


Abstract
Salvia divinorum is a natural occurring hallucinogen that is traditionally used by the Mazatec Indians of central Mexico. The diterpene salvinorin A was identified as an active component of S. divinorum over 20 years ago, but only recently has biochemical screening indicated that a molecular target of salvinorin A in vitro is the κ-opioid receptor. We have examined whether salvinorin A, the C2-substituted derivative salvinorinyl-2-propionate, and salvinorin B can act as κ-opioid receptor agonists in vivo. We found that following intracerebroventricular injection over a dose range of 1 to 30 μg of both salvinorin A and salvinorinyl-2-propionate produces antinociception in wild-type mice but not in a novel strain of κ-opioid receptor knockout mice. Moreover, both salvinorin A and salvinorinyl-2-propionate reduce rectal body temperature, similar to conventional κ-opioid receptor agonists, in a genotype-dependent manner. In addition, we determined that salvinorin A has high affinity for κ1- but not κ2-opioid receptors, demonstrating selectivity for this receptor subclass. Finally, treatment over the same dose range with salvinorin B, which is inactive in vitro, produced neither antinociceptive nor hypothermic effects in wild-type mice. These data demonstrate that salvinorin A is the active component of S. divinorum, selective for κ1-opioid receptors, and that salvinorin A and specific structurally related analogs produce behavioral effects that require the κ-opioid receptor.
Salvia divinorum is a natural occurring hallucinogen that has been used traditionally for divination and other spiritual practices by the Mazatec people of Oaxaca, Mexico (Valdes et al., 1983) and more recently as a legal hallucinogen (Giroud et al., 2000; Sheffler and Roth, 2003). Studies in the 1980s identified an active component of S. divinorum to be salvinorin A (Ortega et al., 1982; Valdes et al., 1983), which was later determined to be the primary psychoactive molecule in the plant (Siebert, 1994). Salvinorin A is a neoclerodane diterpene whose absolute configuration and structure have been determined by NMR and single-crystal X-ray analysis (Ortega et al., 1982; Koreeda et al., 1990; Valdes, 1994), and it is quite distinct from other natural hallucinogens. The putative molecular target of salvinorin A remained elusive until recently when radioligand guanosine 5′-3-O-(thio)-triphosphate and adenylyl cyclase assays all identified salvinorin A to be an agonist at κ-opioid receptors (KORs) in vitro (Roth et al., 2002; Chavkin et al., 2004; Nichols, 2004; Yan and Roth, 2004). It is noteworthy that there is no in vitro agonist activity at the 5-hydroxytryptamine 2A serotonin receptors that mediate actions of most other known hallucinogens (Roth et al., 2002; Chavkin et al., 2004; Nichols, 2004).
These earlier studies established that salvinorin A was a potent and selective KOR agonist. However, it is still unclear whether salvinorin A is selective for κ1- or κ2-opioid receptors. The κ2-opioid receptor was initially defined on the basis of ligand binding and physiological studies (Iyengar et al., 1986; Zukin et al., 1988; Rothman et al., 1990). Although there is no evidence for a gene coding for the κ2-opioid receptor, recent data suggest that the κ2-opioid receptor might result from δ-κ heterodimers (Jordan and Devi, 1999). We have used ligand binding to determine whether salvinorin A shows selectivity for κ1- or κ2-opioid receptors.
Consistent with in vitro data, the reported physiologic effects of salvinorin A are consistent with prospective action through the KOR. For example, documented in vivo effects of S. divinorum and purified salvinorin A include sedation, antinociception, and production of hallucinations (Siebert, 1994; Valdes, 1994; Giroud et al., 2000; Hanes, 2001; Wang et al., 2005; McCurdy et al., 2006). Sedation and antinociception have long been identified as major effects of KOR agonists (Martin et al., 1976; Vonvoigtlander et al., 1983; Leighton et al., 1988), whereas activation of KORs has also been noted to be dysphoric and cause perception alterations (Pfeiffer et al., 1986). Consistent with the dysphoric effects in humans, administration of salvinorin A to mice produces a long-lasting decrease in extracellular dopamine (Zhang et al., 2005) that is consistent with KOR activation (Chefer et al., 2005). Based on the overall similarities between the behavioral effects of salvinorin A and κ agonists, we hypothesized that KOR could be the target for many if not all salvinorin A actions in vivo and thus could potentially mediate additional behavioral responses, such as hypothermia (Baker and Meert, 2002), that result from KOR activation. The most direct way to test the in vivo requirement of KOR is in a knockout (KO) model, so we have compared the responses of several salvinorin-related compounds in both wild-type mice and in a novel mouse strain containing a null mutation of the KOR-1 gene.
Materials and Methods
All experiments were conducted in accordance with the guidelines of the Institutional Care and Use Committees of UMDNJ-RWJMS and the National Institute on Drug Abuse, National Institutes of Health in facilities accredited by the American Association for the Accreditation of Laboratory Animal Care. Adult male mice were used for all initial characterization of the KOR-1 KO mice and were derived from mating of either KOR-1 heterozygous mutant mice maintained on a mixed C57BL6/J×129S6 background or wild-type and KOR-1 mutant mice maintained on an inbred 129S6 background. Subsequent behavioral experiments used wild-type and KOR-1 KO mice of both sexes maintained on the 129S6 background.
Production of KOR-1-Deficient Mice. A digoxigenin-labeled cDNA probe containing murine KOR-1 exons 2 and 3 (Yasuda et al., 1993) was used to screen a genomic library constructed from the 129/SwRe strain. One genomic clone containing exon 3 of KOR-1 was isolated, confirmed by sequencing, and used to construct the targeting vector. A 6-kb NotI/SpeI fragment 5′ of exon 3 and a 1.4-kb EcoRI fragment downstream of exon 3 were subcloned into pBS-KO vector (obtained from Dr. Steven Potter, University of Cincinnati, Cincinnati, OH) containing neomycin (neo) and thymidine kinase selection markers. Replacement of exon 3 with a neo cassette would then occur following successful homologous recombination (Fig. 1A).
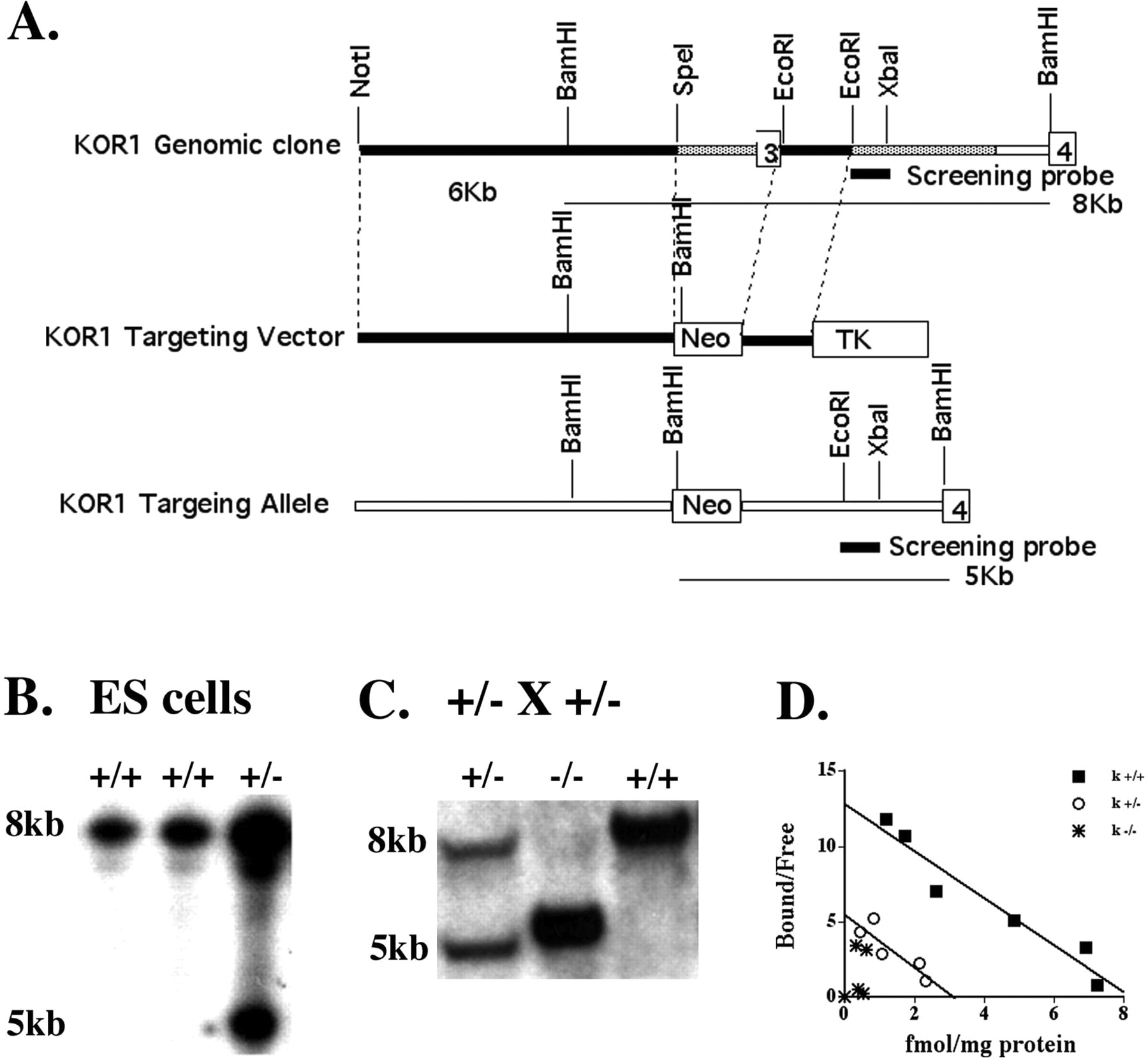
Targeting of KOR-1. A, KOR-1 genomic clone 3 was restriction-mapped, and exon 3 was positioned ∼8 kb from the 5′ end and ∼4 kb from the 3′ end of the clone. The KOR-1-targeting vector was constructed by subcloning ∼6 kb 5′ of exon 3 and 1.4 kb 3′ of exon 3 into the KO vector, such that the neo gene replaced a 2-kb KOR-1 sequence containing exon 3. In the predicted targeted locus, the neo gene would introduce an extra BamHI site. Using a screening probe outside the sequence used for targeting, Southern blot analysis detected an 8-kb wild-type allele and a 5-kb targeted allele after homologous recombination. This screening strategy was used to identify a positively targeted ES cell line after electroporation (B) and to demonstrate mice of all genotypes from offspring of heterozygous mating (C). D, [3H]U69,593 was used to assess κ receptor binding in adult brain homogenates from wild-type, heterozygous, and homozygous KOR-1 littermates. Binding was undetectable in KOR-1 mice and reduced to ∼50% in heterozygous mice. (+/+, Bmax = 8.2 fmol/mg protein; Kd = 0.64 nmol; +/-, Bmax = 3.24 fmol/mg protein; Kd = 0.59; -/-, Bmax = N/A; Kd = N/A (n = 3).
The targeting vector was linearized and electroporated into 129SvEv-derived CCE ES cells (provided by Elizabeth Robertson, Harvard University, Cambridge, MA) as described previously (Schuller et al., 1999). Individual ES colonies that survived the G418 and ganciclovir double selection were screened, and targeted ES cell lines were identified following hybridization with a 0.6-kb screening probe from a genomic region 3′ of the region incorporated into the targeting vector. This probe hybridized to a 8-kb wild-type fragment, and a diagnostic 5-kb band was derived from the targeted allele following BamHI digestion (Fig. 1B). Targeted ES cells were injected into blastocysts, and chimeras were mated with C57BL6/J and 129S6 female mice. Heterozygous mice were identified using the screening strategy described above and used to establish and maintain the mixed C57BL/6×129S6 strain and produce an inbred 129S6 line.
Homogenate Binding Assays. Adult brains from wild-type and homozygous KOR-1 mutant mice were isolated and homogenized in 30 volumes of 50 mM Tris-HCl, pH 7.4, at 4°C and then centrifuged twice at 30,000g for 15 min at 4°C with the supernatant discarded. Brain membranes were suspended in 30 volumes of buffer and incubated for 30 min at 37°C to dissociate bound endogenous ligand before recentrifugation.
Resuspended brain aliquots [400 mg wet weight in 30 ml of Tris buffer, 400-μl aliquots of membrane suspension (∼0.25 mg of protein)] were incubated with eight concentrations of [3H]U69,593 Du-Pont, Wilmington, DE and PerkinElmer Life and Analytical Sciences, Boston, MA) in 50 mM Tris-HCl, pH 7.4, for 90 min. Nonspecific binding was assessed using 10 nM naloxone. The homogenates were filtered under reduced pressure through Whatman GF/B filters (Whatman, Inc., Florham Park, NJ) using a Brandel cell harvester (Brandel, Inc., Gaithersburg, MD). Filters were washed three times with 50 mM Tris-HCl, pH 7.4, at 4°C to remove free radioligands and then assayed by liquid scintillation spectrometry. Protein concentrations were determined by the Lowry procedure (Sigma, St. Louis, MO). Binding affinities and capacities were determined by Scatchard analysis (Munson, 1983).
Chemistry. Salvinorin A was isolated from dried leaves of S. divinorum by the method reported by Valdes et al. (1994). Salvinorin B and salvinorinyl-2-propionate were prepared as detailed previously (Chavkin et al., 2004). Salvinorin B was characterized by 1H NMR, 13C NMR, and high-resolution mass spectrometry (HRMS) and found to be authentic by comparison with literature values (Valdes, 1994). The reported esters were characterized by high-performance liquid chromatography and HRMS. NMR (1H and 13C) spectra were recorded on a Bruker AMX-NMR spectrometer in CDCl3. The HRMS spectra were measured using a Bioapex FT mass spectrometer (Bruker Daltonics, Billerica, MA) with electrospray ionization. High-performance liquid chromatography was conducted on a Waters Deltaprep 4000 system (Waters, Milford, MA) using a Waters Xterra RP18,5 μm, 4.6 × 150-mm column, with mobile phase H2O/acetonitrile (1:1). Thin layer chromatography analyses were carried out on precoated Si gel G254, 250-μm plates, with the developing system hexane/EtOAc (2:1) and visualized with vanillin/H2SO4 in EtOH.
Radioligand Binding Assays. Assays of rat brain κ2-opioid receptor binding sites followed published procedures (Rothman et al., 1992). In brief, membranes prepared from whole-rat brain, pretreated with 1 μM 2-(p-ethoxybenzyl)-1-DEAE-5-isothiocyanatobenzimidazole-HCl (BIT) and 1 μM N-phenyl-N-[1-(2-(p-isothiocyanato)phenylethyl)-4-piperidinyl]propanamide-HCl (FIT) to deplete μ- and δ-opioid receptor binding sites, washed by repeated centrifugation and resuspension, and then assayed with [3H]bremazocine (2.5 nM) in 50 μM potassium phosphate buffer, pH 7.4, for 4 to 6 h at 0°C. The incubations were terminated by rapid filtration over Whatman GF/C filters. Further details of this assay have been published (Rothman et al., 1990).
Drug Administration. All experimental drugs were dissolved in 50, 60, or 100% DMSO. Controls contained equal concentrations of DMSO mixed with saline. Intracerebroventricular (i.c.v.) injections were performed as described elsewhere (Haley, 1957). In brief, mice were exposed to an isoflurane and oxygen combination for approximately 2 to 3 min until full anesthesia was observed. A midline incision was made along the sagittal suture, and a microinjection of 3 μl was administered in the lateral ventricle at 2 mm anterior to the lambda suture and 3 to 3.5 mm lateral to the midline suture that extended 2 mm below the surface of the skull. Nearly complete locomotor/nociceptive recovery from anesthesia was observed within 15 min.
Analgesic Testing. Analysis of salvinorin A, salvinorin B, and salvinorinyl-2-propionate analgesia was performed on wild-type and KOR-1 KO mice of both sexes, maintained on a 129S6 inbred background using the radiant heat tail-flick assay of nociception. Intensity of the beam was adjusted to yield baseline tail-flick latencies between 2 and 3 s, and cutoff of 10 s was employed to reduce tissue damage. Percentage maximal possible effect (%MPE) was determined according to the following formula: [(postinjection latency - preinjection latency)/(10 - preinjection latency)] × 100. Nociceptive thresholds were determined prior to drug administration. Mice were then injected i.c.v. with drug and tested for analgesia 15 min afterward. For time courses, mice were injected i.c.v. with drug and tested for analgesia every 15 min for 1 h. For cumulative dose-response curves, mice were injected i.c.v. with drug and tested for analgesia 15 min afterward. Immediately after testing, mice were injected with the next highest dose. This procedure was repeated until all doses had been administered. Initial experiments were analyzed using one-way ANOVA. Differences were isolated using Fisher's PLSD. Time dependence was evaluated using repeated measures ANOVA with time as the dependent variable and treatment as an independent variable. Individual differences were isolated using one-way ANOVA at individual time points and Fisher's PLSD for post hoc tests. Dose-dependent analgesia was evaluated using repeated measures ANOVA with dose as the dependent variable (p < 0.05). Both salvinorin A and salvinorinyl-2-propionate show a significant effect of dose in wild-type mice. Salvinorin B in wild-type mice and salvinorin A and salvinorinyl-2-propionate in KOR-1 KO mice do not show a significant effect of dose. Individual doses were analyzed using Fisher's PLSD to determine significance between wild-type and KOR-1 KO responses. ED50 values were determined using a nonlinear curve-fitting program for data expressed as both %MPE and percentage mice that became analgesic (Prism; GraphPad Software, Inc., San Diego, CA). Mice were considered analgesic when postinjection latency was greater than twice baseline line tail-flick latency. No difference in drug response was observed between sexes.
Rectal Temperature Measurement. Effects of salvinorin A and salvinorinyl-2-propionate on rectal body temperature was performed on wild-type and KOR-1 KO mice of both sexes maintained on a 129S6 inbred background. All measurements were made with a 2.5-cm rectal temperature probe (Model BAT-12; Physitemp, Clifton, NJ). The probe was inserted 2.5 cm and allowed to reach temperature for 5 to 6 s, and the measurement was then recorded. Rectal temperature was first determined before drug administration. Mice were then injected i.c.v. with drug and then measured for rectal body temperature every 15 min for 2 h. Differences were isolated using one-way ANOVA at individual time points with treatment as the independent factor and Fisher's PLSD for post hoc tests. No difference in drug response was observed between sexes.
Results
Pharmacological Characterization of Salvinorin A, Salvinorin B, and Substituted Salvinorin A Derivatives. Both salvinorin A and salvinorinyl-2-propionate significantly inhibit κ1-opioid receptor binding, whereas neither salvinorin A nor salvinorinyl-2-propionate significantly inhibited κ2-opioid receptor binding (Table 1). In contrast to the weak interaction of salvinorin A with κ2-opioid receptors in rat brain, other agents that activate pharmacologically defined κ2-opioid receptors, such as (-)-ethylketocyclazocine (Sheffler and Roth, 2003), have high affinity for the κ2-opioid receptor site (Iyengar et al., 1986). Thus, based on receptor binding affinity, salvinorin A and its derivatives are κ1-opioid receptor-selective.
Effect of Salvinorin A derivatives on KOR subtype binding and KOR-1-stimulated Ca2+ mobilization KOR-2 binding experiments were done as described under Materials and Methods. The salvinorin A derivatives were screened using a large panel of cloned human receptors, ion channels, and transporters (see Roth et al., 2002 for list) and were inactive (data not shown)

Salvinorin A and salvinorinyl-2-propionate elicit antinociception. A, wild-type mice were injected i.c.v. with 7.5 μg of salvinorin A (black bars), salvinorinyl-2-propionate (dotted bars), and salvinorin B (diagonal striped bars) dissolved in 50% DMSO or 50% DMSO (Control) (white bars); n = 6-18. *, p < 0.05 versus Control. B, wild-type mice were injected i.c.v. with 13 μg of salvinorinyl-2-propionate (dotted bars), salvinorin B (diagonal striped bars) dissolved in 100% DMSO, or 100% DMSO (Control) (white bars); n = 4-9. *, p < 0.05 versus Control.
Production and Pharmacologic Characterization of KOR-1-Deficient Mice. To evaluate the actions of salvinorin-related compounds in vivo, a novel line of KOR-1 KO mice was used. ES cells from one of two KOR-1-targeted ES clones were injected into blastocysts, and germline-transmitting chimeras were identified. Mice heterozygous for the mutant KOR-1 allele were mated, and homozygous mutant mice were identified that were viable and fertile with no obvious morphological abnormalities (Fig. 1C). Genotypes of offspring from heterozygous matings arose in the predicted Mendelian frequency (wild type, 28.2%; heterozygous, 48.4%; and homozygous, 23.4%; n = 273). The effect of the exon 3 KOR-1 gene deletion on binding of the selective κ agonist [3H]U69,593 was determined by homogenate binding assays on adult brain membrane fractions from +/+, +/-, and -/- genotypes. U69,593 binding was absent from KOR-1 KO mice, whereas binding in heterozygous mice was ∼50% that of wild-type levels (Fig. 1D), similar to another line of KOR-1 KO (Simonin et al., 1998) mice in which binding CI-977 binding was measured.
Antinociceptive Effects of Salvinorin A, Salvinorin B, and Salvinorinyl-2-propionate. To determine whether salvinorin A has analgesic potency, we initially injected salvinorin A at a dose of 5 mg/kg i.p. and observed no effect 30 min later (data not shown; also see Wang et al., 2005). We next determined whether salvinorin A has potency when injected supraspinally. Salvinorin A (7.5 μg) injected i.c.v. showed antinociceptive potency when nociception was measured 15 min later (Fig. 2A). We next tested salvinorinyl-2-propionate and salvinorin B supraspinally at the same dose (Fig. 2A). Consistent with the slight reduction in affinity compared with salvinorin A observed in previous studies (Chavkin et al., 2004), salvinorinyl-2-propionate produced a more limited antinociception at 7.5 μg than salvinorin A. In contrast, salvinorin B demonstrated no analgesic potency. We then increased the dose to 13 μg for savinorinyl-2-propionate and salvinorin B (Fig. 2B) and measured nociception 15 min later. At this dose, salvinorinyl-2-propionate produced a significant antinociceptive response, whereas salvinorin B remained inactive. Thus, in vivo analgesic activity could be elicited by only salvinorin A-like compounds active in vitro.
To further characterize salvinorin A-elicited analgesia, time-course studies for the analgesic actions of salvinorin A and salvinorinyl-2-propionate were performed. Intracerebroventricular injection of either 7.5 μg of salvinorin A or 10 μg of salvinorinyl-2-propionate produced significant analgesia as early as 15 min (Fig. 3). By 30 min, the tail-flick latencies for both the salvinorin A and salvinorinyl-2-propionate-injected groups were still elevated but not significantly different from controls. By 45 min after injection, the tail-flick latencies for all treated mice had returned to baseline values. To better compare drug time courses between behavioral paradigms (e.g., hypothermic effects; see Fig. 6), antinociceptive effects were also evaluated after i.c.v. injection of 50 μg of salvinorin A. As seen in Fig. 3, injection of this supermaximal dose of salvinorin A increased the time course for significant drug action. Salvinorin A still acted rapidly and reached 100% MPE within 15 min, but a significant drug effect was maintained for at least 45 min.
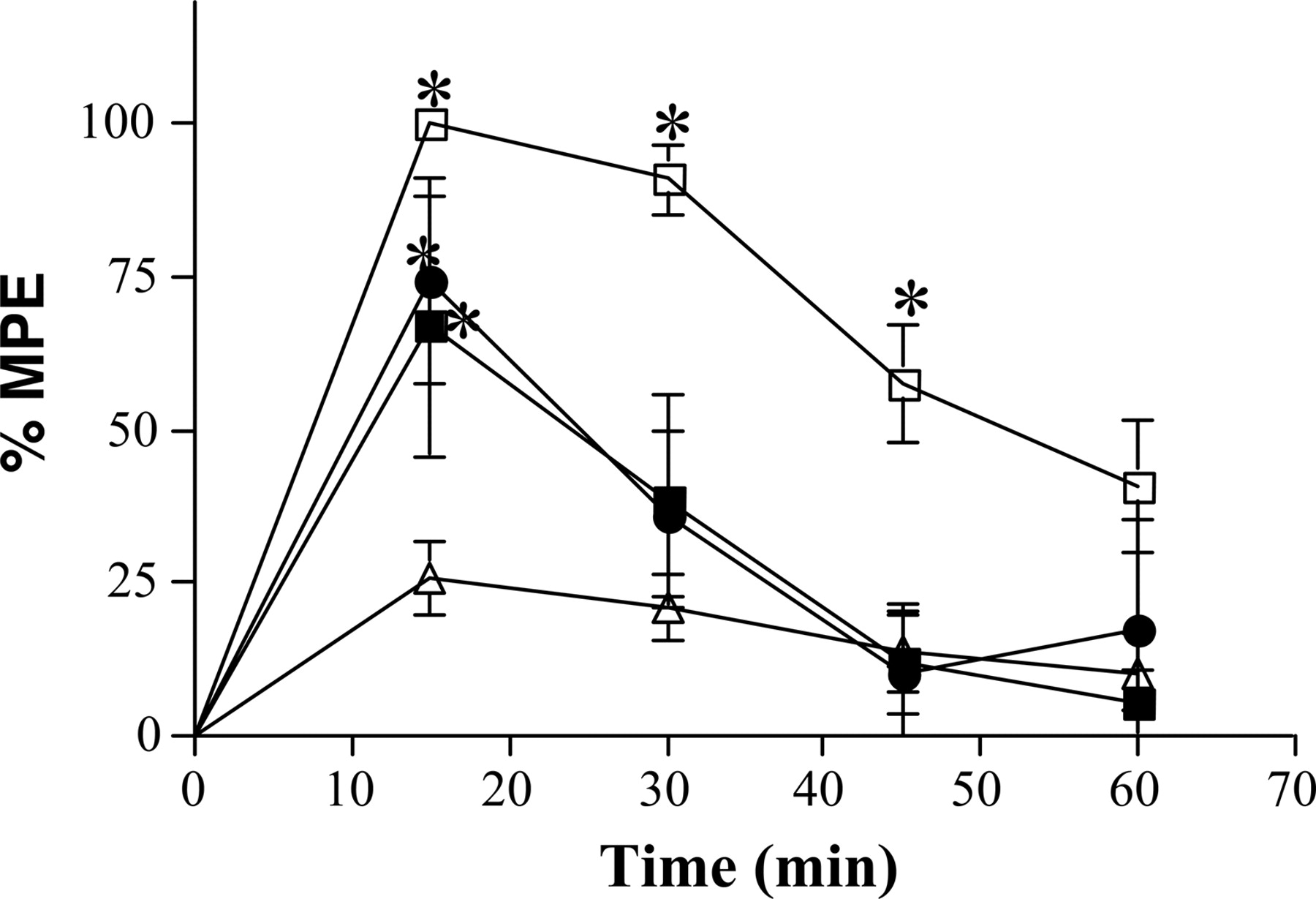
Salvinorin A and salvinorinyl-2-propionate antinociception is short-acting. Wild-type mice were injected i.c.v. with 3 μl of 100% DMSO (Control) (open triangles), 7.5 μg of salvinorin A (closed squares), 50 μgof salvinorin A (open squares), or 10 μg of salvinorinyl-2-propionate (closed circles) dissolved in 100% DMSO. Data are graphed as mean ± S.E.M; n = 6-8. *, p < 0.05 (both 7.5 and 50 μg) salvinorin A versus Control; salvinorinyl-2-propionate versus Control.

Salvinorin A and salvinorinyl-2-propionate produced antinociception through activation of the KOR. A, wild-type (closed squares) or KOR-1 KO (open squares) mice were injected i.c.v. with an escalating dose of salvinorin A (1.5-15 μg) dissolved in 100% DMSO. Data are graphed as mean ± S.E.M. n = 9 for wild-type; n = 8 for KOR-1 KO. *, p < 0.05 wild type versus KOR-1 KO. B, wild-type (closed squares) or KOR-1 KO (open squares) were injected i.c.v. with an escalating dose of salvinorinyl-2-propionate (1-30 μg) dissolved in 100% DMSO. Data are graphed as mean ± S.E.M. n = 8 for wild type; n = 6 for KOR-1 KO. *, p < 0.05 wild-type versus KOR-1 KO. C, wild-type mice (closed squares) were injected i.c.v. with an escalating dose of salvinorin B (1-30 μg) dissolved in 100% DMSO. Data are graphed as mean ± S.E.M.; n = 9. D, wild-type mice were repeatedly injected i.c.v. with either saline (closed circles) or 100% DMSO (open triangles). Data are graphed as mean ± S.E.M.; n = 5 for saline; n = 7 for DMSO.

Repeated i.c.v. injection of DMSO slightly elevates % MPE. Mice were repeatedly injected four times i.c.v. with salvinorin A [n = 9 for wild-type (open bars); n = 8 for KOR-1 KO (closed bars)]; salvinorinyl-2-propionate (n = 8 for wild type; n = 6 for KOR-1 KO) or salvinorin B (n = 9) dissolved in 100% DMSO, 100% DMSO (n = 9), or saline (n = 5). Data presented is the final drug and/or vehicle injection value from Fig. 4, A to D. Data are graphed as mean ± S.E.M. *, p < 0.05 versus wild-type 100% DMSO injected.
Dose-response curves examining the potency of salvinorin A, salvinorinyl-2-propionate, and salvinorin B analgesia were then produced with nociception measured 15 min postinjection. Both salvinorin A and salvinorinyl-2-propionate induced dose-dependent antinociception in wild-type mice with an ED50 of 1.5 and 2.0 μg (for data expressed as %MPE) or 1.4 and 1.7 μg (for data expressed as percentage mice analgesic), respectively (Fig. 4, A and B), whereas salvinorin B was inactive in wild-type mice (Fig. 4C). As further demonstration that salvinorin A and salvinorinyl-2-propionate act specifically on KORs, neither salvinorin A nor salvinorinyl-2-propionate induced analgesia in KOR-1 KO mice (Fig. 4, A and B).
Because i.c.v. injection of salvinorin A and salvinorinyl-2-propionate in KOR-1 KO mice and salvinorin B in wild-type mice increased % MPE slightly at the highest doses of the dose-response curve, we wanted to distinguish whether this effect was an antinociceptive response at high doses of the compound or a nonspecific antinociceptive effect of the vehicle (100% DMSO) in which test compounds were dissolved. Intracerebroventricular injection of 100% DMSO alone consistently stimulated a greater antinociceptive response (∼20%; Fig. 2) than previously seen following i.c.v. injection of saline alone (∼10%; data not shown). We then assessed whether repeated injections of 100% DMSO i.c.v. produced significant antinociception above this baseline value. Figure 4D shows that repeated DMSO injection does induce slight antinociception in wild-type mice after repeated dosing compared with saline injection. However, the level of analgesia observed is significantly lower than that observed following i.c.v. injection of salvinorin A or salvinorinyl-2-propionate in wild-type mice and never reached statistical significance (Fig. 5). Thus, the level of nociception in dose-response curves for salvinorin B in wild-type mice or salvinorin A or salvinorinyl-2-propionate in KOR-1 KO mice after the final i.c.v. injection is comparable with the level of nociception observed after repeated i.c.v. injection of vehicle (100% DMSO) (Fig. 5).
Effects on Rectal Body Temperature. Because KOR agonists are known to cause depression of rectal body temperature, we attempted to ascertain whether salvinorin A and salvinorinyl-2-propionate also affect rectal body temperature. Rectal body temperature is immediately suppressed in both control and drug-treated wild-type mice after i.c.v. injection because of anesthesia used during the procedure (Kushikata et al., 2005). However, rectal body temperature of vehicle-treated mice returns to baseline within 45 to 60 min of injection, whereas rectal body temperature of mice treated with 50 μg of salvinorin A or salvinorinyl-2-propionate shows prolonged suppression at least 120 min post-i.c.v. injection (Fig. 6, A and B). Although a trend for hypothermia after i.c.v. injection of 7.5 μg of salvinorin A is seen at 45 min postinjection, no significant effect on rectal body temperature was observed in wild-type mice (Fig. 6A). Intracerebroventricular injection of salvinorin B does not produce additional suppression of rectal body temperature compared with vehicle injection (Fig. 6C). Finally, to confirm that the reduction in rectal body temperature induced by i.c.v. injection of salvinorin A requires the KOR-1 gene, we injected salvinorin A i.c.v. in KOR-1 KO mice. Similar to the i.c.v. injection of salvinorin B, no difference was seen in rectal body temperature between vehicle and salvinorin A-injected KOR-1 KO mice (Fig. 6D).

Salvinorin A and salvinorinyl-2-propionate reduce rectal body temperature via the κ-opioid receptor. A, wild-type mice injected i.c.v. with 100% DMSO (Control) (closed circles), 7.5 μg of salvinorin A (closed squares), or 50 μg of salvinorin A (open squares) dissolved in 100% DMSO. Data are graphed as mean ± S.E.M.; n = 4-7. *, p < 0.05 salvinorin A versus Control. B, wild-type mice injected i.c.v. with 100% DMSO (Control) (closed circles) or 50 μg of salvinorinyl-2-propionate (closed squares) dissolved in 100% DMSO. Data are graphed as mean ± S.E.M.; n = 7. *, p < 0.05 salvinorinyl-2-propionate versus Control. C, wild-type mice injected i.c.v. with 100% DMSO (Control) (closed circles) or 50 μg of salvinorin B (closed squares) dissolved in 100% DMSO; n = 5. D, KOR-1 KO mice injected i.c.v. with 100% DMSO (Control) (closed circles) or 50 μg of salvinorin A (closed squares) dissolved in 100% DMSO. Data are graphed as mean ± S.E.M.; n = 6.
Discussion
The major finding of the present study is the genetic determination that the in vivo actions of salvinorin A and its propionate-derivative are KOR-1-dependent. Both salvinorin A and its derivative salvinorinyl-2-propionate have antinociceptive and hypothermic effects when injected i.c.v. into wild-type mice (Figs. 2, A and B, and 6, A and B), whereas both drugs were inactive in KOR-1 KO mice (Figs. 4, A and B, and 6D), demonstrating that a principal molecular target of these compounds in vivo is the KOR. The loss of antinociceptive and hypothermic effects of salvinorin A in the KOR-1 KO mouse does not rule out the possibility that salvinorin A has other sites of action for other behavioral responses still to be investigated, but what these other sites of action might be is unclear since prior studies have failed to find any other molecular target for the actions of salvinorin A, despite screening a large number of neurotransmitter receptors, ion channels, and transporters (Roth et al., 2002). Taken together, the most parsimonious explanation is that the effects of salvinorin A in vivo are mediated principally, if not exclusively, by KORs. Indeed, recent reports have suggested that the psychological effects of salvinorin A in humans (Sheffler and Roth, 2003) and discriminative stimulus effects of salvinorin A in monkeys (Butelman et al., 2004) are mediated by KORs.
Consistent with recent studies (Wang et al., 2005; McCurdy et al., 2006) showing that salvinorin A has low potency and a very short half-life in vivo, we demonstrate antinociceptive efficacy only for a transient time following an i.c.v. injection of an ED80 dose (7.5 μg) of salvinorin A (maximal response at 15 min and no effect by 45 min; Fig. 3). Hypothermic effects of salvinorin A are absent after i.c.v. injection of the same 7.5-μg dose of salvinorin A (Fig. 6A). It is possible that salvinorin A may be efficacious at 7.5 μg i.c.v., but the hypothermic effects of the anesthesia required for the i.c.v. surgery may mask these effects at early time points. Intracerebroventricular injection of 25 μg of salvinorin A was also inactive in producing hypothermia (data not shown). We obtained significant hypothermic effects only after i.c.v. injection of 50 μg of salvinorin A (Fig. 6A). The hypothermic efficacy of salvinorin A at this higher dose appears to be relatively longer lasting (maximal effect after 75 min and significant effects out to 120 min) than significant antinociceptive effects. The remaining difference in time course may be due to physiological differences between the two behaviors. Although not well documented, differences in maximal time for κ-opioid agonist action on nociception and rectal temperature have previously been observed. For example, Spencer et al. (1988) demonstrated that, although U50,488H administered i.c.v. to rats shows maximal antinociceptive efficacy after 10 min and all analgesic action is lost after 40 min, the same treatment has maximal efficacy on rectal temperature after 20 min with significant effects still present 40 min later. Similar differences in time course are seen when comparing maximal efficacy for dynorphin A-(1-13) administered i.c.v. Nakazawa et al. (1985) showed a maximal efficacy for i.c.v. administered dynorphin A at 15 min postinjection, whereas dynorphin A-(1-13) effects on rectal body temperature reach maximal effect 60 min postinjection and are still present 2 h after the initial injection (Chen et al., 2005). Exactly why such a difference exists between the time course of effects of κ agonists on nociception and rectal temperature is not known. However, the difference may be due to how activation of κ-opioid receptors influences these behaviors as well as the time required for physiological changes in these behaviors to be manifested. κ-Opioid activation may rapidly affect nociception by directly inhibiting the nociceptive neural circuitry. After degradation of the drug, the effects on nociception disappear as rapidly as they appear. Changes in rectal body temperature may take longer to appear after κ-opioid activation due to the requirement for changes in physiology to lower body temperature and may last longer due to the same requirement for physiological alterations to restore the body temperature following drug clearance.
Finally, although the vehicle DMSO used in these studies has slight behavioral activity, it does not confound interpretation of the data. High concentrations of DMSO (50% or greater) were used to dissolve salvinorin A because of its relative insolubility in common vehicles used for i.c.v. injection. After a single i.c.v. injection of DMSO, we saw ∼20% MPE. Subsequent i.c.v. injections revealed that this effect increased to a maximum of ∼45% following a fourth injection (Fig. 4D). These results are consistent with literature showing that DMSO can have antinociceptive actions (Haigler and Spring, 1981, 1983). Testing salvinorin A in both wild-type and KOR-1 KO mice eliminates any ambiguity in our interpretation of these data. The decrease in the antinociceptive effect of salvinorin A seen in the KOR-1 KO could only be due to the loss of salvinorin A action on the κ-opioid receptor. The remaining antinociceptive effect could then be attributed to DMSO. Therefore, by testing salvinorin A in wild-type and KOR-1 KO mice, we can definitively state that some if not all of salvinorin A antinociceptive effects are due to action at the κ-opioid receptor. In addition, the slight elevation of % MPE in the dose-response curve from wild-type mice injected with salvinorin B is most likely not due to antinociceptive activity of the compound, but rather the action of DMSO vehicle (Fig. 5).
In conclusion, these studies have accomplished several objectives. First, we have confirmed two behavioral responses for salvinorin A and one of its derivatives. These compounds have analgesic efficacy and depress rectal body temperature consistent with their identification as KOR agonists. Second, we show that salvinorin B is inactive at comparable doses, indicating that salvinorin A is, most likely, the main active component of S. divinorum in vivo as it is in vitro. In addition, we provide the first in vivo structure-function studies of salvinorin A at the KOR and have identified salvinorinyl-2-propionate as a novel salvinorin A derivative with appreciable KOR actions in vivo. We also demonstrate that salvinorin A interacts with the κ1-opioid receptor subtype but not the κ2-opioid receptor subtype. Finally, by performing behavioral assays in wild-type and a novel strain of KOR-1 KO mice, we genetically confirm that one in vivo site of action for salvinorin A is the KOR and thus demonstrate that salvinorin A is a functional KOR agonist with behavioral consequences when injected i.c.v.
Footnotes
-
This work was supported by National Institutes of Health (NIH) Grants DA-009040 and DA-015237 (to J.E.P.), the Intramural Research Program of the NIH, and National Institute of Drug Abuse (NIDA) (to R.B.R.), NIH, Grants DA-017204 (to B.L.R.) and MH/AG 19957 and F32 DA-14755 (to M.A.A.).
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.101998.
-
ABBREVIATIONS: KORs, κ-opioid receptors; i.c.v., intracerebroventricularly; KO, knockout; kb, kilobase; neo, neomycin; U69,593, (+)-(5a,7a,8b)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspirodec-8-yl]-benzeneacetamide; HRMS, high-resolution mass spectrometry; BIT, 2-(p-ethoxybenzyl)-1-DEAE-5-isothiocyanatobenzimidazole-HCl; FIT, N-phenyl-N-[1-(2-(p-isothiocyanato)phenylethyl)-4-piperidinyl]propanamide-HCl; DMSO, dimethyl sulfoxide; %MPE, percentage maximal possible effect; U50,488H, (trans)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidiny)-cyclohexyl]-benzeneacetamide; ANOVA, analysis of variance; PLSD, protected least significant difference; ES, embryonic stem.
- Received January 27, 2006.
- Accepted May 2, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}