


Abstract
Recently, extensive behavioral research has been conducted on the benztropine (BZT) analogs with the goal of developing successful therapeutics for cocaine abuse. The present study was conducted to characterize the contribution of dispositional factors in mediating the behavioral differences among the chloro BZT analogs and to identify cytochrome P450 enzymes involved in their metabolism. Bidirectional transport and efflux studies of four of the chloro BZT analogs were conducted. Screening with a panel of human and rat Supersomes was performed for 4′,4″-diCl BZT. In addition, pharmacokinetic and brain distribution studies for 4′-Cl and 4′,4″-diCl BZT in Sprague-Dawley rats were conducted. The permeability of the chloro analogs ranged from 8.26 to 32.23 and from 1.37 to 21.65 × 10–6 cm/s, whereas the efflux ratios ranged from 2.1 to 6.9 and from 3.3 to 28.4 across Madin-Darby canine kidney-multidrug resistance 1 (MDCK-MDR1) and Caco-2 monolayers, respectively. The P-glycoprotein (P-gp) inhibitor verapamil reduced the efflux ratios and enhanced the absorptive transport of the chloro BZT analogs. 4′,4″-diCl BZT was a substrate of human CYP2D6 and 2C19 and rat 2C11 and 3A1. The brain uptake for 4′-Cl and 4′,4″-diCl BZT was comparable and higher than previously reported for cocaine (brain-to-plasma partition coefficient = 4.6–4.7 versus 2.1 for cocaine). The rank order for t1/2 was 4′,4″-diCl BZT ≫ 4′-Cl BZT > cocaine and for steady-state volume of distribution was 4′-Cl BZT > 4′,4″-diCl BZT ≫ cocaine. In conclusion, the chloro analogs differ significantly in their clearance and duration of action, which correlates to their behavioral profiles and abuse liability. Furthermore, these results suggest that the distinctive behavioral profile of these analogs is not due to limited brain exposure.
The psychostimulant effects of cocaine are thought to be mediated mainly through inhibition of dopamine uptake via the dopamine transporter (DAT) (Ritz et al., 1987; Volkow et al., 1997). Accordingly, the DAT has been regarded as one of the key targets for the development of cocaine agonist therapeutics (Carroll et al., 1999; Rothman et al., 2005). Benztropine (BZT) is a potent dopamine uptake inhibitor that is not subject to significant abuse in humans (Rothman, 1990). As such, BZT was used as the pharmacophore for the design of a series of BZT analogs with the purpose of enhancing the potency and selectivity toward DAT (Newman and Kulkarni, 2002; Zou et al., 2003; Kulkarni et al., 2004). The overall goal of these studies has been to better understand the role of DAT in cocaine addiction and to guide the design of ligands that may serve as medications for the treatment for cocaine abuse.
The BZT analogs have shown lower abuse potential in comparison with cocaine (Newman et al., 1994; Woolverton et al., 2001; Katz et al., 2004). However, they vary in terms of their relative efficacies in certain preclinical behavioral assays. In a cocaine discrimination study in rats, 4′-chloro-3α-(diphenylmethoxy)tropane (4′-Cl BZT) had a maximal effect of 54.9% of cocaine-appropriate responses, whereas the maximal effect for 4′,4″-dichloro-3α-(diphenylmethoxy)tropane (4′,4″-diCl BZT) was 17.6% (Katz et al., 1999). It was also reported that 4′-Cl BZT and 3′-chloro-3α-(diphenylmethoxy)tropane (3′-Cl BZT) were the most effective in stimulating locomotor activity of mice in a 1-h observation and in pretreatment studies, followed by 3′,4″-dichloro-3α-(diphenylmethoxy)tropane (3′,4″-diCl BZT), whereas 4′,4″-diCl BZT only marginally increased locomotor activity (Katz et al., 2001). The same report indicated that the maximal stimulation of locomotor activity occurred first for 3′-Cl followed by 4′-Cl and then 3′,4″-diCl BZT. Self-administration studies in rhesus monkeys showed that the rank order for reinforcing effectiveness of the chloro analogs was 3′-Cl BZT = 4′-Cl BZT ≫ 3′,4″-diCl BZT, in agreement with the locomotor data (Woolverton et al., 2001). The fact that the binding and potency profiles of the four chloro analogs at DAT are comparable (Table 1) suggests the involvement of other factors that may explain the discrepancy between binding at DAT and behavior, including possible dispositional factors (Woolverton et al., 2000). These dispositional factors may include differences among the analogs in the rate and/or extent of brain exposure at equivalent doses, differences in their permeability across the blood-brain barrier (BBB), different affinity for efflux transporters expressed at the BBB, or differences in the elimination half-life and consequently the duration of action.
Substitutions, clog P, and binding affinity at DAT of the BZT analogs studied as well as DA uptake inhibition by these analogs
The present study was designed to evaluate the possible contribution of dispositional factors in mediating the reported differences in the behavioral pharmacology of the chloro analogs (compared with one another and with cocaine) and to characterize the metabolic stability as well as the human and rat cytochrome P450 enzymes involved in their metabolism. No previous studies have been reported to characterize the enzymes involved in the metabolism of benztropine or the BZT analogs, even though the metabolites of benztropine were detected in rat urine and bile more than a decade ago (He et al., 1995). Consequently, the human and rat cytochrome P450 enzymes involved in the metabolism of 4′,4″-diCl BZT were screened as an initial step to understand the elimination mechanisms of this class of compounds. In addition, the in vitro permeability and the possible P-glycoprotein (P-gp) interaction of the chloro analogs using two different cell lines was assessed. The goal was to examine whether these analogs differ significantly in permeability and efflux. Based on the results of this evaluation, we selected two analogs and compared their in vivo pharmacokinetic and brain uptake profiles in Sprague-Dawley rats.
Materials and Methods
Materials
Caco-2 cells were purchased from American Type Culture Collection (Manassas, VA). MDCK-MDR1 cells were a gift from Dr. Peter Swaan (University of Maryland at Baltimore, Baltimore, MD). Cell culture supplies [Dulbecco's modified Eagle's medium, phosphate-buffered saline with Ca2+ and Mg2+ (PBS), 100× l-glutamine, nonessential amino acids, fetal bovine serum, 0.25% trypsin-1 mM EDTA, and penicillin G-streptomycin sulfate antibiotic mixture] were purchased from Invitrogen (Carlsbad, CA). [14C]Mannitol (46 mCi/mmol), [14C]paclitaxel (52.3 mCi/mmol), [3H]propranolol (20 mCi/mmol), sodium phosphate dibasic, verapamil HCl, and oxprenolol were purchased from Sigma-Aldrich (St. Louis, MO). Transwell clusters were purchased from Corning Life Sciences (Acton, MA). Microsomes from baculovirus-infected insect cells expressing human CYP1A2, 2A6, 2B6, 2C8, 2C9*1(Arg144), 2C19, 2D6*1, 2E1, and 3A4 and rat CYP1A2, 2B1, 2C11, 2D1, 2E1, and 3A1 (Supersomes) as well as insect cell control and rat P450 reductase insect cell control Supersomes were purchased from BD Gentest (Woburn, MA). The P450s were coexpressed with their corresponding human or rat cytochrome P450 reductase, in addition, human CYP2A6, 2B6, 2C8, 2C9*1, 2C19, 2E1, and 3A4 and rat CYP2B1, 2C11, 2D1, 2E1, and 3A1 were coexpressed with human cytochrome b5. Pooled human liver microsomes, pooled male Sprague-Dawley rat liver microsomes, and NADPH-regenerating systems were also purchased from BD Gentest. Potassium monobasic phosphate and potassium dibasic phosphates were purchased from Fisher Scientific Co. (Fair Lawn, NJ). The BZT analogs (Table 1; Fig. 1) were synthesized as described previously (Newman et al., 1994, 1995; Kline et al., 1997; Katz et al., 2001). All chemicals and solvents were high-performance liquid chromatography (HPLC) grade or American Chemical Society analytical grade.
Cell Culture
Cells were grown at 37°C, 95% relative humidity, and 5% CO2 atmosphere on 12-well Costar inserts (Transwell; 0.4-μm pore polycarbonate filter, 1 cm2 in diameter). Caco-2 cells (passages 41 and 42) were seeded at a density of 80,000 cells/cm2. The cells were grown for 26 to 27 days in 1× Dulbecco's modified Eagle's medium, containing 10% fetal bovine serum, 2% glutamine, 1% nonessential amino acids, 1% penicillin-streptomycin, with the medium changed every other day. MDCK-MDR1 cells were seeded at a density of 425,000 cells/cm2 and grown for 3 to 4 days in a medium similar to that used for Caco-2 cells with a daily medium change.
Characterization of Caco-2 and MDCK-MDR1 Cell Monolayers
Before conducting the transport experiments, monolayer integrity was determined by measuring the transepithelial electrical resistance (TEER) using a Millicell-ERS meter (Millipore Corporation, Billerica, MA) and by determining the permeability of [14C]mannitol and [3H]propranolol. P-gp expression was functionally tested by conducting bidirectional transport studies with [14C]paclitaxel and determining its efflux ratio across Caco-2 and MDCK-MDR1 mono-layers.

Chemical structure of the BZT analogs studied. The chloro substituents (R′ and R″) are presented in Table 1.
BZT Analog Bidirectional Transport and Inhibition Studies
These studies were conducted to evaluate and compare the in vitro permeability and the possible P-gp interaction of the four chloro BZT analogs (Table 1). The goal was to examine whether these analogs differ significantly in permeability and efflux in vitro. Transport experiments for the chloro BZT analogs were performed in both the apical to basolateral (A-B) and the basolateral to apical (B-A) directions across Caco-2 and MDCK-MDR1 monolayers in the presence and absence of the P-gp inhibitor verapamil (Wacher et al., 1995). At the time of experiment, the culture medium was removed from both the apical and basolateral sides of the monolayers and washed twice with PBS. The monolayers (n = 3/group) were incubated with either 200 μM verapamil (Taub et al., 2005) in PBS or PBS for 30 min. Following the preincubation period, mixtures of the 0.1 mM BZT analog with either 200 μM verapamil or with PBS were added to the donor compartments. The receiver compartments solution consisted of either 200 μM verapamil in PBS (transport in presence of verapamil) or PBS (transport in absence of verapamil). For the apical-to-basolateral study, the inserts were moved to new Transwells containing 1.5 ml of the corresponding receiver compartment solution at 30, 60, 90, and 120 min. For the basolateral-to-apical study, samples were drawn from the apical chamber at the same time points and replaced with equivalent volumes of fresh receiver compartment solution. Transport experiments were performed at 37°C with continuous agitation on a plate shaker (75 cycles/min), and samples were stored at –80°C until the time of analysis.
Characterization of Human and Rat Cytochrome P450 Enzymes Involved in the Metabolism of 4′,4″-diCl BZT
Since the four chloro analogs differ only in the position and number of chloro substitutions, we did not expect their in vitro metabolism to differ significantly. Consequently, we selected 4′,4″-diCl BZT as representative of the class, and we screened for the enzymes involved in its metabolism. 4′,4″-diCl BZT was incubated with human CYP1A2, 2A6, 2B6, 2C8, 2C9*1(Arg144), 2C19, 2D6*1, 2E1, and 3A4 as well as rat CYP1A2, 2B1, 2C11, 2D1, 2E1, and 3A1 Supersomes for 60 min. For each enzyme tested, the reaction mixture consisted of 50 pmol/ml P450, NADPH-regenerating system (1.3 mM NADP+, 3.3 mM glucose 6-phosphate, 0.4 U/ml glucose-6-phosphate dehydrogenase, and 3.3 mM magnesium chloride), and 10 μM 4′,4″-diCl BZT in 100 mM potassium phosphate buffer, pH 7.4 (final volume 500 μl). The reactions were initiated by adding ice-cold Supersomes to the prewarmed mixture of buffer, substrate, and cofactors. After a 60-min incubation period at 37°C, the reactions were stopped by the addition of 250 μl of acetonitrile and centrifuged at 10,000g for 5 min. Two hundred microliters of the supernatants was injected onto the HPLC for determination of unchanged 4′,4″-diCl BZT concentrations. Similar incubations with insect cell control and rat P450 reductase insect cell control Supersomes were performed to control for the native activities and non-P450-specific effects. Metabolism incubations were performed in triplicates.
Determination of the Time Course of 4′,4″-diCl BZT Metabolism
The time course of metabolism of 4′,4″-diCl BZT (3 μM final concentration; n = 3) by pooled human liver microsomes, pooled male rat liver microsomes, and the human and rat P450s that significantly reduced the concentration of 4′,4″-diCl BZT in the initial screening experiments was determined. The microsomes were used at a concentration of 0.8 mg/ml. The P450 isoforms, cofactor, and buffer concentrations were similar as described above with a final reaction volume of 1500 μl. The reactions were initiated by adding the drug to the prewarmed reaction mixture. After 0, 5, 10, 20, 30, 40, and 60 min of incubation at 37°C, 200 μl of the reaction mixture was sampled, immediately vortexed with 100 μl of acetonitrile to terminate the reaction, and centrifuged at 10,000g for 5 min. Aliquots of the supernatant were then collected for HPLC analysis.
Animal Pharmacokinetic Studies
Animals. Adult male Sprague-Dawley rats (250–350 g) were used in the study and were purchased from Harlan (Indianapolis, IN). The animal study protocols were approved by the Institutional Animal Care and Use Committee of the School of Pharmacy (University of Maryland Baltimore). Rats were housed in the animal facility at a room temperature of 72 ± 2°F. They were allowed free access to food (Purina 5001 Rodent Chow; Purina, St. Louis, MO) and water ad libitum, and they were maintained on a 12-h light/dark cycle.
Pharmacokinetic Studies. The rats were administered 4′-Cl BZT or 4′,4″-diCl BZT at an i.v. dose of the free base equivalent to 5 or 10 mg/kg hydrochloride salt. 4′-Cl BZT was dissolved in sterile water for injection. 4′,4″-diCl BZT was dissolved in 20% (2-hydroxypropyl)-β-cyclodextrin in sterile water for injection. The dosing for the pharmacokinetic studies was conducted at a volume of 1 ml/kg. A destructive sampling design was adapted where groups of three animals were sacrificed by CO2 asphyxiation predose and postdose at 5, 30, 60, 120, 240, 360, 480, 600, and 1440 min. Blood was collected by heart puncture using heparinized syringes and centrifuged for 10 min at 3000 rpm. The plasma was separated and stored at –80°C until the analysis. Brain tissues were immediately removed, blotted on a filter paper, weighted, and stored at –80°C until the time of analysis.
Analysis of the Transport, Metabolism, and Pharmacokinetic Samples. A previously published UV-HPLC method with few modifications was used for analysis of the BZT analogs (Raje et al., 2002). The chromatographic conditions consisted of a Symmetry C18 column (150 × 4.6 mm; 5 μm) for 4′,4″-diCl BZT or a Supelcosil LC ABZ Plus column (250 × 4.6 mm; 5 μm) (for the remaining BZT analogs), UV detector (λ = 220 nm), mobile phases [methanol/0.05 M Na2HPO4, pH 3.0, 40:60 (v/v) (A) and methanol/0.05 M Na2HPO4,pH 3.0, 80:20 (v/v) (B)], and a flow rate of 1 ml/min pumped using a gradient profile optimized for the different analogs and type of sample.
The transport and metabolism samples were analyzed directly without further processing. For the pharmacokinetic study, the plasma samples were extracted with hexane followed by evaporation and reconstitution in the mobile phase. The brain samples were double extracted with hexane following homogenization with distilled water. Oxprenolol and 4′-Cl BZT were used as internal standards for 4′-Cl BZT and 4′,4″-diCl BZT, respectively.
The calibration curves were linear (r2 ≥ 0.994) in the range of 50 to 10,000, 100 to 10,000, 50 to 5000, and 100 to 20,000 ng/ml for the transport, metabolism, plasma, and brain matrices, respectively. The radioactive samples of the transport markers were analyzed by addition to 5 ml of Universol scintillation cocktail, and radioactivity was measured using a Beckman Coulter LS 6500 multipurpose scintillation counter.
Data Analysis
Transport Data Analysis. The apparent permeability coefficients (in presence and absence of the P-gp inhibitor verapamil) were determined at sink conditions using the following equation:  where dQ/dt is equal to the linear appearance rate of mass the receiver solution, A is the cross-sectional area of the insert filters, and C0 is the donor concentration at time 0. All values are represented as mean and standard deviation from three Transwell inserts. Efflux ratios across the monolayers were calculated using the equation:
where dQ/dt is equal to the linear appearance rate of mass the receiver solution, A is the cross-sectional area of the insert filters, and C0 is the donor concentration at time 0. All values are represented as mean and standard deviation from three Transwell inserts. Efflux ratios across the monolayers were calculated using the equation:  where Papp (B-A) is the permeability from the basolateral to the apical direction (secretory transport) and Papp (A-B) the permeability from the apical-to-basolateral direction (absorptive transport). Enhancement ratio in the apical-to-basolateral transport induced by verapamil was calculated according to the equation:
where Papp (B-A) is the permeability from the basolateral to the apical direction (secretory transport) and Papp (A-B) the permeability from the apical-to-basolateral direction (absorptive transport). Enhancement ratio in the apical-to-basolateral transport induced by verapamil was calculated according to the equation:  The statistical significance of effect of verapamil on the permeability of each of the BZT analogs studied was determined with two-sample Student's t test at α = 0.05 using Microsoft Excel software (Microsoft, Remond, WA). The delta method was used to calculate the standard error of the ratios, and statistical significance was declared when the 95% confidence interval of two ratios did not overlap. Correlation analysis was conducted using SAS for Windows software (SAS Institute, Cary, NC).
The statistical significance of effect of verapamil on the permeability of each of the BZT analogs studied was determined with two-sample Student's t test at α = 0.05 using Microsoft Excel software (Microsoft, Remond, WA). The delta method was used to calculate the standard error of the ratios, and statistical significance was declared when the 95% confidence interval of two ratios did not overlap. Correlation analysis was conducted using SAS for Windows software (SAS Institute, Cary, NC).
Metabolism Data Analysis. Identification of the P450s involved in metabolism. The human and rat P450 isoforms involved in the metabolism of 4′,4″-diCl BZT were identified by analyzing the differences in mean substrate concentrations remaining after 60-min incubations by one-way analysis of variance (ANOVA) followed by Dunnett's multiple comparisons of P450 incubations versus control incubations. We also calculated the percentage of the mean control concentration remaining after 60-min incubation according to the following equation:  where CCYPr, 60 min is the substrate concentration from the rth replicate after 60 min of incubation with a particular P450 Supersome, and Caverage ctrl, 60 min is the average (n = 3) substrate concentration after 60-min incubation with insect cell control Supersomes (for human P450s) or rat P450 reductase insect cell control Supersomes (for rat P450s). The percentages are represented as mean and standard deviation from triplicate reactions.
where CCYPr, 60 min is the substrate concentration from the rth replicate after 60 min of incubation with a particular P450 Supersome, and Caverage ctrl, 60 min is the average (n = 3) substrate concentration after 60-min incubation with insect cell control Supersomes (for human P450s) or rat P450 reductase insect cell control Supersomes (for rat P450s). The percentages are represented as mean and standard deviation from triplicate reactions.
Intrinsic clearance calculation. The intrinsic clearance values were calculated based on the substrate disappearance rate as described previously (Naritomi et al., 2001, 2003). Assuming first-order disappearance of substrate, the disappearance rate constant (Ke) was calculated from the slope of log Ct versus time profile based on the following equation:  where Ct is the concentration of the substrate at the different time points, and C0 is the substrate concentration at time 0. The initial metabolic rate (V0) (picomoles per minute per picomoles of P450 or milligrams of microsomal protein) was calculated from the following equation:
where Ct is the concentration of the substrate at the different time points, and C0 is the substrate concentration at time 0. The initial metabolic rate (V0) (picomoles per minute per picomoles of P450 or milligrams of microsomal protein) was calculated from the following equation:  where PMS is P450 concentration (picomoles per milliliter) or microsomal protein concentration (milligrams per milliliter). V0 can also be described using Michaelis-Menten equation as follows:
where PMS is P450 concentration (picomoles per milliliter) or microsomal protein concentration (milligrams per milliliter). V0 can also be described using Michaelis-Menten equation as follows:  Assuming that C0 ≪ Km, eq. 7 can be written as follows:
Assuming that C0 ≪ Km, eq. 7 can be written as follows:  Accordingly, the intrinsic clearance was calculated based on the formula:
Accordingly, the intrinsic clearance was calculated based on the formula: 
The intrinsic clearance values were calculated separately from each of the replicates and compared statistically using one-way ANOVA followed by Duncan's multiple range test. CLint values are presented as mean ± S.D. from the three replicates performed for each reaction.
Pharmacokinetic Data Analysis. The destructive sampling data obtained from the pharmacokinetic studies were initially analyzed by the naive averaging method. For a given drug, the plasma concentrations from the three animals at each time point for each dose level were averaged. The average concentrations versus time data were then used for compartmental modeling using WinNonlin version 4.1 software (Pharsight, Mountain View, CA). Various compartment models were evaluated to determine the most appropriate model. In addition, various weighting schemes, including equal weight, 1/y, 1/ŷ, 1/y2, and 1/ŷ2 were evaluated, where y is the observed drug concentration, and ŷ is the model-predicted drug concentration. The final model selection was based on the visual inspection of goodness-of-fit plots and on the value of weighted sum of squared residuals, precision of parameter estimates, Akaike's information criteria, and Schwarz criteria.
To be able to estimate the interanimal variability in the pharmacokinetic parameters, nonlinear mixed effect modeling was conducted using NONMEM version 5, level 1.1 (GloboMax LLC, Hanover, MD). For each drug, the data from the two dose levels were compiled and analyzed (data from 42 animals for 4′-Cl BZT and from 54 animals for 4′,4″-diCl BZT). The final parameter estimates from the naive averaging analysis were used as initial estimates for the population analysis. Based on the results from the naive averaging analysis, the population analysis started with a two-compartment structural model for both 4′-Cl BZT and 4′,4″-diCl BZT. Additive, proportional and exponential statistical error models were evaluated to describe the interanimal as well as the residual variability. The analysis was performed using the conventional first-order estimation method. The final model was determined based on inspection of goodness of fit plots, precision of parameter estimates, and the value of the objective function. In addition, the likelihood ratio test (at α = 0.05) was used as a selection criterion between rival hierarchical models (Sheiner and Ludden, 1992). The brain-to-plasma partition coefficient (Ri) was calculated as a measure of brain uptake according to the following formula:  where AUC0–inf (brain) and AUC0–inf (plasma) are the area under the brain and plasma concentrations versus time curves, respectively. The AUC0–inf were calculated using WinNonlin noncompartmental analysis by applying the linear trapezoidal rule for the ascending portions of the concentration versus time profiles and the log linear trapezoidal rule for the descending portions. For each dose, the pharmacokinetic parameters for 4′-Cl BZT and 4′,4″-diCl BZT were compared using a two-sample Student's t test. The pharmacokinetic parameters for the 5-mg/kg dose for the studied analogs were compared with previously published parameters for cocaine at the same dose (Raje et al., 2003) using one-way ANOVA followed by Dunnett's post hoc analysis.
where AUC0–inf (brain) and AUC0–inf (plasma) are the area under the brain and plasma concentrations versus time curves, respectively. The AUC0–inf were calculated using WinNonlin noncompartmental analysis by applying the linear trapezoidal rule for the ascending portions of the concentration versus time profiles and the log linear trapezoidal rule for the descending portions. For each dose, the pharmacokinetic parameters for 4′-Cl BZT and 4′,4″-diCl BZT were compared using a two-sample Student's t test. The pharmacokinetic parameters for the 5-mg/kg dose for the studied analogs were compared with previously published parameters for cocaine at the same dose (Raje et al., 2003) using one-way ANOVA followed by Dunnett's post hoc analysis.
Results
Characterization of Cell Lines. Across MDCK-MDR1 monolayers, mannitol Papp ranged from 3.89 to 4.46 × 10–6 cm/s, and the TEER values were >600 Ω · cm2. Propranolol Papp was 24.33 × 10–6 cm/s, and paclitaxel efflux ratio was 101, indicating high level of P-gp expression. Across Caco-2 monolayers, mannitol Papp ranged from 0.22 to 0.28 × 10–6 cm/s, and the TEER values ranged from 450 to 500 Ω · cm2, indicating high monolayer integrity. Propranolol permeability was 13.34 × 10–6 cm/s, indicating higher transcellular resistance compared with MDCK-MDR1 cells. Paclitaxel efflux ratio ranged from 40 to 43 across Caco-2 cells.
MDCK-MDR1 Chloro BZT Analog Permeability. Across MDCK-MDR1 monolayers, the chloro analogs have shown permeability values ranging from 8.26 to 32.23 × 10–6 cm/s with the following rank order: 3′ Cl BZT > 4′ Cl BZT > 3′,4″-diCl BZT > 4′,4″-diCl BZT (Table 2). All of the analogs showed polarized transport with higher B-A permeability than the A-B permeability. The efflux ratio ranged from 2.1 to 6.9 with the highest efflux observed with 4′,4″-diCl BZT and the lowest with the mono-substituted chloro analogs (Table 2).
Permeability values and efflux ratios of the chloro-BZT analogs across MDCK-MDR1 and Caco-2 monolayers at a concentration of 0.1 mM
Papp values are expressed as mean ± S.D. (n = 3).
Caco-2 BZT Permeability. The studied analogs also displayed polarized transport across Caco-2 monolayers. The permeability values ranged from 1.37 to 21.65 × 10–6 cm/s with rank order similar to that observed with MDCK-MDR1 cells, i.e., 3′-Cl BZT > 4′-Cl BZT > 3′,4″-diCl BZT > 4′,4″-diCl BZT (Table 2). The efflux ratio ranged from 3.3 to 28.4 with the highest efflux observed with 4′,4″-diCl BZT, replicating the efflux pattern observed with the MDCK-MDR1 cells (Table 2).
Effect of Verapamil on BZT-Polarized Transport. Verapamil was used to verify the involvement of P-gp in mediating the observed efflux of the BZT analogs. Verapamil increased the A-B permeability of the BZT analogs in both MDCK-MDR1 and Caco-2 cell lines (Fig. 2, A and B, respectively). The increase in the A-B permeability was statistically significant (p < 0.05) for all of the analogs except for 3′-Cl BZT (Fig. 2, A and B) where statistical significance was not reached; probably due to the high variability. The enhancement ratio of the A-B transport induced by verapamil ranged from 1.1 for 3′-Cl BZT to 3.2 for 4′,4″-diCl BZT across MDCK-MDR1 monolayers (Fig. 2A) and from 1.3 for 3′-Cl BZT to 4.3 for 4′,4″-diCl BZT across Caco-2 monolayers (Fig. 2B). Except for 3′-Cl BZT in MDCK-MDR1 cells, verapamil reduced the efflux ratios of all of the BZT analogs across both cell lines, and the reduction was statistically significant in most instances (Fig. 2, C and D). At 200 μM, MTS and XTT cytotoxicity assays indicated that verapamil did not affect either MDCK-MDR1 or Caco-2 cell viability (p < 0.05) over 2.5 h of exposure at conditions identical to those in which the transport experiments were conducted (data not shown).
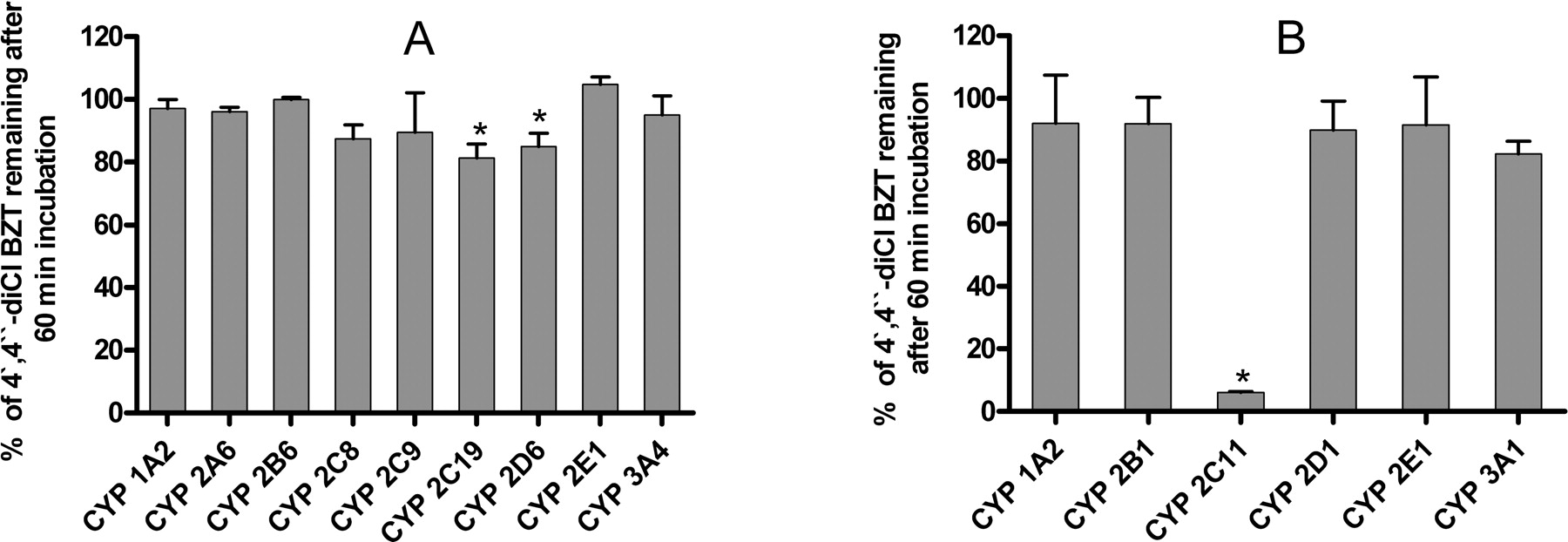
Characterization of Human and Rat Cytochrome P450 Enzymes Involved in the Metabolism of 4′,4″-diCl BZT. The objective of these studies was to screen for the enzymes involved in the metabolism of 4′,4″-diCl BZT as a compound representative of the class. Among the P450 isoforms tested, human CYP2C19 and CYP2D6 and rat CYP2C11 resulted in statistically significant disappearance of 4′,4″-diCl BZT after 60-min incubation compared with the control (p < 0.05) (Fig. 3, A and B). Based on the post hoc analysis, the effect of rat CYP3A1 was not statistically significant, although a t test comparison to the control have shown a p value less than 0.05. The most extensive substrate disappearance was observed with 4′,4″-diCl BZT using rat CYP2C11 (6.05 ± 0.26% of the parent compound remaining at the end of incubation period) (Fig. 3B).
Intrinsic Clearance of 4′,4″-diCl BZT in Human and Rat Recombinant P450s and Pooled Human and Male Rat Liver Microsomes. Under the experimental conditions, 4′,4″-diCl BZT metabolism followed first-order reaction with the concentration of the substrate declining monoexponentially with time. The calculated intrinsic clearance values of 4′,4″-diCl BZT are reported in Table 3. The preference for metabolizing 4′,4″-diCl BZT was as follows: CYP2C19 > CYP2D6 for human P450s and CYP2C11 ⋙ CYP3A1 for rat P450s. The differences in the intrinsic clearance between human CYP2D6, human CYP2C19, and rat CYP3A1 were not statistically significant (Table 3). The intrinsic clearance of 4′,4″-diCl BZT was 1.68-fold higher in pooled male rat liver microsomes than in human liver microsomes.
Intrinsic clearance values for 3 μM 4′,4″-diCl BZT in human and male Sprague-Dawley rat-pooled liver microsomes (0.8 mg/ml) and in human and rat recombinant P450s (50 pmol/ml)
The intrinsic clearance values in the recombinant P450s that share the same letter are not significantly different at α = 0.05.
Pharmacokinetics of 4′-Cl and 4′,4″-diCl BZT. Two analogs were selected for the pharmacokinetics and brain distribution study as they represent the two extremes among the chloro BZT analogs in terms of their behavioral profiles (Katz et al., 1999). In addition, they differed significantly in their in vitro transport and their P-gp-mediated efflux. Figure 4A represents the observed and the predicted plasma concentrations of 4′-Cl BZT and 4′,4″-diCl BZT upon i.v. administration to male Sprague-Dawley rats at two dose levels (5 and 10 mg/kg) based on the best fit achieved with WinNonlin. Figure 4B represents the corresponding brain concentrations versus time profiles. The brain concentrations were higher than the plasma concentrations at each time point. The pharmacokinetics of 4′-Cl BZT and 4′,4″-diCl BZT were best described with a two-compartmental model. The final parameter estimates from WinNonlin were used as initial estimates for the population analysis. The final population model consisted of a two-compartmental structural model parameterized in terms of clearance (CL), volume of the central compartment (Vc), volume of the peripheral compartment (Vp), and intercompartmental clearance (Q) (AD-VAN3 TRANS4 NONMEM subroutines). The interanimal variability (IAV) in Vc, Vp, and Q for 4′-Cl BZT and in CL, Vc, and Q for 4′,4″-diCl BZT were best described with an exponential pharmacostatistical model. Based on the likelihood ratio test, the interanimal variability in CL for 4′-Cl BZT and Vp for 4′,4″-diCl BZT were not significantly different from zero (p > 0.05). Consequently, they were omitted from the final model. The residual random error was best described with a proportional model. The residual error was estimated to be 13.4% for 4′-Cl BZT and was fixed to 11.8% for 4′,4″-diCl BZT based on a sensitivity analysis and the value selected was the one resulting in the lowest objective function and the highest precision in the estimation of the interanimal variability.

Effect of 200 μM verapamil on permeability and P-gp-mediated efflux of the chloro BZT analogs across MDCK-MDR1 and Caco-2 monolayers. A and B, Apical-to-basolateral permeability in absence and in presence of verapamil across MDCK-MDR1 and Caco-2 monolayers, respectively (data represented as mean ± S.D.). C and D, efflux ratios across MDCK-MDR1 and Caco-2 monolayers in absence and in presence of verapamil (data represented as the ratio of the mean permeability values ± S.E.M. calculated using delta method). *, p < 0.05. Black bar, in absence of verapamil; and gray bar, in presence of verapamil.

Metabolism of 4′,4″-diCl BZT (a representative analog of the class) by the human recombinant P450s (A) and the rat recombinant P450s (B). 4′,4″-diCl BZT (10 μM) was incubated with the Supersomes (50 pmol/ml) and cofactors for 60 min followed by termination of reactions and determination of unchanged substrate. Data are presented as mean percentage of the average control ± S.D. from triplicate reactions. *, p < 0.05 based on Dunnett's test.

Plasma and brain pharmacokinetic profiles of 4′-Cl BZT and 4′,4″-diCl BZT (5 and 10 mg/kg i.v.) in male Sprague-Dawley rats (n = 3/time point/dose). A, observed (mean ± S.D.) and predicted plasma concentration versus time profiles based on the naive averaging analysis conducted using WinNonlin. B, observed brain concentrations (mean ± S.D.) versus time profiles.
Figure 5 represents the relevant diagnostic plots for the final population models for 4′-Cl BZT and 4′,4″-diCl BZT and indicates a lack of any systematic bias in describing the pharmacokinetic profiles. The population pharmacokinetic parameters along with their associated interanimal variability and the precision of the parameter estimates (expressed as coefficient of variation) are reported in Table 4. The secondary pharmacokinetic parameters of interest along with their associated standard deviation were calculated from the post hoc estimates of the individual animal parameters provided by NONMEM and are reported in Table 5. Data obtained after a single 5-mg/kg i.v. dose of cocaine to rats are presented for comparison in Table 5 (Raje et al., 2003). Both 4′-Cl BZT and 4′,4″-diCl BZT had significantly longer t1/2 and larger steady state volume of distribution (Vss) compared with cocaine at the same dose (p < 0.05). When comparing the two BZT analogs across the same doses, the elimination half-life of 4′,4″-diCl BZT was significantly longer than 4′-Cl BZT (p < 0.001). The Vss for 4′,4″-diCl BZT was significantly lower than that of 4′-Cl BZT (p < 0.001). The difference in steady-state volume of distribution resulted mainly from the difference in the volume of the peripheral compartment (Table 4). For a given analog, the effect of the dose on the Vss and t1/2 was not statistically significant except for 4′,4″-diCl BZT, where the t1/2 for the 10-mg/kg dose was significantly longer than the 5-mg/kg dose (p < 0.05). Based on the results of the noncompartmental analysis, the brain uptake for 4′-Cl BZT and 4′,4″-diCl BZT seemed to be comparable and higher than cocaine (Table 5). In addition, the brain t1/2 for each BZT analog was considerably longer than cocaine (Table 5). In general, the pharmacokinetics of the two BZT analogs tested seems to be almost linear over the dose range studied (Table 5; Fig. 4).
NONMEM estimated population pharmacokinetic parameters for 4′-Cl BZT (n = 42) and 4′,4″-diCl BZT (n = 54) along with their associated interanimal variability upon i.v. administration to Sprague-Dawley rats
Values in parentheses are % CV.
Secondary pharmacokinetic parameters (calculated from the NONMEM empirical Bayes estimates of the individual animal-specific PK parameters) and brain-to-plasma partition coefficient and brain t1/2 (based on the noncompartmental analysis performed with WinNonlin) for 4′-Cl BZT and 4′,4″-diCl BZT in comparison with previously published data from our laboratory for cocaine (Raje et al., 2003)
Discussion
Several reports have indicated that the benztropine analogs possess behavioral profiles that are significantly different from cocaine despite their high in vitro affinity and selectivity to DAT and their high potency as DA uptake inhibitors. However, not all benztropine analogs are equally devoid of cocaine-like effects, but rather they produce dissimilar behavioral profiles. These behavioral differences are not necessarily explainable by the binding and affinity profiles at DAT. Consequently, several hypotheses have been suggested to explain such differences (Katz et al., 2001; Woolverton et al., 2001; Desai et al., 2005; Li et al., 2005).
It is well recognized that the rate and extent of brain uptake and consequently DAT occupancy are important determinants of the behavioral profiles and the abuse liability of the DA uptake inhibitors. Rapid brain uptake and fast association with DAT as well as a certain level of DAT occupancy has been predicted to cause high abuse liability (Volkow et al., 1997; Gorelick, 1998). In addition, the short duration of action of some drugs of abuse (e.g., cocaine) is thought to contribute to the craving and the need for repeated administration (Quinn et al., 1997). Consequently, the present studies were conducted to delineate the contribution of dispositional factors in mediating the behavioral differences of the chloro BZT analogs.
The chloro BZT analogs are basic lipophilic molecules that contain oxygen and nitrogen atoms that can function as hydrogen bond acceptors. These characteristics suggest possible interaction with the efflux transporter P-gp, which may modulate their brain uptake. In the present study, the BZT analogs have shown polarized transport across the P-gp-overexpressing MDCK-MDR1 cells and Caco-2 cells, in agreement with the apical localization of P-gp. The efflux ratios of the chloro analogs were in the reverse order of their permeability values with 4′,4″-diCl BZT having noticeably high efflux followed by 3′,4″-diCl BZT followed by 4′-Cl and 3′-Cl BZT. The higher efflux of the dichloro analogs mediated their reduced in vitro permeability and may be a result of their higher lipophilicity compared with the monosubstituted analogs (Table 1). In addition, by examining the efflux ratios of the chloro analogs, it seems that the para chloro substitution enhances the affinity of the BZT analogs to P-gp over the meta-substitution. Interestingly, the in vitro permeability and efflux data correlated well with the reported behavioral differences aforementioned in the Introduction.
The P-gp inhibitor verapamil enhanced the A-B permeability and reduced the efflux ratios of the chloro BZT analogs, verifying that P-gp was responsible for the observed efflux. However, verapamil did not abolish it. Taking into account that verapamil inhibits P-gp competitively; the attenuated efflux in the presence of verapamil may be a result of the relatively high concentration of the BZT analogs used in the experiments (100 μM) or of the high affinity of these analogs for P-gp.
In vivo, 4′,4″-diCl BZT, in spite of its higher in vitro P-gp efflux, showed a comparable brain-to-plasma partition coefficient to 4′-Cl BZT. The higher lipophilicity of 4′,4″-diCl BZT (Table 1) may have resulted in offsetting the effect of its higher P-gp efflux, resulting in comparable brain uptake to 4′-Cl BZT. Similar pattern was previously observed with the BZT analog JHW 007 (clog P = 5.53) (Raje et al., 2003). JHW 007 had high brain uptake (Ri = 5.6) despite its high observed P-gp efflux (efflux ratio of 8 across bovine brain microvessel endothelial cell monolayers). However, the volume of the peripheral compartment was lower for 4′,4″-diCl BZT in comparison with 4′-Cl BZT, which could be a result of P-gp affecting the distribution of 4′,4″-diCl BZT to other tissues. Overall, the higher brain uptake of the two chloro analogs in comparison with cocaine excludes the possibility that limited extent of permeation across the BBB is responsible for their lack of cocaine-like effects or for their behavioral differences (compared with one another). In addition, it indicates that P-gp plays a very minor role, if any, in limiting the extent of BBB transport of this class of compounds. However, it should be noted that the time resolution of our sampling technique did not allow us to capture the entry phase to the brain, because the brain Cmax was observed at the first sampling time (5 min postdose). Consequently, we were not able to determine whether these two analogs differ from one another and from cocaine in their rate of brain entry within the first 5 min of exposure and whether P-gp has any role in modifying that rate. In addition, our pharmacokinetic studies were conducted via i.v. drug administration whereas the behavioral evaluations, except for the self-administration procedures, where conducted using the i.p. route. Consequently, the possibility that these analogs could differ in their rate or extent of absorption from the peritoneal cavity should not be discounted.

Diagnostic plots for the population analysis of the plasma concentrations of 4′-Cl BZT (n = 42 rats) and 4′,4″-diCl BZT (n = 54 rats) conducted using NONMEM. For each drug, the data from the two dose levels tested (5 and 10 mg/kg i.v.) were compiled before the analysis. A, population predicted versus observed plasma concentrations. B, individual animal predicted versus observed plasma concentrations. C, weighted residuals versus population predicted plasma concentrations.
The pharmacokinetic study also indicated that 4′-Cl BZT and 4′,4″-diCl BZT are characterized by larger Vss compared with cocaine (Vss rank order 4′-Cl BZT > 4′,4″-diCl BZT ⋙ cocaine). This is partially because of the higher brain uptake for the BZT analogs. Both 4′-Cl BZT and 4′,4″-diCl BZT are characterized by longer elimination t1/2 compared with cocaine (t1/2 rank order 4′,4″-diCl BZT ⋙ 4′-Cl BZT ≫ cocaine). Cocaine undergoes rapid nonenzymatic as well as enzymatic hydrolysis by the plasma and liver esterases, and this is responsible for its short duration of action (Quinn et al., 1997). The long t1/2 relative to cocaine explains the previously reported prolonged duration of stimulation of locomotor activity caused by the chloro BZT analogs compared with cocaine (Katz et al., 1999, 2001) as well as the prolonged elevation of brain dopamine level caused by 4′-Cl BZT in comparison with cocaine (Tanda et al., 2005).
The clearance of 4′,4″-diCl BZT from the brain was slower than 4′-Cl BZT, which was much slower than cocaine as can be seen from the brain half-lives (Table 5). The high brain uptake and the slow clearance coupled with the high affinity of the chloro analogs to DAT could result in saturation of DAT for a long period upon administration of the first few doses. Consequently, this will interfere with the frequent dosing in the self-administration procedures, since any subsequent doses of these analogs will not be as rewarding due to the possible DAT saturation. 4′,4″-diCl BZT was not evaluated in self-administration studies, but assuming the similarity in the pharmacokinetic profile between 3′,4″-diCl BZT and 4′,4″-diCl BZT, due to the minimal structural difference, the difference in the brain clearance may explain the rank order for reinforcing effectiveness observed in self-administration studies in rhesus monkeys (cocaine > 3′-Cl BZT = 4′-Cl BZT ≫ 3′,4″-diCl BZT) as reported previously (Woolverton et al., 2001).
Although benztropine (Cogentin) has been on the market for many years, the enzymes involved in its metabolism have not been characterized. Accordingly, little is known about the metabolism of its analogs. In general, the structure and the pharmacokinetic data suggest higher metabolic stability for the BZT analogs in comparison with cocaine. The results of our metabolism study indicate that the human and rat CYP2D and CYP2C subfamilies are probably the key players in the metabolism of the BZT analogs. The P450 screening indicated that 4′,4″-diCl BZT is a substrate of human CYP2D6. This is in agreement with the clinical reports that indicated that CYP2D6 inhibitors augment the side effects of benztropine and increased its serum levels (Roth et al., 1994; Armstrong and Schweitzer, 1997). 4′,4″-diCl BZT is also a substrate for CYP2C19 with intrinsic clearance comparable with CYP2D6 (Table 3). The rat P450 screening indicated that 4′,4″-diCl BZT is a substrate for CYP2C11 and CYP3A1 (with CYP 2C11 resulting in the most extensive metabolism among the entire panel tested). Our data also indicate that the clearance of 4′,4″-diCl BZT is faster in pooled rat liver microsomes than in pooled human liver microsomes. This may be indicative of longer duration of action in humans compared with rats, which may result in even lower abuse potential in humans. The validity of the last conclusion will depend on the fraction metabolized versus excreted unchanged and how these in vitro data will extrapolate to the in vivo situation.
In conclusion, the results presented in this article indicate that the chloro BZT analogs possess high and comparable brain uptake despite their differences in lipophilicity and in interaction with P-gp. In addition, there are significant differences among the chloro analogs in clearance and duration of action. These differences may explain some aspects of the behavioral differences observed among the chloro BZT analogs and between them and cocaine. The present results also provide the first report on the enzymes involved in the metabolism of the BZT analogs. This will help in predicting the possible drug-drug interactions and can be regarded as the initial step to understanding the metabolic profile of the BZT analogs, a series of compounds to which extensive research has been recently devoted to and from which a successful cocaine therapeutic may emerge.
Acknowledgments
We are grateful to Dr. Jonathan Katz for critical reading of an earlier version of this article. We also thank Dr. Santosh Kulkarni and J. Cao for synthesizing multigram quantities of the BZT analogs used in this study.
Footnotes
-
This work was supported by National Institutes of Health Grant R01 DA16715-03 from the National Institute on Drug Abuse–Intramural Research Program.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.111245.
-
ABBREVIATIONS: DAT, dopamine transporter; BZT, benztropine; 4′-Cl BZT, 4′-chloro-3α-(diphenylmethoxy)tropane; 4′,4″-diCl BZT, 4′,4″-dichloro-3α-(diphenylmethoxy)tropane; 3′-Cl BZT, 3′-chloro-3α-(diphenylmethoxy)tropane; 3′,4″-diCl BZT, 3′,4″-dichloro-3α-(diphenylmethoxy)tropane; P-gp, P-glycoprotein; MDR, multidrug resistance; PBS, phosphate-buffered saline; HPLC, high-performance liquid chromatography; TEER, transepithelial electrical resistance; A-B, apical to basolateral; B-A, basolateral to apical; ANOVA, analysis of variance; AUC, area under the curve; Papp, apparent permeability coefficient; Ri, brain-to-plasma partition coefficient; CL, clearance; Vc, volume of the central compartment; Vp, volume of the peripheral compartment; Q, intercompartmental clearance; IAV, interanimal variability; Vss, steady-state volume of distribution; DA, dopamine; BBB, blood brain barrier; JHW 007, N-(n-butyl)-(bis-fluorophenyl)methoxytropane; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl-2H-tetrazolium, inner salt; XTT, 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazolium hydroxide.
- Received July 20, 2006.
- Accepted September 25, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}