


Abstract
Previous studies identified partial inhibitors of serotonin (5-HT) transporter and dopamine transporter binding. We report here on a partial inhibitor of 5-HT transporter (SERT) binding identified among a group of 1-[2-[bis(4-fluorophenyl)methoxy]ethyl]-4-(3-phenylpropyl)piperazine analogs (4-[2-[bis(4-fluorophenyl)-methoxy]ethyl]-1-(2-trifluoromethyl-benzyl)-piperidine; TB-1-099). Membranes were prepared from rat brains or human embryonic kidney cells expressing the cloned human dopamine (hDAT), serotonin (hSERT), and norepinephrine (hNET) transporters. β-(4′-125Iodophenyl)tropan-2β-carboxylic acid methyl ester ([125I]RTI-55) binding and other assays followed published procedures. Using rat brain membranes, TB-1-099 weakly inhibited DAT binding (Ki = 439 nM), was inactive at NET binding ([3H]nisoxetine), and partially inhibited SERT binding with an extrapolated plateau (“A” value) of 20%. Similarly, TB-1-099 partially inhibited [125I]RTI-55 binding to hSERT with an extrapolated plateau (A value) of 14%. Upon examining the effect of increasing concentrations of TB-1-099 on the apparent Kd and Bmax of [125I]RTI-55 binding to hSERT, we found that TB-1-099 decreased the Bmax in a dose-dependent manner and affected the apparent Kd in a manner well described by a sigmoid dose-response curve. TB-1-099 increased the Kd but not to the magnitude expected for a competitive inhibitor. In rat brain synaptosomes, TB-1-099 noncompetitively inhibited [3H]5-HT, but not [3H]dopamine, uptake. Dissociation experiments indicated that TB-1-099 promoted the rapid dissociation of a small component of [125I]RTI-55 binding to hSERT. Association experiments demonstrated that TB-1-099 slowed [125I]RTI-55 binding to hSERT in a manner unlike that of the competitive inhibitor indatraline. Viewed collectively, these results support the hypothesis that TB-1-099 allosterically modulates hSERT binding and function.
The serotonin (5-HT) transporter (SERT) is a member of the twelve transmembrane-spanning transporter family that transports 5-HT across cell membranes in a sodium-dependent manner (Amara and Kuhar, 1993; Uhl and Johnson, 1994). SERT, in the central nervous system, is an important target for a wide range of medications used to treat a variety of psychiatric conditions such as anxiety, depression, and obsessive-compulsive disorder (Gorman and Kent, 1999; Zohar and Westenberg, 2000).
Drugs that interact with transporters generally interact with this protein in two distinct ways. Reuptake inhibitors bind to transporter proteins but are not transported. These drugs elevate extracellular concentrations of transmitter by blocking transporter-mediated uptake of transmitters from the synapse. Substrate-type releasers bind to transporter proteins and are subsequently transported into the cytoplasm of nerve terminals. Releasers elevate extracellular transmitter concentrations by a two-pronged mechanism: 1) they increase cytoplasmic levels of transmitter by disrupting storage of transmitters in vesicles, and 2) they promote non-exocytotic release of transmitters by a process of carrier-mediated exchange (Rudnick and Clark, 1993). Although most therapeutic agents that target the SERT inhibit its function, an effect termed uptake inhibition, there is emerging evidence that SERT substrates may offer unique therapeutic effects (Rothman and Baumann, 2002a,b).
Several different types of assays can be used to characterize the interaction of test agents with the biogenic amine transporters. These include nonfunctional ligand binding assays, where compounds are tested for their ability to inhibit the binding of a radioactive transporter inhibitor such as 3β-(4′-125iodophenyl)tropan-2β-carboxylic acid methyl ester ([125I]RTI-55) to the transporter and functional assays where compounds are tested for their ability to inhibit the reuptake of 3H-neurotransmitter by synaptosomes. For the sake of brevity, we refer in this study to the transporter ligand binding assays, which measure the affinity of a test compound for the transporter inhibitor site of the transporter, as SERT, NET (norepinephrine transporter), and DAT (dopamine transporter) binding assays. Similarly, we refer to the uptake inhibition assays as either SERT, NET, and DAT uptake assays or [3H]5-HT, [3H]NE, or [3H]dopamine (DA) uptake assays.
As part of our ongoing studies of the biogenic amine transporters and efforts to develop novel agents to test the DA hypothesis of cocaine addiction, we have characterized hundreds of compounds at SERT using both ligand binding assays and functional assays (Prisinzano et al., 2004). Until recently, active compounds inhibited [125I]RTI-55 binding to the SERT in a manner consistent with mass action. Recently, however, we reported an agent, SoRI-6238, that partially inhibited SERT binding, allosterically modulated the dissociation rate of [125I]RTI-55 binding from rat brain SERT, and allosterically modulated SERT function (Nandi et al., 2004), as indicated by noncompetitive inhibition of the [3H]5-HT uptake. Previously, we reported a novel compound, SoRI-9804 (4-[(diphenylmethyl)amino]-2-phenylquinazoline), that partially inhibited [125I]RTI-55 binding to the DAT and [3H]DA uptake by rat brain synaptosomes (Rothman et al., 2002). In the present report we describe a GBR12909 (1-[2-[bis(4-fluorophenyl)methoxy]ethyl]-4-(3-phenylpropyl)piperazine) analog, TB-1-099 (4-[2-[bis(4-fluorophenyl)methoxy]ethyl]-1-(2-trifluoromethyl-benzyl)-piperidine), that appears to allosterically modulate certain properties of hSERT expressed in HEK cells, such as SERT binding and SERT uptake.
Materials and Methods
Animals. Male Sprague-Dawley rats (300–450 g) used for 3H-neurotransmitter uptake assays were obtained from Charles River Laboratories (Wilmington, MA). The animal housing facilities were fully accredited by the American Association of the Accreditation of Laboratory Animal Care (AAALAC), and all experiments were performed within the guidelines delineated in the Institutional Care and Use Committee of the National Institute on Drug Abuse (NIDA), Intramural Research Program (IRP).
Tissue Preparation. The transporter binding assays used membranes prepared from frozen rat brains (Nandi et al., 2004). Briefly, frozen rat brains (Pel Freeze, Rogers, AZ) were each thawed in 20 ml of ice-cold binding buffer (BB; 55.2 mM sodium phosphate buffer, pH 7.4) for 15 min and then homogenized with a Brinkmann Polytron (Brinkmann Instruments, Westbury, NY; setting 6 for 20 s). The homogenates were centrifuged twice at 20,000g for 15 min, with resuspensions in equal volumes of ice-cold BB, and then resuspended in 10 ml/brain ice-cold binding buffer. The membranes were pooled, mixed, and then separated into 1-ml aliquots in polypropylene microcentrifuge tubes. Each aliquot was centrifuged in a TOMY refrigerated microcentrifuge (model MTX 150; Tomy Seiko, Tokyo, Japan) at maximum speed for 5 min; the supernatant was discarded. The aliquots were then stored at -80°C until needed. When used in the experiment, each aliquot was diluted in 120 ml of binding buffer.
HEK cells expressing hNET, hDAT, or hSERT were grown to confluency using standard methods on plates, medium was removed, and the plates were stored at -80°C until the day of the assay. The plates were thawed, and the cells scraped off and rinsed with BB and homogenized with a polytron at setting 6 for 10 s. The homogenate was centrifuged twice at 30,000g for 10 min, with resuspensions in phosphate buffer. The final pellets were resuspended in a final volume of 300 ml/plate with BB.
Transporter Binding Assays. For rat brain transporter binding assays, experiments were carried out in 12 × 75-mm polystyrene tubes that were prefilled with 100 μl of drug, 100 μl of radioligand, and 50 μl of a blocker or buffer (Rothman et al., 1998). For assay of the SERT and DAT, the blockers were either 100 nM 4-(2-benzhydryloxy-ethyl)-1-(4-nitro-benzyl)piperidine oxalate to block the DAT or 100 nM citalopram to block SERT. [125I]RTI-55 was made in a protease inhibitor cocktail (BB with 25 μg/ml chymostatin, 25 μg/ml leupeptin, 1 μM EGTA, and 1 μM EDTA). The drugs and blockers were made up in BB with 1 mg/ml bovine serum albumin at pH 7.4. The experiment was initiated with the addition of 750 μl of membranes in BB. Samples were incubated in a final volume of 1 ml, at 4°C for 18 to 24 h (steady state). After incubation the samples were filtered with a Brandel cell harvester (Brandel, Gaithersburg, MD) over Whatman GF/B filters (Brandel) presoaked in wash buffer (ice-cold 10 mM Tris/HCl, and 150 mM NaCl, pH 7.4) containing 2% poly(ethyleneimine). For assay of the rat NE transporter (NET), we used [3H]nisoxetine (American Radiolabeled Chemicals, St. Louis, MO) as described elsewhere (Rothman et al., 1998). Nonspecific binding was measured using 10 μM nisoxetine. It was not possible to test SoRI-6238 concentrations greater than 10-4 M. Typical total and nonspecific cpms observed for the rat brain transporter binding assays were: SERT (4000; 80), DAT (4000; 90), and NET (1500; 300). Assays using HEK cells expressing hSERT, hNET, or hDAT were conducted as described above, except that no blockers were used, [125I]RTI-55 was used as the radioligand for all three transporters, and nonspecific binding was determined with 1 μM indatraline. For the sake of brevity, in the study, hSERT, hNET, or hDAT binding assays refer to assays conducted with HEK cells. Typical total and nonspecific cpms observed for the HEK cell transporter binding assays were: hSERT (4000; 80), hDAT (2000; 75), and hNET (2000; 200).
Kinetic Experiments. Kinetic experiments were conducted with minor modifications of published methods (Nandi et al., 2004). For SERT dissociation experiments, [125I]RTI-55 (0.01 nM) was incubated for 2 h at 25°C (steady state). At that point, 100 μl of RTI-55 (final concentration 1 μM) or buffer was added. Ten minutes later, test drug (TB-1-099, 25 μM; SoRI-6238, 10 μM) was added. This design insured that any effect of the test drug could not be due to interactions with the SERT binding site, since this site would be completely occupied by RTI-55. Triplicate samples were filtered at various time points after the addition of TB-1-099: 10, 30, 50, 80, and 110 min. For the purpose of data analysis, the 100% of control point was “time 0 min” for conditions that did not receive test drug and “time 10 min” for conditions that received test drug.
In other kinetic experiments we followed methods described by Contreras et al. (1986). Briefly, we measured the association [125I]RTI-55 (0.01 nM) to SERT in the absence and presence of competing drugs. Triplicate samples were filtered at eight time points, from 5 min to 2 h. [125I]RTI-55 binding surfaces were generated at the same time to permit calculation of the Kd and Bmax of SERT in the same preparation of membranes used for the time course. The time course data were fit to the equations described by Contreras for the best-fit estimates of the association (kon) and dissociation rates (koff) (Contreras et al., 1986).
Neurotransmitter Uptake Assays. Neurotransmitter uptake inhibition assays were conducted as described elsewhere (Rothman et al., 2001). Freshly removed caudate ([3H]DA uptake) or whole brain minus caudate ([3H]5-HT uptake) were homogenized in 10% ice-cold sucrose with 12 strokes of a handheld Potter-Elvehjem homogenizer followed by centrifugation at 1000g for 10 min. The supernatants were saved on ice and used immediately. The assay buffer used was Krebs-phosphate buffer containing 154.4 mM NaCl, 2.9 mM KCl, 1.1 mM CaCl2, 0.83 mM MgCl2, 5 mM glucose, 1 mg/ml ascorbic acid, and 50 μM pargyline. Nonspecific uptake was measured by incubating in the presence of 1 μM GBR12909 ([3H]DA uptake) or 10 μM fluoxetine ([3H]5-HT). The reactions were stopped after 15-min (for [3H]DA) or 30-min (for [3H]5-HT) incubations by filtering with a Brandel cell harvester over Whatman GF/B filters presoaked in wash buffer (10 mM Tris/HCl, pH 7.4). [3H]NE assays were conducted as described (Rothman et al., 2001). Retained tritium was measured with a Trilux liquid scintillation counter after overnight extraction in 0.6 ml of liquid scintillation cocktail. Typical total and nonspecific cpms observed for the rat brain transporter uptake inhibition assays were: SERT (16,000; 800), DAT (18,000; 1000), and NET (5000; 1300).
Experimental Design. Inhibition curves were generated by displacing a single concentration of [125I]RTI-55 (0.01 nM) by 8 to 10 concentrations of drug. For binding surface experiments (Rothman, 1986; Rothman et al., 1991), two different concentrations of radioligand were each displaced by 8 to 10 concentrations of test agents in the absence or presence of various blockers. Similarly, surfaces for [3H]5-HT uptake were generated by displacing two concentrations of [3H]5-HT (2 and 22 nM) each by nine concentrations of 5-HT (2–766 nM) in the absence and presence of TB-1-099. Surfaces for [3H]DA uptake were generated by displacing two concentrations of [3H]DA (10 and 55 nM) by DA (4–1532 nM) in the absence and presence of TB-1-099. The higher concentrations of [125I]RTI-55, [3H]DA, or [3H]5-HT were obtained by adding RTI-55, DA, or 5-HT to the radioligand. Dissociation and association experiments were conducted with minor modification of published procedures (Rothman et al., 1991).
Data Analysis and Statistics. Data analysis proceeded as described elsewhere (Rothman et al., 1998). IC50 values were calculated with the MLAB-PC program (Civilized Software, Bethesda, MD) using the two- or three-parameter logistic equation. The two-parameter logistic equation is:  and the three-parameter logistic equation is:
and the three-parameter logistic equation is:  where A is the plateau, and N is the slope factor.
where A is the plateau, and N is the slope factor.
For drugs that produced inhibition curves without apparent plateaus, the data were fit to the two parameter logistic equation for the best-fit estimates of the IC50 and N values. For curves with apparent plateaus, the data were fit first to the three-parameter logistic equation to determine the best-fit value of the plateau (A value). Using this value, the inhibition curve was normalized according to the following equation:  The data were then fit to the two-parameter logistic equation (eq. 1).
The data were then fit to the two-parameter logistic equation (eq. 1).
Graphs were generated with KaleidaGraph 3.6 software (Abelbeck/Synergy, Reading, PA). Binding surfaces were fit to one- and two-site binding models using MLAB-PC as described elsewhere (Rothman et al., 1991). Dissociation experiments were fit to one- or two-component dissociation models (Nandi et al., 2004). Statistical significance among binding models was determined using the F-test with a threshold of P < 0.01.
Chemicals. [3H]Nisoxetine (SA = 80 Ci/mmol) was obtained from American Radiolabeled Chemicals. [3H]DA (SA = 31.8 Ci/mmol) and [3H]5-HT (SA = 23.7 Ci/mmol) were from PerkinElmer Life and Analytical Sciences (Boston, MA). Thomas Prizansano kindly provided the highly selective DA uptake inhibitor 4-(2-benzhydryloxy-ethyl)-1-(4-nitro-benzyl)piperidine oxalate, synthesized as described (Greiner et al., 2003). SoRI-6238 was synthesized as described (Temple Jr. et al., 1982). The sources of other chemicals are published (Rothman et al., 1998).
Results
Using rat brain membranes, initial studies with TB-1-099 indicated that it weakly inhibited DAT binding (Ki = 440 nM) and was inactive at NET binding, determined with [3H]nisoxetine. As reported in Fig. 1, TB-1-099 partially inhibited SERT binding, with an extrapolated plateau (A value) of 21 ± 4%. In contrast, TB-1-101, which differed from TB-1-099 only in the “R” substituent, completely inhibited SERT binding. SoRI-6238, reported previously (Nandi et al., 2004), also partially inhibited SERT binding, with an extrapolated A value of 34.5%. It was not possible to test higher concentrations of SoRI-6238 because of solubility issues.
As reported in Fig. 2A, TB-1-099 inhibited hSERT binding with a plateau (A = 13 ± 2%) with an IC50 value of 1215 ± 122 nM. Fitting the hSERT binding data to a three-parameter logistic equation significantly improved the goodness of fit (F = 55, p < 0.0001). Much as the TB-1-099 plateau was lower with hSERT binding as compared with rat brain SERT binding, the SoRI-6238 plateau was lowered with the hSERT binding to the point where the extrapolated A value was not significantly different from zero (3%). In contrast, indatraline potently inhibited hSERT binding with no discernable plateau (KI = 3.9 ± 0.2 nM). TB-1-099 inhibited [125I]RTI-55 binding to hDAT (IC50 = 783 ± 69 nM) and hNET (IC50 = 1017 ± 117 nM) with similar potencies (Fig. 2B). Fitting these data to the three-parameter logistic equation did not significantly improve the goodness of fit as compared with the two-parameter logistic equation, indicating that TB-1-099 did not produce a plateau in the hDAT and hNET binding assays.

Effect of TB-1-099, TB-1-101, and SoRI-6238 on [125I]RTI-55 binding to SERT to rat brain membranes. [125I]RTI-55 (0.01 nM) was displaced by the indicated concentrations of test drug. Each value is the mean of three determinations, which varied by less than 10%. The data for SoRI-6238 are taken from Nandi et al., 2004.

A, effect of indatraline, SoRI-6238, and TB-1-099 on [125I]RTI-55 binding to hSERT expressed in HEK cells. B, effect of TB-1-099 on [125I]RTI-55 binding to hSERT, hDAT, and hNET expressed in HEK cells. [125I]RTI-55 (0.01 nM) was displaced by the indicated concentrations of TB-1-099. Each value is the mean of three determinations that differed by less than 10%. Only the hSERT curve produced a statistically significant plateau (A = 13 ± 2%, F = 55, p < 0.0001). Each point is the mean ± S.E.M. (n = 3).

Effect of TB-1-099 on [125I]RTI-55 binding to hSERT expressed in HEK cells. A, effect of TB-1-099 on the Kd and Bd of [125I]RTI-55 binding to hSERT. Two concentrations of [125I]RTI-55 (0.01 and 1 nM) were each displaced by 10 concentrations of RTI-55 (0.1 to 35.2 nM) in the absence and presence of various concentrations of TB-1-099 (1, 5, and 25 μM). The data of three experiments were combined and analyzed simultaneously with the control data (144 data points) for the best-fit estimates of the one-site model. The control Bmax = 6910 ± 430 fmol/mg protein and the control Kd = 0.42 ± 0.03 nM. The data for the Kd and Bmax values are reported as a percentage of control. *, p < 0.01 when compared with control (F-test). Each value is ±S.D. B, the effect of TB--099 on the Kd of [125I]RTI-55 binding to hSERT. The data are shown as a percentage of increase compared with the control Kd value of 0.42 nM. The inset shows the effect of TB-1-099 on the apparent Kd assuming that TB-1-099 competitively inhibited hSERT binding with a KI = 1.2 μM.
We next determined the effect of increasing concentrations of TB-1-099 on the apparent Kd and Bmax of [125I]RTI-55 binding to hSERT. It is expected that increasing the concentration of a competitive inhibitor would have no effect on the Bmax but would increase the apparent Kd proportionally according to the equation:  The results showed that TB-1-099 decreased the Bmax in a dose-dependent manner (Fig. 3A). The effect of TB-1-099 on the apparent Kd was not compatible with competitive inhibition (Fig. 3B). Assuming a competitive KI of 1.2 μM (the IC50 value for TB-1-099 for hSERT binding reported in Fig. 2), TB-1-099 is predicted to increase the apparent Kd of [125I]RTI-55 binding to hSERT in a linear fashion (inset to Fig. 3B). However, the effect of TB-1-099 on the apparent Kd was well described by a sigmoid dose-response curve. Moreover, the magnitude of the TB-1-099-induced increase in the apparent Kd was much lower than predicted according to a competitive inhibition model. In contrast to the effects of TB-1-099, and as expected for a competitive inhibitor, 5 nM indatraline increased the Kd for [125I]RTI-55 binding to hSERT from 0.25 ± 0.1 to 1.0 ± 0.1 nM without changing the Bmax (data not shown, based on three independent experiments with a total number of 132 data points). The calculated Ki of indatraline was 1.7 nM.
The results showed that TB-1-099 decreased the Bmax in a dose-dependent manner (Fig. 3A). The effect of TB-1-099 on the apparent Kd was not compatible with competitive inhibition (Fig. 3B). Assuming a competitive KI of 1.2 μM (the IC50 value for TB-1-099 for hSERT binding reported in Fig. 2), TB-1-099 is predicted to increase the apparent Kd of [125I]RTI-55 binding to hSERT in a linear fashion (inset to Fig. 3B). However, the effect of TB-1-099 on the apparent Kd was well described by a sigmoid dose-response curve. Moreover, the magnitude of the TB-1-099-induced increase in the apparent Kd was much lower than predicted according to a competitive inhibition model. In contrast to the effects of TB-1-099, and as expected for a competitive inhibitor, 5 nM indatraline increased the Kd for [125I]RTI-55 binding to hSERT from 0.25 ± 0.1 to 1.0 ± 0.1 nM without changing the Bmax (data not shown, based on three independent experiments with a total number of 132 data points). The calculated Ki of indatraline was 1.7 nM.

Scatchard plots of the data reported in Fig. 3. As described under Results, the data (264 data points) described in Fig. 3 were fit to the two-site binding model for the best-fit parameter estimates. These parameter estimates were then used to generate predicted Scatchard plots (solid lines) for [125I]RTI-55 binding to hSERT in the presence of 0, 1000, 5000, and 25,000 nM TB-1-099. Scatchard plots of the actual data were generated after the data of the three experiments described in Fig. 3 were averaged and converted to Scatchard format. The best-fit straight lines (dashed) of the actual data for 0 (▪), 1000 (×), 5000 (+), and 25,000 nM (□) TB-1-099 are shown above.
Since the presence of a plateau in the hSERT binding assay could also result from the presence of two sites and/or states, we conducted an additional analysis of the data set reported in Fig. 3. The entire data set was pooled (264 data points) and fit to the two site-binding model for the best-fit estimates of the Bmax, Kd, and KI values. According to this two-site fit, [125I]RTI-55 labeled two binding sites with Bmax values (±S.D.) of 4850 ± 322 and 1070 ± 260 fmol/mg protein. The corresponding Kd and KI values (±S.D.) (site 1 and site 2) were RTI-55 (0.39 ± 0.03 nM, 0.32 ± 0.06 nM) and TB-1-099 (970 ± 170 nM, 1225 × 103 ± 8729 × 103 nM). The extremely high S.D. associated with the KI value of TB-1-099 for the low-affinity binding site reflects the apparent ultra-low affinity of TB-1-099 for this putative binding site. To assess the validity of the two-site model, we plotted the averaged data in Scatchard format and compared the observed data with the curves predicted by the two-site model (Fig. 4). The actual data points of the [125I]RTI-55 binding saturation binding curves in the absence and presence of TB-1-099 were each well described by a straight line (dashed). In contrast, the curves predicted by the two-site model (solid lines) only described the [125I]RTI-55 binding saturation binding curve in the absence of TB-1-099 well. Importantly, the predicted curves in the presence of 5000 and 25,000 nM TB-1-099 were markedly curvilinear, whereas the observed data were well described by a straight line (see Fig. 4, inset).
The next series of experiments determined the effect of TB-1-099 on the dissociation kinetics of [125I]RTI-55 binding to hSERT. Since our previous study showed that SoRI-6238 slowed the dissociation rate of [125I]RTI-55 binding to rat brain SERT, we also included SoRI-6238 in these experiments as a positive control. Table 1 reports the experiment design used for these experiments. Briefly, the addition of 1 μM RTI-55 initiated dissociation of [125I]RTI-55 hSERT binding, after which test drugs were added so that any effect of the test agent could not be due to an effect at the transporter binding sites, as these were prebound with RTI-55. As reported in Fig. 5A, the dissociation of [125I]RTI-55 from hSERT initiated by 1 μM RTI-55 proceeded in a monotonic manner and was well described by a single-component dissociation model (koff = 0.03 ± 0.002 min-1). The addition of SoRI-6238 after the addition of RTI-55 slowed the dissociation rate (koff = 0.01 ± 0.0007 min-1). The dissociation of [125I]RTI-55 initiated by the addition of SoRI-6238 alone was well described by a two-component dissociation model. However, the parameter values had extremely large S.D. values, for example, koff = 8.6 × 10-19 ± 0.0006, indicating that the data were in fact too complex or too limited to produce a meaningful fit to a two-component model. Unlike SoRI-6238, TB-1-099 failed to alter RTI-55-induced dissociation of [125I]RTI-55 from hSERT (Fig. 5B).
Experimental design for kinetic dissociation experiments Binding of [125I]RTI-55 (0.01 nM) to hSERT proceeded for 2 h at 25°C. At this point (time 0), 1 μM RTI-55 was added to conditions 2 and 4. Ten minutes later, test drugs were added to conditions 3 and 4. For the purpose of data analysis, the 100% of control point was ′time 0 min′ for conditions 1 and 2 and ′time 10 min′ for conditions 3 and 4. Each data point is the mean ± S.D. of three experiments. The data were fit to a one- or two-component exponential decay model to calculate the k-1 constant and half-life.
We used the method described by others (Motulsky and Mahan, 1983; Contreras et al., 1986) to compare the effect of SoRI-6238 and indatraline on the association kinetics of [125I]RTI-55 binding to hSERT. Indatraline was chosen as an example of a competitive inhibitor. According to this method, a time course of radioligand binding is generated in the absence and presence of competing drug. When the data for radioligand binding in the absence of a competing drug is fit to the appropriate single-ligand equation, it is possible to determine the kinetic constants (kon and koff) of the radioligand. The Kd value, determined at the same time by equilibrium binding experiments, should equal the value calculated from the kinetic constants (koff/kon). At equilibrium, a competing drug will increase the apparent Kd of the radioligand by a factor equal to (1 + [drug]/KI). According to Contreras et al. (1986) and Motulsky and Mahan (1983), a competing drug slows the apparent association rate of the radioligand, decreasing the apparent value of kon. Moreover, the value of koff must change such that the kinetically calculated apparent Kd (koff/kon) increases. In the context of the present experiments, we postulated that indatraline, as a competitive inhibitor of SERT binding, would behave in this way and that TB-1-099, as an allosteric modulator, would not.

hSERT kinetic dissociation experiments. The experimental design is described in Table 1. Each point is the mean ± S.D. (n = 3). The curves shown are generated by a one-component dissociation model except for the curves for SoRI-6238 alone (A) and TB-1-099 alone (B), which were fit to a two-component dissociation model.

hSERT kinetic association experiments. [125I]RTI-55 (0.01 nM) binding to SERT was assessed at various time points in the absence and presence of the indicated test drugs, as illustrated for 1.5 and 15 nM TB-1-099 (A). Similar time courses were run using 2 and 10 nM indatraline (B). Each value is the mean ± S.D. (n = 3). See Table 2 for the data analysis.
The association experiments showed that indatraline behaved like a competitive inhibitor (see Fig. 6B and Table 2). It decreased the apparent kon and, when indatraline was increased from 2 to 10 nM, the kinetically calculated Kd increased 3.6-fold, which is similar to the predicted 4.8-fold increase. Moreover, the apparent Ki of indatraline calculated from the association experiments is 1.4 nM, similar to the value determined by direct binding studies (1.7 nM, see above). In contrast, TB--099 did not behave like a competitive inhibitor (Fig. 6A and Table 2). As noted above, a competitive inhibitor is expected to decrease the apparent kon in a dose-dependent manner and increase the kinetically calculated Kd (koff/kon) by a factor of (KI + [inhibitor-2])/(KI +[inhibitor-1]), where inhibitor-2 is the higher and inhibitor-1 is the lower inhibitor concentration. Thus, assuming a KI of 1.2 μM for TB-1-099 (Fig. 3), 15 μM TB-1-099 should have increased the kinetically calculated Kd 6-fold above that calculated for 1.5 μM TB-1-099 rather than the 2-fold increase actually observed.
Summary of association experiment results using hSERT [125I]RTI-55 association time courses were conducted and analyzed as described under Materials and Methods. The indatraline and TB-1-099 experiments were done separately, requiring separate control conditions. The apparent kinetic Kd = Koff/Kon and the actual Kd were determined by binding surfaces conducted at the same time and using the same membranes as the association experiments.
As reported in Fig. 7, TB-1-099 inhibited [3H]DA uptake (IC50 = 4190 ± 340 nM) about 10 times more potently than [3H]5-HT uptake (IC50 = 56,600 ± 5540 nM), without any evidence of a plateau. TB-1-099 was inactive at inhibiting [3H]NE uptake (IC50 > 75 μM, data not shown). The KI values for indatraline, determined at the same time as a positive control, were [3H]DA uptake (7.4 ± 0.5 nM), [3H]5-HT uptake (4.5 ± 0.4 nM), and [3H]NE uptake (11 ± 1 nM). To determine whether TB-1-099 inhibited [3H]neurotransmitter uptake in a competitive manner, uptake surfaces were generated as described in Materials and Methods, allowing us to determine the effect of TB-1-099 on the Vmax and Km of [3H]DA and [3H]5-HT uptake. TB-1-099 competitively inhibited [3H]DA uptake, as indicated by a statistically significant increase in the apparent Km without any change in the Vmax (Table 3). The calculated Ki of TB-1-099 for inhibiting [3H]DA uptake was 405 nM. In contrast, 50 μM TB-1-099 decreased the Vmax for [3H]5-HT uptake by about 50%, with a minimal but significant increase in the Km value. Converting these data to Scatchard format (Fig. 8) clearly showed that TB-1-099 noncompetitively inhibited [3H]5-HT uptake but not [3H]DA uptake. Indatraline, included as a positive control, acted as a competitive inhibitor, with a calculated KI value of 2.1 nM.
TB-1-099 noncompetitively inhibits [3H]5-HT uptake Uptake binding surfaces were generated as described under Materials and Methods in the absence and presence of the indicated concentrations of TB-1-099. The data of three experiments (132 data points) were pooled and fit to the one-component uptake equation for the best-fit estimates of the Vmax and Km. The two [3H]5-HT experiments were run several months apart, each with its own control. Each value is ±S.D., n = 3.
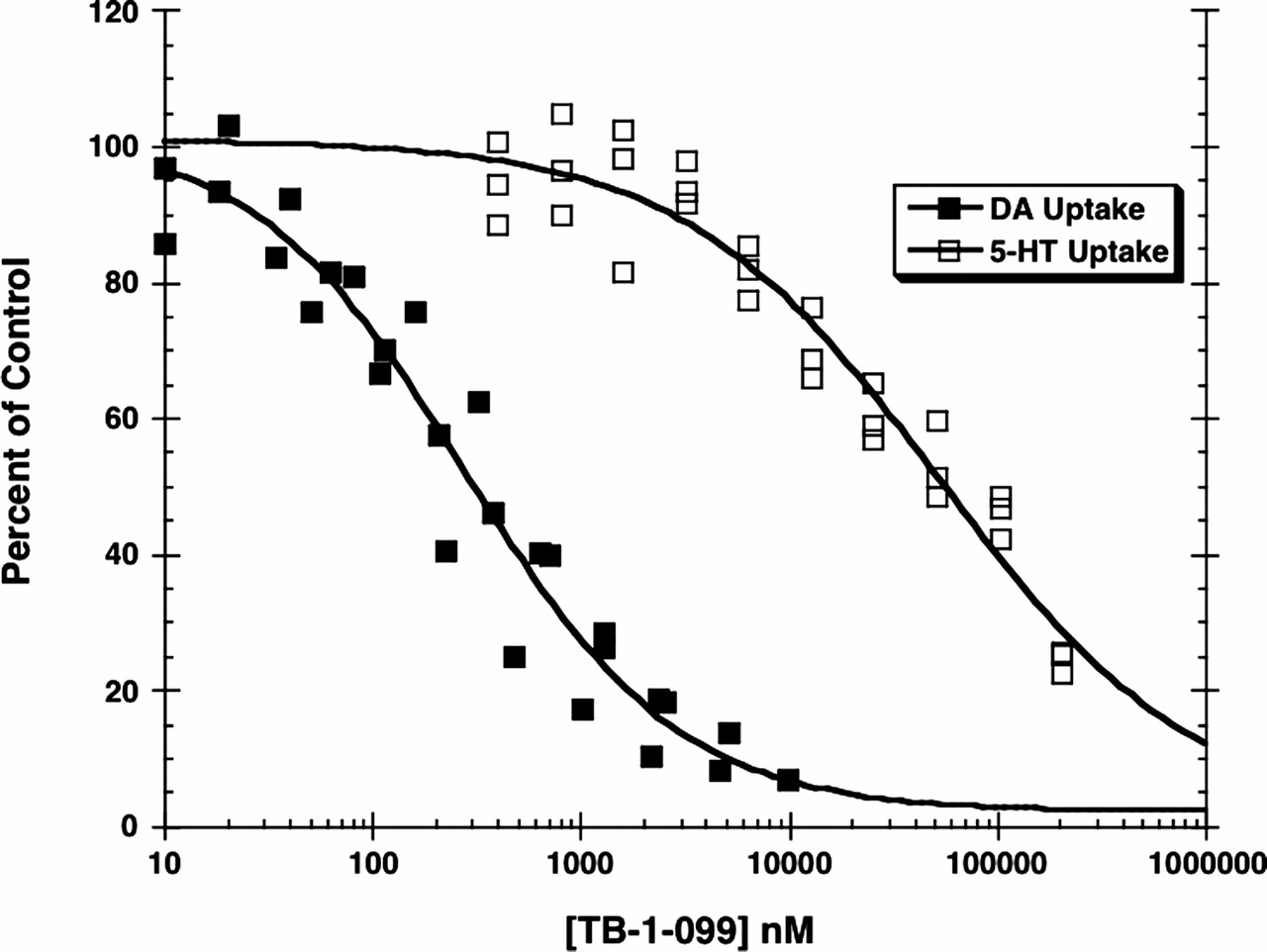
TB-1-099-mediated inhibition of [3H]DA (IC50 = 318 ± 30 nM) and [3H]5-HT (IC50 = 56,600 ± 5540 nM) uptake by rat brain synaptosomes. Uptake assays were conducted as described in Materials and Methods. The individual data points derived from three experiments are plotted above.
Discussion
Based on our experience screening hundreds of compounds in biogenic amine transporter binding assays (Prisinzano et al., 2004), with few exceptions most agents inhibited rat brain SERT binding with slope factors (Hill coefficients) close to 1, a finding strongly suggestive of a binding process governed by simple mass action kinetics. In some cases, we observed that a few compounds had slope factors in the range of 0.80 and were able to resolve two apparent binding SERT binding sites in rat brain cortex using [125I]RTI-55 (Silverthorn et al., 1995). SoRI-6238 was the first compound we came across that inhibited [125I]RTI-55 binding to rat brain SERT in a qualitatively different manner (Nandi et al., 2004) by virtue of a prominent partial inhibitory profile. Although this pattern might be consistent with the existence of two binding sites, the other lines of evidence that SoRI-6238 allosterically modulated rat brain SERT binding indicated that it would be incorrect to interpret the data in this manner.
Early hints of allosteric interactions at the biogenic amine transporters included our finding that pretreatment of guinea pig membranes with paroxetine increased the dissociation rate of [3H]cocaine from SERT (Akunne et al., 1992). Using rat SERT expressed in HEK cells, Sur et al. (1998) presented evidence that imipramine allosterically modulated the ability of citalopram to inhibit [3H]5-HT transport. Others reported apparent allosteric interactions between 5-HT and [3H]paroxetine binding to human platelet SERT (Andersson and Marcusson, 1989), as well between β-estradiol and SERT (Chang and Chang, 1999). More recently, we reported novel allosteric modulators of both DAT (Rothman et al., 2002) and SERT (Nandi et al., 2004), and Chen et al. reported evidence for allosteric modulation of [3H](S)-citalopram binding (Chen et al., 2005).
We initially examined the interaction of TB-1-099, a GBR12909 analog, with rat brain biogenic amine transporters as part of our routine screening efforts (Prisinzano et al., 2004). The fact that TB-1-099 partially inhibited rat brain SERT binding (Fig. 1) provided the first clue that this compound behaved differently than most agents at SERT. Moreover, the observation that changing the R substituent from CF3 to CH3“normalized” the SERT inhibition curve suggests that it may be possible, in the future, to determine the structural requirements for the type of partial inhibition observed with TB-1-099. At the present time, we do not have sufficient structure-activity data to hypothesize why such a small change in the structure of TB-1-099 changes its interaction the SERT.
Having gathered initial evidence that TB-1-099 could partially inhibit rat brain SERT binding, we conducted the rest of the study using hSERT expressed in HEK cells, since this model system reduces the probability that the partial inhibition might be due to the existence of two distinct types of SERT binding sites. Although TB-1-099 partially inhibited [125I]RTI-55 to hSERT, the plateau was lower than observed in rat brain. This also occurred with SoRI-6238, which did not display a plateau over the concentration examined. The reason for these differences will require additional research. The ability of TB-1-099 to partially inhibit [125I]RTI-55 binding to hSERT appeared to be selective for hSERT, since TB-1-099 partially inhibited [125I]RTI-55 binding to hSERT but not to hDAT or to hNET (Fig. 2). Interestingly, TB-1-099 was inactive at inhibiting [3H]nisoxetine binding to the rat brain NET and much more potent at [125I]RTI-55 binding to hNET. This likely reflects the fact that [3H]nisoxetine underestimates the potency of compounds at NET (Reith et al., 2005).
Saturation binding experiments of [125I]RTI-55 binding to hSERT, in the absence and presence of various concentrations of TB-1-099, demonstrated that TB-1-099 noncompetitively inhibited SERT binding (Figs. 3A and 4). Fitting the data to a two-site binding model failed to produce a set of best-fit parameter estimates that described the observed data well, both quantitatively and qualitatively. In particular, the two-site model clearly predicts curvilinear Scatchard plots in the presence of 5000 and 25,000 nM TB-1-099 that extrapolate to the same x-intercept as the control Scatchard plot. This was not observed. Moreover, TB-1-099 increased the apparent Kd but not to the degree expected if the interaction was directly competitive. Indeed, increasing the TB-1-099 concentration from 5 to 25 μM increased the Kd by only 30%, rather than the expected 422%. These observations support the hypothesis that the interaction of TB-1-099 with [125I]RTI-55 binding to hSERT is not consistent with competitive inhibition at either one or two binding sites, and supports the hypothesis that TB-1-099 allosterically modulates [125I]RTI-55 binding to hSERT.

Scatchard plots of TB-1-099-mediated inhibition of [3H]DA (B) and [3H]5-HT (A) uptake. “V” refers to the rate of uptake, measured in fmol/mg protein min-1. The data of the uptake binding experiments described in Table 3 were averaged and converted to Scatchard format. The standard deviations of the averaged V values were less than 10% of the mean. For the [3H]5-HT data, the 5-HT concentration of the 14 data points ranged from 3.64 to 108 nM. For the [3H]DA data, the DA concentration of the 20 data points ranged from 14 to 1587 nM.
Dissociation experiments are classically used to detect allosteric effects, specifically comparing the dissociation rate observed when dissociation is initiated by dilution versus the addition of an excess of a competing ligand. As described in our previous study (Nandi et al., 2004), we could not use the dilution method because a 100-fold dilution produced an initial dissociation followed by a reassociation of [125I]RTI-55 binding to hSERT. Thus, we determined the ability of a test agent to alter [125I]RTI-55 dissociation initiated by the addition of 1 μM RTI-55. The test agents (TB-1-099 or SoRI-6238) were added 10 min after the RTI-55, insuring that any effect observed could not be due to interactions at the RTI-55 hSERT binding site (see Table 1). Unlike SoRI-6238, which slowed the dissociation of [125I]RTI-55 from hSERT, TB-1-099 had no effect. The results reported here for SoRI-6238 extend that of our previous study, which used rat brain membranes.
We have used an experimental approach to association binding experiments pioneered by others (Motulsky and Mahan, 1983; Contreras et al., 1986) to detect the presence of allosteric interactions. This method permits the calculation, from an association time course, of the kinetic constants kon and koff. When the time course is conducted in the presence of a competitive inhibitor, the competing drug slows the apparent association rate of the radioligand, decreasing the apparent value of kon. Moreover, the value of koff must change such that the kinetically calculated apparent Kd (koff/kon) increases. The kinetically calculated apparent Kd can then be compared with the actual apparent Kd determined at the same time. For a competitive inhibitor, the kinetically calculated Kd and the actual Kd should agree fairly well. This is what we observed here for the competitive inhibitor indatraline (Table 2) and in our previous study using citalopram (Nandi et al., 2004). TB-1-099 did not behave like a competitive inhibitor, since increasing its concentration 10-fold increased the kinetically calculated Kd only 2.2-fold, not the expected 6-fold. Although these data support the hypothesis that TB-1-099 does not interact with hSERT binding in a competitive manner, it does not represent direct evidence of an allosteric interaction.
A final piece of evidence that supports an allosteric interaction between TB-1-099 and SERT is that TB-1-099 noncompetitively inhibited [3H]5-HT uptake. As observed in the binding assays (Fig. 2), the apparent allosteric effect appeared to be selective for SERT, since TB-1-099 competitively inhibited [3H]DA uptake. The fact that TB-1-099 noncompetitively inhibits [125I]RTI-55 binding to hSERT and [3H]5-HT uptake suggests that TB-1-099, acting via an allosteric site, regulates the availability of SERT binding sites and therefore the functional capacity of the transporter. The relationship between oligomerization of hSERT and the phenomena observed here remains to be determined (Schmid et al., 2001).
As noted in the Introduction, reports over the years hint of an allosteric binding site associated with SERT. Although compelling evidence for an allosteric modulatory site associated with SERT will likely await the development of a high-affinity ligand for the site, the data presented here and in our previous study (Nandi et al., 2004) present strongly suggestive evidence for such an allosteric modulatory site. The implications of such a site for the actions of antidepressants and drugs of abuse, such as cocaine, remains to be determined.
Acknowledgments
HEK cells expressing hNET and hDAT were kindly provided by Dr. Randy Blakely (Vanderbilt University School of Medicine). The hNET cells originated in the laboratory of Dr. Susan Amara (University of Pittsburgh School of Medicine). The hDAT cells originated in the laboratory of Dr. Marc Caron (Duke University Medical Center). The hSERT cells were kindly provided by Dr. Aaron J. Janowsky (Oregon Health and Science University). We acknowledge the expert technical assistance of John H. Partilla, who prepared the [125I]RTI-55, and also Dr. F. Ivy Carroll (Research Triangle Institute International), who provided the precursor used in the preparation of [125I]RTI-55.
Footnotes
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.105.084376.
-
ABBREVIATIONS: 5-HT, 5-hydroxytryptamine, serotonin; SERT, 5-HT transporter; RTI-55, 3β-(4′-iodophenyl)tropan-2β-carboxylic acid methyl ester; hNET, cloned human norepinephrine transporter; hDAT, cloned human dopamine transporter; NE, norepinephrine; DA, dopamine; SoRI-6238, ethyl 5-amino-3-(3,4-dichlorophenyl)-1,2-dihydropyrido[3,4-b]pyrazin-7-ylcarbamate; SoRI-9804, 4-[(diphenylmethyl)-amino]-2-phenylquinazoline; GBR12909, 1-[2-[bis(4-fluorophenyl)methoxy]ethyl]-4-(3-phenylpropyl)piperazine; TB-1-099, 4-(2-[bis(4-fluorophenyl)methoxy]ethyl)-1-(2-trifluoromethyl-benzyl)-piperidine; hSERT, cloned human 5-hydroxytryptamine transporter; HEK, human embryonic kidney; BB, binding buffer; SA, specific activity; TB-1-101, 4-{2-[bis-(4-fluoro-phenyl)-methoxy]-ethyl}-1-(2-methyl-benzyl)-piperidine oxalate.
- Received February 1, 2005.
- Accepted April 20, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}