


Abstract
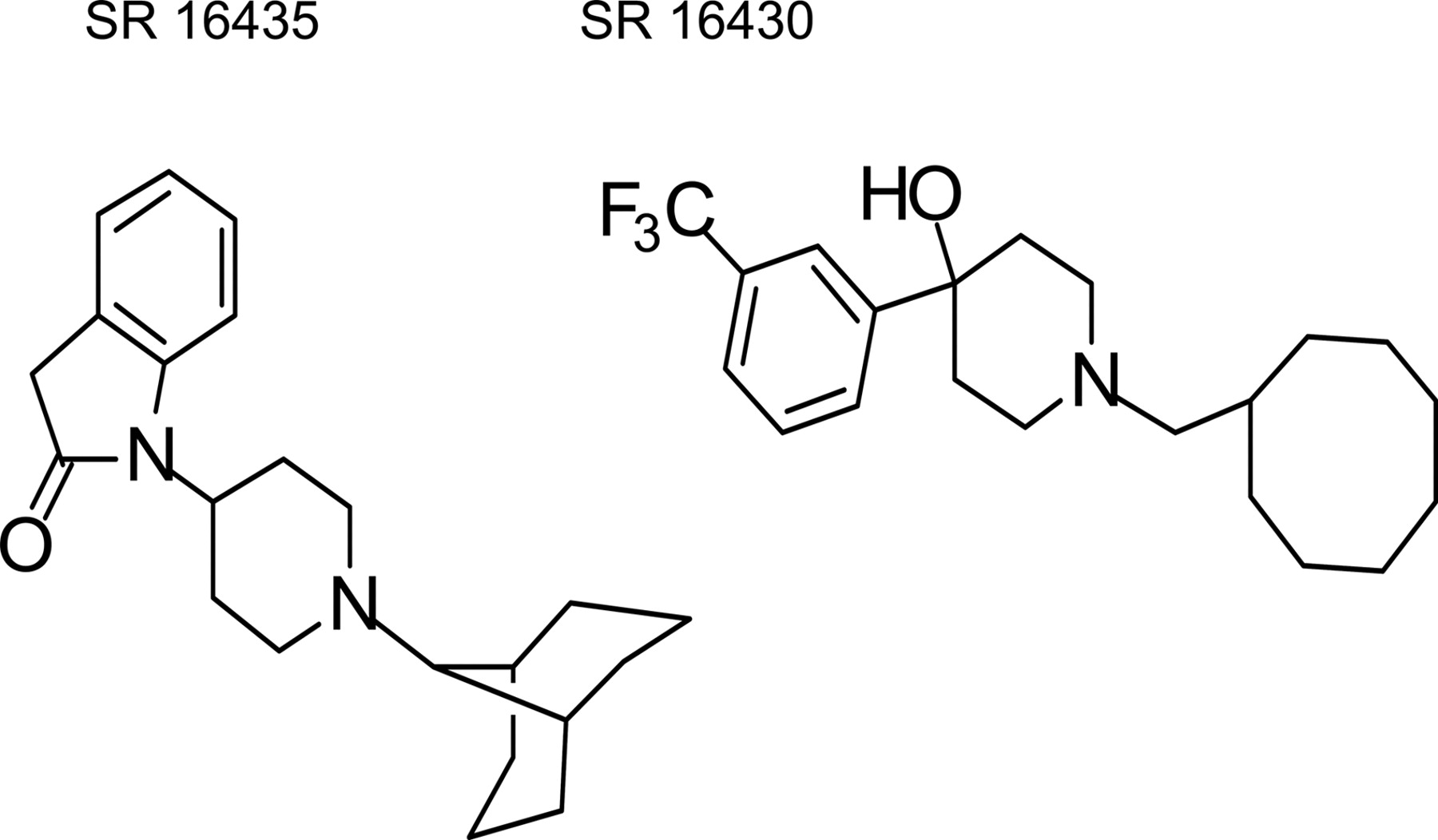
We identified a novel nociceptin/orphanin FQ (NOP)/μ-opioid receptor agonist, SR 16435 [1-(1-(bicyclo[3.3.1]nonan-9-yl)piperidin-4-yl)indolin-2-one], with high binding affinity and partial agonist activity at both receptors. It was hypothesized that SR 16435 would produce antinociception and yet, unlike morphine, would have diminished rewarding properties and tolerance development. Antinociception was assessed in mice using the tail-flick assay, whereas behavioral and rewarding effects were assessed using the place conditioning (PC) paradigm. PC was established by pairing drug injections with a distinct compartment. Behavioral effects were measured after acute and repeated drug administration, and the test for PC was carried out 24 h after four drug- and vehicle-pairing sessions. SR 16435 produced an increase in tail-flick latency, but SR 16435-induced antinociception was lower than that observed with morphine. Given that naloxone blocked SR 16435-induced antinociception, it is highly likely that this effect was mediated by μ-opioid receptors. Compared with morphine, chronic SR 16435 treatment resulted in reduced development of tolerance to its antinociceptive effects. SR 16435-induced conditioned place preference (CPP) was evident, an effect that was probably mediated via μ-opioid receptors, as it was reversed by coadministration of naloxone. NOP agonist activity was also present, given that SR 16435 decreased global activity, and this effect was partially reversed with the selective NOP antagonist, SR 16430 [1-(cyclooctylmethyl)-4-(3-(trifluoromethyl)phenyl)piperidin-4-ol]. Naloxone, however, also reversed the SR 16435-induced decrease in activity, indicating that both opioid and NOP receptors mediate this behavior. In summary, the mixed NOP/μ-opioid partial agonist SR 16435 exhibited both NOP and μ-opioid receptor-mediated behaviors.
The NOP receptor [formerly known as the opioid receptor-like receptor (ORL1)] was discovered simultaneously by several groups on the basis of homology with the δ-opioid receptor (Bunzow et al., 1994; Mollereau et al., 1994; Wang et al., 1994). This receptor has nucleotide and amino acid homology to the three opioid receptors; however, NOP receptors transfected into mammalian cells do not seem to bind opiates with high affinity. N/OFQ, the natural ligand for the NOP receptor, is a 17-amino acid neuropeptide (Meunier et al., 1995; Reinscheid et al., 1995). Although this peptide is in the opioid peptide family, N/OFQ has a different behavioral profile than that of typical opioid ligands. N/OFQ modulates nociception differentially, depending upon site of administration. N/OFQ delivered i.c.v. has been shown to have no effect or to be pronociceptive and can block morphine-induced analgesia (Meunier et al., 1995; Reinscheid et al., 1995; Mogil et al., 1996; Tian et al., 1997). However, i.t. administration of N/OFQ produces antinociception and potentiates morphine analgesia (Tian et al., 1997).
This complicated profile of N/OFQ and its receptor, NOP, in pain has been further examined using novel selective agonists and antagonists. High-affinity and selective peptide antagonists such as [Nphe]N/OFQ(1–13)NH2 and UFP-101 are antinociceptive when injected i.c.v. in acute thermal pain models (Calo' et al., 2000, 2002). High-throughput screening and classic medicinal chemistry have led to the identification of several small-molecule ligands that are active after systemic administration and could potentially be developed as analgesic medications for therapeutic use (Zaveri, 2003). Systemic administration of the highly selective NOP antagonists J-113397 and SB-612111 has no effect on acute thermal pain (Ozaki et al., 2000; Zaratin et al., 2004), whereas the nonselective antagonist JTC-801 (only 4-fold selective for NOP relative to μ-opioid receptors) produced naloxone-resistant antinociception (Shinkai et al., 2000; Yamada et al., 2002). These discrepancies could be due to different selectivities for the NOP versus opioid receptors and/or pharmacokinetic distribution in vivo. Ro 64-6198, an NOP agonist with 100-fold selectivity for NOP receptors over μ-opioid receptors, possesses anxiolytic activity but has no analgesic activity alone and inhibits morphine-induced antinociception when given systemically (Jenck et al., 2000; Kotlinska et al., 2003).
Extended treatment with opioid analgesics is usually accompanied by undesirable side effects such as tolerance development and dependence. The role of the NOP receptor in modulating these opioid receptor-mediated effects has been examined previously. N/OFQ, administered i.c.v., attenuates development of tolerance to morphine antinociception (Lutfy et al., 2001). Small-molecule NOP antagonists J-113397 (i.t. administration) and SB-612111 (i.v. administration) also block morphine-induced tolerance to antinociception (Ueda et al., 2000; Zaratin et al., 2004). In addition, i.c.v. administration of N/OFQ and systemic administration of the NOP agonist Ro 64-6198 attenuate the rewarding effects of morphine in the PC paradigm (Murphy et al., 1999; Shoblock et al., 2005). Although NOP receptor agonists can attenuate morphine-induced CPP, they do not induce CPP or CPA when administered alone (Devine et al., 1996; Le Pen et al., 2002).
Therefore, it seems that stimulation of NOP receptors can reduce morphine-induced tolerance development and reward. Because μ-opioid receptor stimulation produces powerful antinociception, a compound that stimulates both NOP and μ-opioid receptors may act as a potent analgesic but be less susceptible to the development of abuse liability and tolerance to antinociception. In the course of our program to develop NOP ligands, we have synthesized compounds showing agonist activity at both NOP and μ-opioid receptors. SR 16435 is a dihydroindolinone-based small-molecule ligand with high binding affinity at both NOP and μ-opioid receptors (Zaveri et al., 2004). In the [35S]GTPγS functional assay, SR 16435 is a potent partial agonist at both sites (see Table 1). Buprenorphine, a high-affinity μ-opioid partial agonist has significant antinociceptive potency with reduced addiction liability (Jasinski et al., 1978; Jacob et al., 1979). Accordingly, we sought to test the hypothesis that a μ-opioid partial agonist such as SR 16435, with equally high affinity for both NOP and μ-opioid receptors and partial agonist activity at both receptors, could have a particularly beneficial behavioral profile.
Binding affinities and functional activities of SRI International compounds at NOP and opioid receptors compared with N/OFQ, buprenorphine, and morphine
Receptor binding and [35S]GTPγS binding was measured in Chinese hamster ovary cell membranes containing the appropriate human receptor, as described in Dooley et al. (1997). Binding constants are shown as Ki ± S.D. values for each compound, which are derived from at least two individual experiments conducted in triplicate. An EC50 value of >10,000 for [35S]GTPγS binding indicates no significant stimulation at that concentration.
In the following experiments we examined whether SR 16435, a mixed NOP/μ-opioid partial agonist, possesses analgesic activity with reduced tolerance development and diminished reward in mice. The tail-flick assay was used to examine acute and repeated effects of SR 16435 on thermal nociception. The PC paradigm was used to assess whether SR 16435 had any rewarding properties and to determine how similar and/or different the behavioral profile was relative to that of morphine.
Experimental Procedures
Animals
Male ICR mice weighing 20 to 25 g at the start of the experiment were used. Animals were group-housed under standard laboratory conditions and were kept on a 12:12-h day/night cycle (lights on at 8:00 AM). Animals were handled for at least 2 to 3 days before the experiments were conducted. For thermal nociception experiments, animals were transported to the testing room and acclimated to the environment for 1 h. Mice were maintained in accordance with the guidelines of SRI International and of the Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research (National Research Council, 2003).
Drugs
SR 16435 [1-(1-(bicyclo[3.3.1]nonan-9-yl)piperidin-4-yl)indolin-2-one] and SR 16430 [1-(cyclooctylmethyl)-4-(3-(trifluoromethyl)phenyl)piperidin-4-ol] were synthesized in our laboratories, as hydrochloride salts (Zaveri et al., 2004). SR 16435, morphine hydro-chloride (Eli Lilly & Co., Indianapolis, IN), and naloxone (Sigma-Aldrich, St. Louis, MO) were dissolved in water. SR 16430 was dissolved in 1% dimethyl sulfoxide and 0.5% aqueous hydroxypropylcellulose. Drugs were injected in a volume of 0.1 ml/25 g (s.c.). Controls received 0.1 ml/25 g of the appropriate vehicle.
Assessing Acute Thermal Nociception
Tail-Flick Assay. Acute nociception was assessed using the tail-flick assay with an analgesia instrument (Stoelting, Wood Day, IL) that uses radiant heat. This instrument is equipped with automatic quantification of tail-flick latency, and a 15-s cutoff to prevent damage to the animal's tail. During testing, the focused beam of light was applied to the lower half of the animal's tail, and tail-flick latency was recorded. Baseline values for tail-flick latency were determined before drug administration in each animal. The mean ± S.E.M. basal tail-flick latency was 5.1 ± 0.1 s.
After baseline measures, animals received s.c. injections of their assigned dose of drug and were tested for tail-flick latencies at 10, 30, and 60 min postinjection. Animals that were controls received an injection of vehicle before testing.
Drug Regimen. Animals (n = 10–12/group) received s.c. injections of SR 16435 alone (3–30 mg/kg) or vehicle. Higher doses of SR 16435 were not tested because of drug insolubility. In a follow-up experiment to examine whether the effects of SR 16435 were reversed by naloxone, animals (n = 10/group) received s.c. injections of SR 16435 (10 and 30 mg/kg) coadministered with naloxone (1 mg/kg). The dose of naloxone was chosen because it has been shown to reverse the effects of morphine.
To compare the effects of SR 16435 with those of morphine, a morphine dose-response curve for acute thermal antinociception was conducted using parameters similar to those described above. Animals (n = 10–12/group) received s.c. injections of morphine alone (1–30 mg/kg) or vehicle.
Statistical Analyses. Antinociception [% maximal potential effect (%MPE)] was quantified by the following formula: % MPE = 100 × [(test latency – baseline latency)/(15 – baseline latency)]. If the animal did not respond before the 15-s cutoff, the animal was assigned a score of 100%.
Behavioral results were analyzed using ANOVAs with drug treatment (SR 16435, morphine, and naloxone) as between-group variables and postdrug injection time (10, 30, and 60 min) as the repeated measure followed by Student Newman-Keuls post hoc tests where appropriate. The level of significance was set at P < 0.05.
Assessing Tolerance Development to Thermal Antinociception
Injection Regimen and Testing. Animals received s.c. injections of either vehicle, SR 16435 (30 mg/kg, n = 15), or morphine (10 mg/kg, n = 10) once daily for 9 days. The 30 mg/kg dose of SR 16435 was chosen because it produced the greatest level of antinociception of the doses tested, whereas the 10 mg/kg dose of morphine was chosen because it produced the same levels of antinociception as 30 mg/kg SR 16435. On days 1 to 5 and 9, animals were tested for tail-flick latencies using the equipment and methods described above. Baseline tail-flick latencies were taken. The mean ± S.E.M. basal tail-flick latencies were consistent across test days (day 1, 6.0 ± 0.2 s; day 2, 5.7 ± 0.1 s; day 3, 5.5 ± 0.2 s; day 4, 5.4 ± 0.2 s; day 5, 5.5 ± 0.2 s; and day 9, 5.3 ± 0.2 s). Animals then received an injection of their assigned dose of drug and were tested once 30 min after their injection. The 30-min time point was chosen because the same effects were seen at the 30- and 60-min testing time points (in the acute analgesia assessment described above).
Statistical Analyses. %MPE was calculated as described above, and a repeated ANOVA with drug treatment (morphine and SR 16435) as the between-group variable and injection day (days 1 to 5 and 9) as the repeated measure was used. To examine tolerance development, after a significant overall and repeated-measures ANOVAs (for each drug tested), paired t tests were used to compare data obtained on days 2 to 5 and 9 with data from day 1. To compare drug effects, after significant effects, Student Newman-Keuls post hoc tests were performed as described above. The level of significance was set at P < 0.05.
Assessing the Rewarding Effects and Behavioral Effects of SR 16435 Using the PC Paradigm
PC Apparatus. The apparatus consisted of rectangular Plexiglas chambers divided into two distinct equal-sized compartments (19 cm × 2.8 cm × 18 cm high, Lafayette Instruments, Lafayette, IL). One compartment had cedar-scented bedding underneath a bar grid floor, and all walls but the front wall were black. The other compartment had pine-scented bedding beneath a mesh floor, and all walls but the front wall were white. The front walls were transparent so that the animal's behavior was monitored. A removable partition divided the two compartments. During conditioning, the compartments were divided by a solid partition. On the PC test day, the solid partition was replaced with a partition that had an opening allowing the animal free access to both compartments. A video camera that was linked to a computer was mounted above the chambers and tracked the animals' movement. Previous experiments using this experimental setup have indicated that the apparatus is unbiased as animals do not show a preference for one compartment over the other (T. V. Khroyan et al., unpublished data).
PC Training. Conditioning trials were composed of two sessions conducted over 2 consecutive days. During the drug session, animals received s.c. injections of their respective drug and were confined to one of the compartments for 20 min. On the other day, the vehicle session, animals received injections of saline and were confined to the alternate compartment for 20 min. In addition, a group of mice received saline injections in both compartments and served as controls. These two sessions were repeated over 8 consecutive days such that animals received four drug sessions and four vehicle sessions. The particular compartment paired with the drug and the order of placement into the drug-paired versus saline-paired compartment were counterbalanced across groups.
Acute and Repeated Behavioral Measures. During conditioning, the behavioral effects of acute and repeated drug injection were defined as follows: sniffing—directed sniffing of the floor or wall; rearing—the animal's front two paws being off the ground; and grooming—animal licking/biting or brushing itself. Each effect was measured by recording its presence every 10 s throughout the session. These measures were recorded immediately after the first and last drug administration by an observer unaware of the animals' treatment group. The overall global activity of the animals was captured by the Spontaneous Motor Recording and Tracking software system (SMART; Panlab S.L., Barcelona, Spain), a color image-capturing system that works in real time and can track the animal's behavior for a given amount of time via a video camera connected to the computer.
PC Test Day. Twenty-four hours after the last conditioning session, the animals were given access to both compartments simultaneously for 15 min, and the amount of time the animals spent in each compartment was recorded by the SMART tracking system.
Dose Regimen. Animals were assigned to groups receiving s.c. injections of either vehicle (n = 15), morphine (15 mg/kg; n = 8), or one of two doses of SR 16435 (10 or 30 mg/kg; n = 8/group) immediately before placement in the drug-paired compartment. Because SR 16435 produced a significant CPP, a follow-up experiment examined whether naloxone could block the effects of SR 16435. Animals (n = 8/group) were randomly assigned to groups receiving 10 mg/kg SR 16435 alone or coadministered with 1 mg/kg naloxone (s.c.). Another group of animals served as controls and received vehicle injections. PC testing was carried out as described above.
Given that acute administration of SR 16435 suppressed global activity, an effect possibly involving NOP receptor activation, we tested whether SR 16430 (a NOP antagonist) (see Fig. 1; Table 1) could reverse the suppressive effects of SR 16435. Animals (n = 6–8/group) were randomly assigned to groups receiving 10 mg/kg SR 16435 alone or coadministered with 30 mg/kg SR 16430 (s.c.). Another group of animals served as controls and received vehicle injections. Animals received their respective injection of drug and were immediately tested for global activity in the PC apparatus for 20 min.
Statistical Analyses. Drug-induced behaviors were analyzed using ANOVAs with SR 16435, morphine, naloxone, and SR 16430 as between-subjects measures and injection day (first versus fourth) as a repeated measure. Significant interactions were further analyzed with one-way ANOVAs and post hoc tests. To examine sensitization effects, after a significant overall ANOVA, t tests were used to compare data after the fourth injection relative to the first. For the PC test day data, the percentage of time animals spent in their drug-paired compartment relative to saline-treated controls was analyzed using one-way ANOVAs, and significant effects were further analyzed with post hoc tests. A CPP was evident if animals spent significantly more time in their drug-paired compartment relative to control animals, whereas a CPA was evident if animals spent significantly less time in their drug-paired compartment. The level of significance was set at P < 0.05.
Results
Chemical structures of the NOP partial agonist SR 16435 and antagonist SR 16430 used in this study are shown in Fig. 1. Table 1 shows in vitro binding affinities and functional activities of these compounds, as well as morphine and N/OFQ, at the NOP, μ-opioid, and κ-opioid receptors. As seen in Table 1, SR 16435 has high affinity at both NOP and μ-opioid receptors and partial agonist activity, as measured by [35S]GTPγS stimulation, at both sites. In contrast, SR 16430 has ∼10-fold selectivity for the NOP receptor over μ-opioid receptors and is an antagonist at the NOP receptor.

Structures of SR 16435 and SR 16430.
Effects of SR 16435 on Tail-Flick Latency and Reversal by Coadministration of Naloxone. The effects of SR 16435 on tail-flick latency are shown in Fig. 2A. The overall ANOVA indicated that there was no significant interaction effect, and only the main effect of drug [F(3,48) = 9.8, P < 0.05] was significant, indicating that regardless of postdrug injection time (10, 30, and 60 min) SR 16435 produced the same effects. Thus, looking at the dose effects averaged across postdrug injection time, post hoc tests indicated that SR 16435 (10 and 30 mg/kg) produced significant increases in tail-flick latency relative to vehicle controls (P < 0.05). SR 16435 produced a dose-dependent increase in tail-flick latency because animals that received the 30 mg/kg dose produced a greater increase in tail-flick latency relative to animals that received the 10 mg/kg dose (P < 0.05).
The effects of morphine on tail-flick latency are shown in Fig. 2B. The overall ANOVA indicated that there was no significant interaction effect, and only the main effect of dose [F(5,62) = 32.8, P < 0.05] was significant, indicating that regardless of postdrug injection time (10, 30, and 60 min) the effects of morphine did not change. Thus, looking at the dose effects averaged across postdrug injection time, post hoc tests indicated that 3 to 30 mg/kg morphine produced significant dose-dependent increases in tail-flick latency relative to vehicle (P < 0.05). Compared with 15 and 30 mg/kg morphine, antinociception produced by SR 16435 never reached maximal levels (Fig. 2).
The antinociceptive activity of SR 16435 was blocked by the opioid antagonist naloxone (Fig. 3), suggesting that this activity was mediated by the μ-opioid component of SR 16435. The overall ANOVA indicated that there was a significant SR 16435 × naloxone effect [F(2,70) = 3.1, P < 0.05] regardless of postinjection time (10, 30, or 60 min). Averaged across postinjection time, naloxone significantly decreased SR 16435-induced antinociception at both doses of SR 16435 tested (10 mg/kg, F(1,28) = 11.28, P < 0.05; 30 mg/kg, F(1,24) = 8.0, P < 0.05). Naloxone administered alone did not alter tail-flick latency because animals that received naloxone exhibited similar levels of %MPE relative to vehicle control animals (N.S., P > 0.05).
Effects of SR 16435 on Tolerance Development to Thermal Antinociception. The effect of SR 16435 on thermal antinociception across repeated test days is shown in Fig. 4. The overall ANOVA indicated a significant drug × injection day interaction [F(10, 185) = 2.64, P < 0.05]. Examining the one-way ANOVAs for each day tested (days 1 to 5 and 9), we found a significant effect of dose. On test days 1 to 9, SR 16435 (30 mg/kg) produced an increase in tail-lick latency that was significantly greater than that observed in vehicle controls, whereas morphine produced an increase in tail-flick latency relative to controls on days 1 to 5 (P < 0.05) but not on day 9. To examine the effects of tolerance development, levels of %MPE on test days 2 to 5 and 9 were compared with those on the first test day. As expected, tolerance developed to the antinociceptive effects of 15 mg/kg morphine [F(5, 45) = 4.3, P < 0.05]. Compared with day 1 (63% MPE), morphine produced a significant reduction in %MPE on days 2, 4, 5, and 9 (P < 0.05; 15% MPE by last test day). SR 16435 also produced a significant decrease in %MPE across days [F(5,70) = 2.9, P < 0.05]; however, there was only a significant difference for days 5 and 9 compared with day 1 (P < 0.05). The decrease in antinociception observed after repeated administration of SR 16435 was not as dramatic as that for morphine (day 1 = 62.5% MPE; day 9 = 34.5% MPE).
Effects of SR 16435 on Place Conditioning. The effect of SR 16435 on place conditioning is shown in Fig. 5A. The overall ANOVA indicated a significant main effect of dose [F(3, 34) = 8.22, P < 0.05]. Animals that received morphine and 10 and 30 mg/kg SR 16435 exhibited a significant CPP (P < 0.05) relative to vehicle controls. The 10 mg/kg dose of SR 16435 was also tested in combination with naloxone, and the results indicated that naloxone not only blocked the rewarding effects of SR 16435 but also produced a significant CPA when coadministered with SR 16435 (P < 0.05) (Fig. 6A).
Behavioral Effects of SR 16435 After Acute and Repeated Administration. The effect of SR 16435 on global activity is shown in Fig. 5B. The overall ANOVA indicated that there was a significant dose × injection day interaction [F(3,35) = 6.45, P < 0.05]. As expected, morphine administration produced an increase in global activity after the first injection relative to vehicle controls (P < 0.05). Furthermore, sensitization of morphine-induced global activity was also evident as an increase in activity after the fourth injection relative to the first injection (P < 0.05). Conversely, SR 16435 (10.0 mg/kg) administration produced a significant decrease in global activity relative to vehicle controls after the first injection (P < 0.05) that was not evident after the fourth drug injection (P > 0.05).

The effects of a range of doses of SR 16435 (A) and morphine (B) on tail-flick latency compared with vehicle controls. Data are mean %MPE ± S.E.M. *, P < 0.05, significant difference from vehicle controls (Student Newman-Keuls test). SC, subcutaneous.
The effect of naloxone on the SR 16435-induced decrease in global activity is shown in Fig. 6B. The overall ANOVA indicated that there was a significant drug × injection day interaction [F(2,27) = 5.3, P < 0.05]. After the first injection, naloxone coadministered with SR 16435 did not produce a decrease in activity and in fact produced a significant increase in activity relative to vehicle controls (P < 0.05). After the fourth injection, the effects of SR 16435 alone and coadministered with naloxone were not significantly different from vehicle controls.
Further experiments were performed to examine whether the acute suppression of global activity induced by SR 16435 was an NOP receptor-mediated effect. The effect of the NOP antagonist SR 16430 on the SR 16435-induced decrease in global activity is given in Table 2. The overall ANOVA indicated that there was a significant SR 16435 × SR 16430 interaction [F(1,23) = 5.8, P < 0.05]. After acute drug injection, the suppressive effects of SR 16435 were partially reversed by SR 16430. SR 16435 coadministered with SR 16430 produced an increase in global activity relative to animals that received SR 16435 alone; however, the levels of activity never reached the levels observed in vehicle controls (P < 0.05). As seen in Table 2, levels of global activity produced by SR 16430 administration were not significantly different relative to vehicle controls.
Effects of SR 16430 on SR 16435-induced decrease in global activity
Other behaviors examined included grooming, sniffing, and rearing (Table 3). The overall ANOVAs indicated that there was a significant main effect of dose for grooming [F(3,35) = 51.9, P < 0.05] and sniffing [F(3,35) = 337.3, P < 0.05] and a significant drug × injection day interaction for rearing [F(3,35) = 7.33, P < 0.05]. Compared with vehicle controls, morphine administration produced the same incidence of sniffing, but a significantly lower incidence of grooming and rearing (P < 0.05). After repeated administration, morphine produced a significant increase in the incidence of rearing; however, the incidence of rearing was still 50% less than that observed in vehicle controls. SR 16435 alone produced a dramatic decrease in the incidence of all the behaviors examined relative to vehicle controls (P < 0.05). In most of the animals receiving SR 16435, these behaviors were not present. These animals were lying very still in one corner and in most cases had their eyes open (not quantified). This nonexistence of exploratory behavior/grooming was also present after repeated drug administration. Naloxone coadministered with SR 16435 reversed the behavior-dampening effects of SR 16435 and reinstated the same levels of grooming, sniffing, and rearing relative to vehicle controls (P < 0.05) (Table 3).
Behaviors elicited after acute and repeated drug administration
Discussion
Significant efforts to develop NOP ligands to further elucidate the role of this receptor in pain are ongoing. NOP-selective peptide antagonists administered i.c.v. have antinociceptive effects to thermal stimuli (Calo' et al., 2000, 2002), whereas systemic or i.c.v. administration of selective nonpeptide antagonists do not (Ozaki et al., 2000; Zaratin et al., 2004). NOP agonists have also been shown to have antinociceptive properties. Intrathecal but not i.c.v. injections of N/OFQ produce antinociception in acute pain models and are effective in models of chronic pain (Tian et al., 1997). High-affinity hexapeptide NOP agonists are also effective in animal models of chronic pain (T. V. Khroyan et al., 2007). The nonpeptide small-molecule agonist Ro 64-6198 is not antinociceptive when administered alone, reverses morphine antinociception, and yet is antiallodynic when administered alone in a neuropathic pain model (Jenck et al., 2000; Kotlinska et al., 2003; Obara et al., 2005). Thus, it still remains unclear whether NOP ligands can be developed as clinically useful analgesics.
In the present study, we examined the behavioral effects of a mixed NOP/μ-opioid agonist, SR 16435. SR 16435 has a high affinity for both NOP and μ-opioid receptors and is a partial agonist at both these receptors as determined by the [35S]GTPγS binding assay (Table 1) (Zaveri et al., 2004). SR 16435 produced an increase in tail-flick latency after acute administration. However, the slope of the dose-response curve for SR 16435 was considerably shallower than that of morphine, as would be expected for a compound with low intrinsic efficacy (O'Callaghan and Holtzman, 1975). Naloxone blocked the antinociceptive effect of SR 16435, suggesting that the analgesic effect is mediated by μ-opioid receptors.

The effect of naloxone (1 mg/kg) on SR16435-induced antinociception collapsed across postinjection time (10, 30, or 60 min). Data are %MPE (mean ± S.E.M.). *, P < 0.05, significant difference from vehicle controls (Student Newman-Keuls test); †, P < 0.05, significant difference from SR 16435 alone (Student Newman-Keuls test). SC, subcutaneous.

To measure tolerance development, the effect of SR 16435 (30 mg/kg) was compared with the effect of morphine (10 mg/kg) by examining tail-flick latency tested across 9 days. Data are %MPE (mean ± S.E.M.). *, P < 0.05, significant difference from vehicle controls (Student Newman-Keuls test); †, P < 0.05, significant difference from the first injection (Student Newman-Keuls test); ▵, P < 0.05, significant difference from morphine alone (Student Newman-Keuls test).
We had hypothesized that a compound containing both NOP and μ-opioid agonist activities would have reduced tolerance development to its antinociceptive effects. SR 16435 produced a significant, albeit not dramatic, decrease in tolerance development to its antinociceptive effects. SR 16435 produced a 45% decrease in %MPE on day 9 relative to day 1, whereas morphine produced a 76% decrease in %MPE, resulting in similar levels of %MPE as those in controls by day 9. Decreased tolerance development does not appear to be a function of partial agonist activity of SR 16435 at μ-opioid receptors, because low-efficacy μ-opioid agonists, such as buprenorphine, have been shown to display greater tolerance than higher-efficacy compounds (Grecksch et al., 2006). Previous studies have shown that both NOP agonists and antagonists can attenuate tolerance to morphine-induced antinociception (Rizzi et al., 2000; Lutfy et al., 2001; Zaratin et al., 2004). These conflicting studies highlight the complicated relationship between NOP receptor activation and μ-opioid-mediated tolerance.
N/OFQ blocks morphine-, cocaine-, and alcohol-induced CPP (Ciccocioppo et al., 1999, 2002; Murphy et al., 1999; Sakoori and Murphy, 2004), although it has no rewarding or aversive effects when administered alone (Devine et al., 1996). In addition, Ro 64-6198, when administered systemically 15 min before morphine, also attenuates morphine-induced CPP (Shoblock et al., 2005). Accordingly, in theory, if a compound has agonist activity at both NOP and μ-opioid receptors, the NOP agonist activity would attenuate the rewarding effects of the μ-opioid receptor-mediated component, leading to a compound with reduced addiction liability. However, in the present study SR 16435 still produced CPP, thereby demonstrating that even with relatively equal affinities at the two receptors, a decrease in CPP is not evident when both μ-opioid and NOP partial agonist activities are present together in the same molecule. There are two potential explanations for the disparate findings as to why SR 16435 is rewarding, whereas Ro 64-6198 diminished morphine-induced reward. It is possible that the temporal relationship between μ-opioid and NOP receptor activation is important, such that activation of the two receptors simultaneously by SR 16435 is not sufficient for the NOP receptor to attenuate the actions of the μ-opioid receptor, whereas activation of NOP receptors by Ro 64-6198 before morphine administration was sufficient. Another possibility is that SR 16435, a partial agonist at the NOP receptor, does not have efficacy as high as Ro 64-6198 to modulate the μ-opioid receptor-mediated reward. This possibility is being investigated with additional compounds from our series of compounds that have higher efficacy at NOP.

The effect of SR 16435 alone on (A) place conditioning and (B) global activity. Data are means ± S.E.M. for (A) time (seconds) spent in the drug-paired compartment or (B) activity (centimeters) after the first and last drug injections. *, P < 0.05, significant difference from vehicle controls (Student Newman-Keuls test); †, P < 0.05, significant difference from first injection (Student Newman-Keuls test). SC, subcutaneous.
Given that NOP receptor activation itself does not produce CPP, SR 16435-induced CPP is probably mediated by μ-opioid receptor activation. This is further substantiated by the observation that naloxone reversed SR 16435-induced CPP and produced the typical CPA when coadministered with SR 16435. Previous studies have shown that the presence of μ-opioid receptors is crucial for acquisition of naloxone-induced CPA because μ-opioid receptor knockout mice do not exhibit naloxone-induced CPA (Skoubis et al., 2001). In addition, it seems that partial agonist activity at μ-opioid receptors is sufficient to produce CPP because buprenorphine produces CPP (Tzschentke, 2004). Collectively, these findings suggest that the μ-opioid receptor is a critical component in mediating the oppositional CPP and CPA produced by opioid agonists and antagonists, respectively. In addition, activation of the NOP receptors may modulate reward, taking into consideration the temporal relationship between μ-opioid and NOP receptor activation and also efficacy at the NOP receptor (Murphy et al., 1999; Shoblock et al., 2005).
SR 16435 produced a decrease in global activity after the first injection but not after repeated administration. NOP agonists administered i.c.v. (UFP-102) and systemically (Ro 64-6198) produce a decrease in global activity (Kuzmin et al., 2003; Carra et al., 2005). In the present study, systemic administration of the NOP antagonist SR 16430 partially reversed the inhibitory effects of SR 16435. However, naloxone also reversed the inhibitory effects of SR 16435, as well as the decrease in exploratory and grooming behavior. It is highly unlikely that SR 16435-mediated suppression of activity was due to its ability to stimulate μ-opioid receptors because morphine produces an increase in global activity in mice (Fig. 5B). Although κ-opioid agonists produce a decrease in locomotor activity (Kuzmin et al., 2000), SR 16435 has no κ-opioid agonist activity as measured by the [35S]GTPγS binding assay (Table 1). Furthermore, a more selective NOP agonist SR 14150 [1-(1-cyclooctyl-piperidin-4-yl)1,3-dihydroindol-2-one], from our series (Zaveri et al., 2004), also produced suppression in global activity after the first and fourth injection (T. V. Khroyan et al., unpublished data). Therefore, it seems likely that the activity-inhibitory effects of SR 16435 are mediated through the NOP receptor.

The effect of naloxone (1 mg/kg) on SR 16435-induced conditioned place preference (A) and inhibition of global activity (B). Data are means ± S.E.M. for (A) time (seconds) spent in the drug-paired compartment or (B) activity (centimeters) after the first and last drug injections. *, P < 0.05, significant difference from vehicle controls (Student Newman-Keuls test). †, P < 0.05, significant difference from SR 16435 alone (student Newman-Keuls test). SC, subcutaneous.
As indicated above, the SR 16435-induced decrease in global activity is naloxone-reversible. The reversal by naloxone of the SR16435-mediated decrease in global activity is not the first demonstration of naloxone inhibiting an aspect of NOP-mediated behavior. Indeed, naloxone blocks spinal N/OFQ-mediated analgesic activity in chronic pain models (Courteix et al., 2004) and also inhibits the hyperphagic effects of N/OFQ (Polidori et al., 2000). However, the mechanisms underlying naloxone-induced antagonism of NOP-mediated behaviors are unclear. It could be speculated that opioid receptors may interact with NOP receptors and enable NOP-mediated behaviors. However, naloxone could also have a general inhibitory effect independent from that of any system that may be controlling activity or food intake. Further experiments with more selective agonists and antagonists could shed some light on the interaction of NOP and opioid receptor systems.
In conclusion, the mixed NOP/μ-opioid ligand, SR 16435, has high binding affinity and is a partial agonist at both sites. SR 16435 has antinociceptive activity and has reduced tolerance development to its antinociceptive effects. However, SR 16435 induces CPP, suggesting that the NOP receptor-mediated component was unable to attenuate the reward induced by the μ-opioid receptor activity. It may be possible, however, to shift the functional balance between NOP and μ-opioid receptor stimulation to favor NOP versus μ-opioid receptor-mediated effects by using a compound with full agonist activity at NOP. Such a compound may produce antinociceptive effects with reduced rewarding properties. Compounds with this profile from our series of bifunctional NOP/μ-opioid ligands are currently being examined in ongoing studies in our laboratory.
Acknowledgments
We thank Rajesh Khanna for help with the graphics and Drs. Girolamo Calo' and Barbara Spagnolo for helpful suggestions.
Footnotes
-
This research was supported by National Institute on Drug Abuse Grant DA14026 (to N.T.Z.).
-
Preliminary reports of these data were presented at the following annual meetings: Khroyan TV, Zaveri NT, Polgar WE, Orduna J, Olsen C, Jiang F, and Toll L (2006) Differential effects of mixed NOP/mu receptor ligands in antinociception and reward in mice, in College on Problems of Drug Dependence Abst.; 2006 June 17–22; Phoenix, AZ. College on Problems of Drug Dependence, Philadelphia; Khroyan TV, Zaveri N, Polgar WE, Orduna J, and Toll L (2004). Rewarding and analgesic effects of a novel mu-opioid, ORL1 agonist, in College on Problems of Drug Dependence Conference; 2004 June 12–17; San Juan, Puerto Rico. College on Problems of Drug Dependence, Philadelphia; and Khroyan TV, Zaveri N, Polgar WE, Orduna J, and Toll L (2004) Examining the analgesic and rewarding effects of SR16435, a novel mu-opioid/NOP agonist, in Society for Neuroscience Abst; 2004 Oct 23–27; San Diego, CA. Society for Neuroscience, Washington, DC.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.111997.
-
ABBREVIATIONS: NOP receptor, nociceptin/orphanin FQ receptor, opioid receptor-like receptor (ORL1); N/OFQ, nociceptin/orphanin FQ; i.c.v., intracerebroventricular; i.t., intrathecal; UFP-101, [Nphe1,Arg14,Lys15]N/OFQ-NH2; PC, place conditioning; J-113397, (1-[3R,4R)-1-cyclooctymethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3,-dihydro-2H-benzimidazol-2-one; SB-612611, (–)-cis-1-methyl-7-[[4-(2,6-dichlorophenyl)piperidin-1-yl]methyl]-6,7,8,9-tetrahydro-5H-benzocyclohepten-5-ol; JTC-801, N-(4-amino-2-methylquinolin-6-yl)-2-(4-ethylphenoxymethyl) benzamide monohydrochloride; Ro 64-6198, 1S,3aS-8–2,3,3a,4,5,6-hexahydro-1H-phenalen-1-yl-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one; PC, place conditioning; CPP, conditioned place preference, CPA, conditioned place aversion; GTPγS, guanosine 5′-O-(3-thio)triphosphate; SR 16435, 1-(1-(bicyclo[3.3.1]nonan-9-yl)piperidin-4-yl)indolin-2-one; SR 16430, 1-(cyclooctylmethyl)-4-(3-(trifluoromethyl)phenyl)piperidin-4-ol; %MPE, % maximal potential effect; ANOVA, analysis of variance; SR 14150 1-(1-cyclooctyl-piperidin-4-yl)1,3-dihydro-indol-2-one.
- Received August 3, 2006.
- Accepted November 21, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}