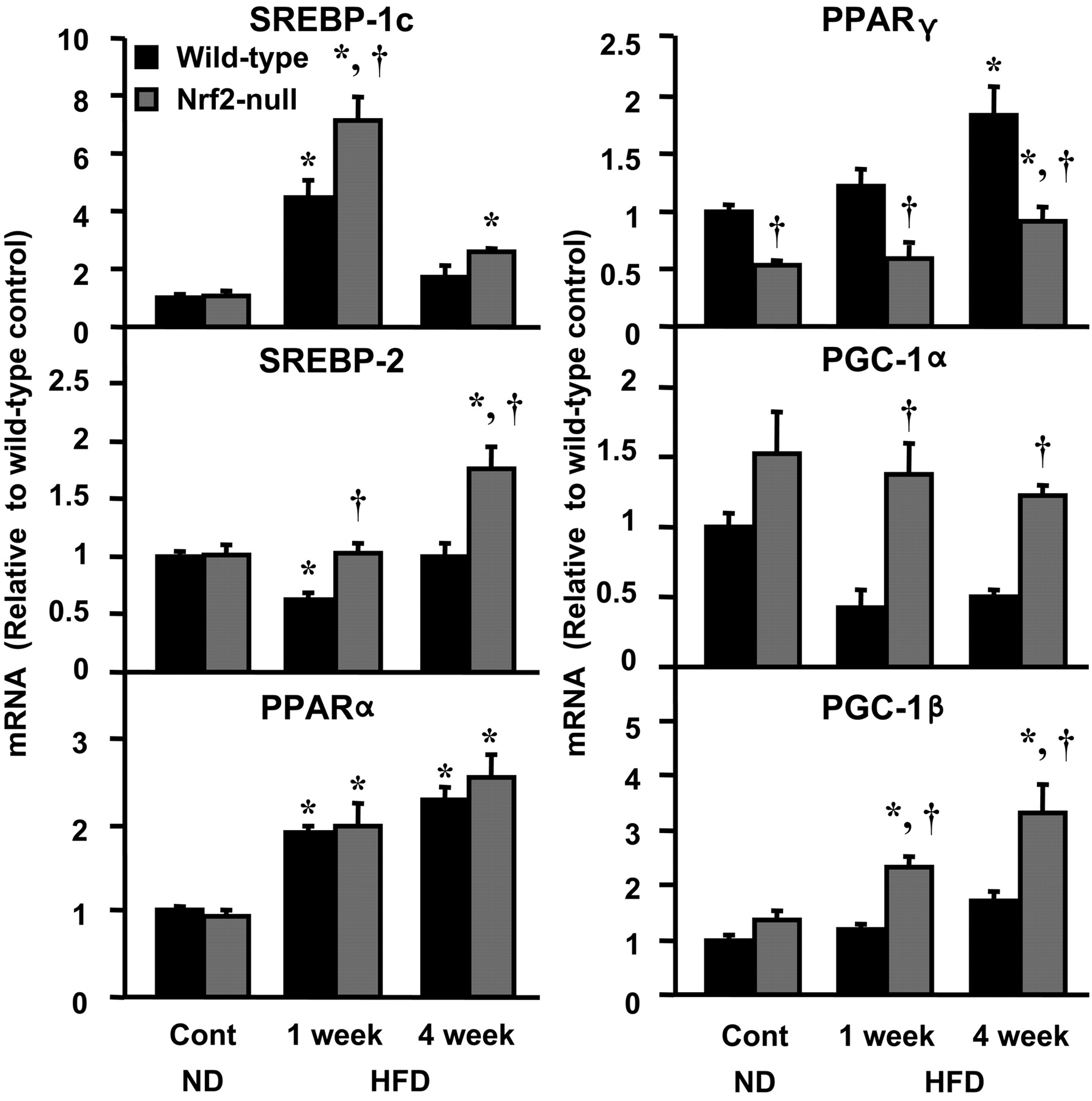
Coregulator and transcription factor mRNA expression in wild-type and Nrf2-null mice after HFD feeding. Total hepatic RNA was isolated from both mice fed a ND and a HFD, and it was analyzed as described under Materials and Methods. Data are presented as mean ± S.E.M. (each group, n = 5–10 animals). Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. Asterisks (*) represent statistically significant difference (p < 0.05) compared with either wild-type or Nrf2-null mice fed a ND, respectively; daggers (†) represent a statistically significant difference (p < 0.05) from the matched wild-type control mice within the same diet group.
The present study demonstrates for the first time that feeding a HFD reduces expression of Nrf2 and its target genes in mouse liver (Fig. 1). This is opposite of what is typically observed in rodent models of chemical-induced hepatotoxicity and hepatic carcinogenesis, in which Nrf2 is activated and the target gene Nqo1 is up-regulated (Chan et al., 2001; Okawa et al., 2006; Yates et al., 2006). It is not clear at present what biochemical events are responsible for Nrf2 down-regulation in response to a HFD. In a previous microarray analysis, a HFD (36% fat) increased Nrf2 and Nrf2 target genes, such as Gstm2 and Gstm6 (Kim et al., 2004). One explanation for this discrepancy may be due to the duration of feeding a HFD; 12 weeks in the study by Kim et al. (2004), whereas 1 and 4 weeks in this study. It is possible that induction of Nrf2 and its target genes by long-term feeding a HFD may be an adaptive response to oxidative stress induced by chronic fat accumulation.
Upon discovering that Nrf2 is down-regulated in wild-type mice after a HFD, subsequent experiments characterized serum and hepatic levels of cholesterol, fatty acids, triglycerides, and MDA equivalents. The HFD elevated serum cholesterol and free fatty acids in both genotypes, with a trend toward higher levels in wild-type mice. The absence of Nrf2 expression in the null mice was associated with greater increases in hepatic lipids and MDA equivalents (by-product of lipid peroxidation). Although the changes of mRNA expression do not necessarily point to functional endpoints, these data suggest that wild-type mice with reduced levels of Nrf2 were unable to prevent lipid accumulation in serum when fed a HFD, but that the absence of Nrf2 leads to more hepatic retention of lipids. Elevated MDA equivalents in Nrf2-null mice are probably secondary to fatty acid retention and generation of oxidative stress from mitochondria, cytochrome P450 2E1 in the endoplasmic reticulum, and peroxisomes in the liver.
Numerous genes are involved in the synthesis, uptake, catabolism, and export of cholesterol and fatty acids. Expression of these genes in wild-type and Nrf2-null mice fed a HFD was quantified in an attempt to reveal the biochemical mechanisms, possibly contributing to the observed serum and hepatic lipid profiles. Nrf2-null mice fed a HFD for 4 weeks exhibited higher mRNA levels of the cholesterol uptake and synthesis genes SR-B1, LDL receptor, HMG CoA synthase, and HMG CoA reductase compared with wild-type mice, suggesting that expression of these genes is inversely related to the amount of Nrf2. The transcription factor SREBP-2 regulates expression of LDL receptor, HMGCoA reductase, and HMGCoA synthase (Cuthbert and Lipsky, 1992). In the present study, expression of SREBP-2 was higher in Nrf2-null mice after 1 and 4 weeks of a HFD. It is attractive to speculate that SREBP-2 is activated upon consuming a HFD, leading to increased transcription of cholesterol synthesis genes preferentially in Nrf2-null mice. However, this hypothesis needs to be tested to establish a causal relationship. Despite higher expression of cholesterol synthesis genes in Nrf2-null mice fed a HFD, there was no difference in hepatic cholesterol levels (Fig. 2). Longer exposure to a HFD (>4 weeks) or an atherogenic diet might elevate hepatic cholesterol levels in Nrf2-null mice, with minimal change in wild-type mice.
It is interesting to note that the mRNA expression of Cyp7a1 and Cyp8b1 is oppositely regulated when consuming a HFD. Cyp7a1 is the rate-limiting enzyme in synthesizing the two primary bile acids, cholic acid and chenodeoxycholic acid. Cyp8b1 catalyzes the 12-hydroxylation of chenodeoxycholic acid to form cholic acid. The mechanism of the opposite regulation of these two genes by a HFD is unknown.
LXR contributes to the regulation of hepatic cholesterol homeostasis (Repa et al., 2002). However, expression of LXRα and its target genes (Abca1, Abcg5, and Abcg8) were unchanged in both genotypes after a HFD, suggesting little involvement of this nuclear receptor in regulating cholesterol excretion in the present study (data not shown).
As shown in Fig. 7, expression of fatty acid oxidation genes (Cpt-1, Cyp4a10, and Cyp4a14) was reduced to a similar extent in both genotypes after a HFD, which may have contributed in part to the elevated serum and hepatic fatty acid levels, by preventing their catabolism. In contrast, fatty acid synthesis gene expression (FAS, ACC1, FAE) was higher in Nrf2-null mice after 4 weeks on a HFD compared with their wild-type counterparts. Differential regulation of these genes in the absence of Nrf2 may be responsible for higher hepatic free fatty acid concentrations in Nrf2-null mice fed the high-fat diet, although this hypothesis still needs to be proven experimentally.
FAS, ACC1, and FAE genes are known to be positively regulated by SREBP-1c (Bené et al., 2001; Moon et al., 2001; Matsuzaka et al., 2002). Although the exact profiles of SREBP-1c, FAS, ACC1, and FAE mRNA do not parallel each other, there is a general trend of higher expression of these genes in the Nrf2-null mice. It is interesting to note that FAS and Nrf2 mRNA decreased in wild-type mice fed a HFD over time, suggesting that Nrf2 participates in FAS regulation, or that both of these two genes are under the control of another transcription factor.
Up-regulation of ACC1 and FAE in Nrf2-null mice on a HFD may also be regulated by other transcription factors. LXR and the coactivator PGC-1α can regulate ACC1 expression (Oberkofler et al., 2003; Talukdar and Hillgartner, 2006). LXRα mRNA was unchanged in the present study after feeding a HFD to both genotypes (data not shown). Therefore, regulation of ACC1 mRNA in response to a HFD may be affected by both SREBP-1c and PGC-1α. Likewise, FAE is also known to be regulated by SREBP-2 (Moon et al., 2001; Matsuzaka et al., 2002). Thus, regulation of FAE mRNA while consuming a HFD could be mediated by both SREBP-1c and SREBP-2.
CD36 and L-FABP are involved in fatty acid uptake. Recent studies showed that increased expression of these two genes plays a major role in steatosis after consumption of a diet rich in polyunsaturated fats (Baumgardner et al., 2008). In the present study, CD36 was unchanged in mice fed a HFD. Prior research has shown that PGC-1β interacts with PPARα and activates the transcription of L-FABP (Spann et al., 2006). In the present study, mRNA of these three genes were increased in response to a HFD, suggesting that PGC-1β and PPARα may be in part regulating L-FABP induction in mice with reduced (wild-type) and no (Nrf2-null) expression of Nrf2 (Figs. 6 and 8). Additional experiments are necessary to establish causal roles of these transcription factors in regulating L-FABP overexpression in this model. In contrast, two known PPARα target genes (Cyp4a10 and Cyp4a14) were decreased in both genotypes in response to a HFD (Fig. 7). This is contrary to previous research that showed up-regulation of these fatty acid oxidation genes after feeding a HFD for 12 weeks (Kim et al., 2004; Patsouris et al., 2006). Similar to Nrf2 target genes, regulation of Cyp4a isoforms in mice fed a HFD may be biphasic with reduced expression initially (up to 4 weeks) and up-regulation with longer feeding (beyond 4 weeks).
MTP participates in the assembly and secretion of triglyceride-rich, apolipoprotein B-containing lipoproteins, such as VLDL and chylomicrons. MTP is constitutively lower in Nrf2-null mice, and it is reduced by feeding a HFD, which may predispose these mice to accumulation of lipoproteins. Hepatic regulation of MTP occurs via activation of PPARα (Améen et al., 2005). However, in the present study, PPARα mRNA was induced in both wild-type and Nrf2-null mice on a HFD diet. These opposing gene expression profiles suggest that alternate transcription factors (possibly Nrf2) may regulate MTP mRNA levels.
PGC-1α is a major regulator of oxidative metabolism and mitochondrial biogenesis, and it is involved in the transcriptional regulation of the mitochondrial antioxidant defense system in vascular endothelial cells (Valle et al., 2005). It should be noted that a HFD tended to decrease PGC-1α mRNA in wild-type mice, but it did not change levels in Nrf2-null mice, indicating that the down-regulation of PGC-1α is dependent upon Nrf2 expression (Fig. 8). Previous research has also demonstrated that a HFD decreases PGC-1α in mouse skeletal muscle fed a HFD for 3 weeks (Sparks et al., 2005).
Oxidative stress plays an important role in the transition of steatosis to NASH (Sakaida and Okita, 2005). An early decline in Nrf2 expression upon feeding a HFD probably enhances this progression. Additional research is necessary to determine whether pharmacological activation of Nrf2 in mice fed a HFD can prevent the decline in Nrf2 mRNA expression, interfere with lipogenesis, and block the transition from fatty liver to NASH.
In summary, these results show that a HFD decreases expression of Nrf2 and its target genes in wild-type mice. In addition, reduced (wild-type mice) and no (Nrf2-null mice) expression of Nrf2 is associated with elevated hepatic and serum cholesterol and fatty acid levels during feeding a HFD. Hepatic MDA equivalents were higher in Nrf2-null mice than wild-type mice after feeding a HFD, suggesting that Nrf2 inhibits oxidative stress that might be induced by free fatty acid accumulation. Complete absence of Nrf2 function was associated with induction of genes involved in lipogenic and cholesterologenic pathways, as well as coregulators (SREBP-1c, SREBP-2, PGC-1α, and PGC-1β). Taken together, these data suggest that Nrf2 may be a novel therapeutic target for the prevention and treatment of fatty liver and NASH.
Acknowledgments
We thank Youcai Zhang for technical assistance.
Footnotes
-
This work was supported by National Institutes of Health Grants ES09649, ES09716, ES07079, and RR021940.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.107.135822.
-
ABBREVIATIONS: NASH, nonalcoholic steatohepatitis; SREBP, sterol regulatory element-binding protein; FAS, fatty acid synthase; ACC-1, acetyl-CoA carboxylase 1; FAE, fatty acid elongase; SCD1, stearoyl-CoA desaturase-1; PPAR, peroxisome proliferator-activated receptor; AOX, acyl-CoA oxidase; Cpt-1, carnitine palmitoyltransferase-1; Cyp4a, cytochrome P450 4a; Cyp4a10, cytochrome P450 4a10; Cyp4a14, cytochrome P450 4a14; LDL, low-density lipoprotein; LXR, liver X receptor; Cyp7a1, cholesterol 7-α hydroxylase; Cyp8b1, sterol 12-α hydroxylase; MTP, microsomal triglyceride transfer protein; VLDL, very low-density lipoprotein; Nrf2, NF-E2-related factor 2; Keap1, Kelch-like ECH-associated protein 1; GST, glutathione transferase; Nqo1, NAD(P)H:quinone oxidoreductase 1; HFD, high-fat diet; MDA, malondialdehyde; ND, normal diet; PCR, polymerase chain reaction; PGC, peroxisome proliferator-activated receptor γ coactivator; TBARS, thiobarbituric acid reactive substances; L-FABP, liver fatty acid-binding protein; Gstm6, Gst mu 6; SR-B1, scavenger-receptor class B, type 1; FAT, fatty acid translocase.
-
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material. - Received December 21, 2007.
- Accepted February 14, 2008.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}