


Abstract
Depsipeptide FK228 [(E)-(1S,4S,10S,21R)-7[(Z)-ethylideno]-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,20,23-tetraazabicyclo[8,7,6]-tricos-16-ene-3,6,9,22-pentanone], a novel histone deacetylase (HDAC) inhibitor, previously was reported to be a P-glycoprotein (Pgp) substrate. We now expand the investigation to demonstrate that FK228 is a substrate for Pgp and multidrug resistance-associated protein 1 (MRP1). Transport of FK228 across the Caco-2 cell monolayer in apical to basolateral (AP→BL) and basolateral to apical (BL→AP) directions in the absence and presence of Pgp and MRP inhibitors were investigated. An in vitro uptake study in human red blood cells (RBCs) and a cytotoxicity assay in MRP1(-) HL60 and MRP1(+) HL60Adr cells were conducted to show that FK228 is an MRP1 substrate. An FK228-resistant cell line (HCT15R) was developed from HCT15 colon carcinoma and characterized using a 70-oligomer cDNA microarray, reverse transcription-polymerase chain reaction, Western blot analysis, histone acetyltransferase (HAT) and HDAC activity assays, and cytotoxicity assays. FK228 showed a nearly unidirectional flux across the Caco-2 cell monolayer, with the BL→AP apparent permeability coefficient (Papp) 32 times that of AP→BL without apparent saturation. Pgp inhibition decreased the BL→AP Papp and increased the AP→BL Papp. RBC showed a concentration-dependent uptake and saturable efflux of FK228. HL60Adr cells were 4-fold more resistant to FK228 than HL60 cells, and the resistance was reversed by MRP inhibition. Up-regulation of Pgp, but not changes of MRPs or HAT/HDAC enzymatic activities, was the major mechanism for the acquired FK228 resistance. These studies demonstrate that FK228 is a substrate for Pgp and MRP1, and reversible Pgp up-regulation is predominantly involved in FK228 resistance in vitro.
Depsipeptide FK228, formerly FR901228 and NSC-630176 (Fig. 1), is a novel, naturally occurring bicyclic peptide with a disulfide linkage isolated as a fermentation product of Chromobacterium violaceum (Shigematsu et al., 1994; Ueda et al., 1994a,b). It originally was developed as an anti-ras compound (Ueda et al., 1994c; Wang et al., 1998), later was found to interfere with mitogen-induced signaling pathways (Rajgolikar et al., 1998; Sandor et al., 2000a,b), and more recently has been found to be a potent histone deacetylase (HDAC) inhibitor (Nakajima et al., 1998; Yoshida and Horinouchi, 1999; Yoshida et al., 2001). Because of its observed preclinical antitumor activity, selectivity, and passage in its preclinical toxicology evaluation, FK228 has entered into several phase I clinical trials against various refractory cancers (Marshall et al., 2002; Sandor et al., 2002), and phase II clinical trials are now being planned.

Structure of depsipeptide FK228.
Preclinical pharmacokinetics studies in rats showed a low oral bioavailability of FK228 (Chan et al., 1997; Li and Chan, 2000). It later was reported that FK228 is a P-glycoprotein (Pgp) substrate, but not a Pgp inhibitor, during a National Cancer Institute screening project (Scala et al., 1997). However, because of the screening nature of the study, no kinetic or quantitative transport data of FK228 were reported.
Multidrug resistance-associated protein 1 (MRP1) is highly expressed in red blood cell (RBC) membrane, responsible for the efflux of oxidized glutathione to maintain a reducing intracellular environment (Rychlik et al., 2000). MRP1 also is associated with drug resistance and functions as an efflux pump in cancer cells for drug glutathione conjugates and a variety of structurally unrelated drugs, including some neutral compounds (Paul et al., 1996; Rychlik et al., 2000; Wijnholds, 2002). The overlap of substrates between Pgp and MRP1 (van Zuylen et al., 2000) suggests the possibility of FK228 being an MRP1 substrate. Moreover, expression of MRP2 on the apical membrane of the Caco-2 monolayer (Hirohashi et al., 2000; Sun et al., 2002; Cooper et al., 2004) provides a model to investigate whether FK228 shares affinity for MRP2. Herein, we aimed to further characterize Pgp- and/or MRP1-mediated FK228 transport and uptake kinetics using multiple models, including the Caco-2 cell monolayer, human RBCs (Rychlik et al., 2000), and Pgp(-)/MRP1(-) HL60 and Pgp(-)/MRP1(+) HL60Adr cell lines (Gollapudi and Gupta, 1992).
HDAC inhibitors cause histone hyperacetylation (Weidle and Grossmann, 2000) and up-regulate the expression of various genes, including MDR1 (or ABCB1, both gene names of Pgp) (Jin and Scotto, 1998; El-Osta et al., 2002). Because FK228 is a Pgp substrate and potent HDAC inhibitor, we were interested in determining whether FK228 induces Pgp and MRP1 expression, which in turn leads to FK228 resistance. For this reason, we used the human colon carcinoma HCT15 cell line and its resistant daughter cell line HCT15R, developed in our laboratory, to study the nature of FK228 resistance. Both cell lines were characterized using a custom 70-oligomer cDNA microarray, reverse transcription-polymerase chain reaction (RT-PCR), Western blot analysis, cytotoxicity assay, and histone acetyltransferase (HAT)/HDAC activity assays. Reversibility of the Pgp induction and cross-resistance of HCT15R cells to other Pgp substrate drugs also were studied.
Materials and Methods
Materials. Nonformulated depsipeptide FK228 (Fig. 1; purity, >99%) was supplied by the Drug Synthesis and Chemistry Branch, National Cancer Institute (Bethesda, MD), and used without further purification. N-t-Boc-Met-Leu-Phe (purity, >97%) and indomethacin was purchased from Sigma-Aldrich (St. Louis, MO). Potassium phthalate buffer (pH 4, 50 mM) was obtained from Van Water and Rogers Scientific (Chicago, IL). MK571 (LL6607), a specific MRP inhibitor, was purchased from BIOMOL Research Laboratories (Plymouth Meeting, PA). Cyclosporin A (CsA), (±)-verapamil hydrochloride (Ver), and Lucifer Yellow CH were purchased from Sigma-Aldrich. All organic solvents were obtained from Fisher Scientific Co. (Pittsburgh, PA) and were of high-performance liquid chromatography (HPLC) grade. HPLC-grade water (>18 mΩ) was generated with an E-pure water purification system (Barnstead, Dubuque, IA). RPMI 1640 medium, Dulbecco's modified Eagle's medium nonessential amino acids, sodium penicillin G, streptomycin, HEPES buffer, Hanks' balanced salt solution (HBSS), Dulbecco's phosphate-buffered saline (DPBS), and fetal bovine serum (FBS) were purchased from Invitrogen (Carlsbad, CA). Caco-2 and HCT15 cells were obtained from American Type Culture Collection (Manassas, VA). HL60 and HL60Adr cells were a gift from Dr. Hans Hinderman (Roswell Park Cancer Institute, Buffalo, NY).
Cell Cultures. Caco-2 cells were cultured in high glucose Dulbecco's modified Eagle's medium, supplemented with 10% FBS, 25 mM HEPES, 0.1 mM nonessential amino acid, 4 mM l-glutamine, 100 U/ml sodium penicillin G, and 100 μg/ml streptomycin. HCT15, HL60, and HL60Adr cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 100 U/ml sodium penicillin G, and 100 μg/ml streptomycin. Cells were grown in 75-cm2 tissue culture flasks at 37°C in a 5% CO2 atmosphere, and the culture medium was replaced every other day. The adhesive cells were harvested at 80% confluence by exposure to a trypsin-EDTA solution (0.25% trypsin and 0.002% EDTA in HBSS). For transport studies, Caco-2 cells of passages 44 to 46 were seeded on collagen-coated six-well Transwell inserts (0.4-μm pore size, 4.7 cm2 growth area; Costar, Cambridge, MA) at densities of 5 × 104 cells/cm2. The medium was changed every other day for the first week and every day thereafter until the transport study (24–27 days postseeding).
FK228 Transport. The Caco-2 cell monolayers were washed twice with warm (37°C) DPBS (1×, pH 7.4). For FK228 transport studies without Pgp inhibition, the DPBS was replaced by the warm HBSS bathing solution [1× HBSS containing 25 mM d-glucose and 10 mM HEPES (pH 7.4)]. The volumes of HBSS bathing solution at the apical and basolateral sides of the cell layer were 1.5 and 2.5 ml, respectively. The Transwell plates were returned to the incubator to equilibrate at 37°C for 30 min. The HBSS bathing solution was removed, and the Transwell inserts were transferred to a clean six-well plate. Warm HBSS bathing solution alone or HBSS bathing solutions containing FK228 at appropriate concentrations were added to the acceptor and donor chamber, with 1.5 ml on the apical side and 2.5 ml on the basolateral side, respectively, followed by incubation of the plate at 37°C. A 200-μl sample was taken from the acceptor side at preselected time points of 0, 15, 30, 45, 60, 90, 120, 150, and 180 min, and 200 μl of warm blank HBSS bathing solution was added each time to the acceptor side to maintain the constant volume. FK228 concentrations in the donor chamber evaluated for apical to basolateral (AP→BL) and basolateral to apical (BL→AP) transport were 0.5, 1, 2, 5, 10, and 20 μM. This range included the plasma steady-state concentrations in patients receiving 4-h FK228 infusion at 13 mg/m2 (Sandor et al., 2002). Three inserts were used for each treatment. By measuring the transepithelial electrical resistance before and after the transport, the integrity of the cell monolayer was monitored. Only monolayers with transepithelial electrical resistance values >800 Ω · cm2 were used. Lucifer Yellow CH was used as a paracellular transport marker during the experiments, and no leak was observed. For FK228 transport with pretreatment and coincubation with the Pgp (CsA and Ver) or MRP (MK571 and indomethacin) inhibitors, the same procedures as described above were followed, except that the HBSS bathing solutions were added to 5 μM CsA, 100 μM Ver, 50 μM MK571, or 20 and 40 μM indomethacin during the pretreatment and transport study. FK228 concentrations were determined by HPLC/tandem mass spectrometry as described previously (Li and Chan, 2000).
Calculation of Apparent Permeability Coefficient Papp. Apparent permeability coefficients (Papp) were calculated from concentration-time profiles as measured in the receiver compartment according to Fick's first law using the following equation:  where dC/dt represents the appearance of FK228 in the receiver chamber (picomoles per milliliter per minute), V is the volume of the receiver compartment (milliliters), A is the cross-section area, and C0 is the initial donor concentration (picomoles per milliliter) at time = 0. The flux across the monolayer was determined by linear regression from individual FK228 concentration versus time curves. Comparison of Papp in the BL→AP direction (Papp, BL→AP) with Papp in the AP→BL direction (Papp, AP→BL) was used to assess efflux transport efficiency (Teff = Papp, BL→AP/Papp, AP→BL). The statistical significance of differences between treatments was evaluated using two-tailed, paired, Student t tests.
where dC/dt represents the appearance of FK228 in the receiver chamber (picomoles per milliliter per minute), V is the volume of the receiver compartment (milliliters), A is the cross-section area, and C0 is the initial donor concentration (picomoles per milliliter) at time = 0. The flux across the monolayer was determined by linear regression from individual FK228 concentration versus time curves. Comparison of Papp in the BL→AP direction (Papp, BL→AP) with Papp in the AP→BL direction (Papp, AP→BL) was used to assess efflux transport efficiency (Teff = Papp, BL→AP/Papp, AP→BL). The statistical significance of differences between treatments was evaluated using two-tailed, paired, Student t tests.
Uptake Studies. Freshly obtained heparinized blood samples from eight healthy volunteers were used to test FK228 uptake at two concentrations, 1.8 and 18 μM. At 1.8 μM FK228, the uptake study in the presence of 50 μM of MRP inhibitor MK571 also was carried out. Following addition of appropriate drugs, the blood samples were incubated at 37°C, and the plasma FK228 concentrations were followed for up to 60 min.
Development of FK228-Resistant HCT15R Cell Line. HCT15 cells were treated with FK228 started from a sub-IC50 concentration, 100 nM. The medium was changed every other day with an increment of 100 nM FK228 each time. Within 1 month, the HCT15 cells acquired FK228 resistance and were cultured in a medium containing 1000 nM FK228 thereafter. The daughter cell line was designated HCT15R.
To study the reversibility of the FK228-induced Pgp up-regulation, HCT15R cells were cultured in either medium containing 1000 nM FK228 or FK228-free medium for 1 to 6 weeks. After 6 weeks, the cells then were treated with 500 nM FK228 for 1 week. Cells were harvested and analyzed for Pgp expression by Western blot analysis.
Cytotoxicity Assay. HCT15 and HCT15R cells were seeded on 96-well plates at 2000 cells/well and allowed to adhere overnight before drug treatment. The cells were treated with FK228, paclitaxel, or doxorubicin at a series of concentrations alone or in combination with 5 μM CsA or 50 μM MK571. After 24 h of treatment, the medium was replaced by fresh drug-free medium, followed by a 72-h incubation. The cytotoxicity was determined by standard sulforhodamine B assay (Skehan et al., 1990) for attached cells. HL60 and HL60Adr cells also were seeded on 96-well plates at 5000 cells/well and treated with FK228 at appropriate concentrations in the absence or presence of 50 μM MK571 continuously for 72 h. The cytotoxicity was determined by standard 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxyanilide assay, which generally is used for suspension cells (Goodwin et al., 1995). The IC50 was defined as the concentrations of the tested compounds at which the number of living cancer cells was reduced by 50% compared with the untreated controls. To avoid possible intracellular retention of FK228 from the resistance development, HCT15R cells were cultured in FK228-free medium for 3 days before seeding. All cytotoxicity assays were conducted in triplicate.
Custom cDNA Microarray. We used a 70-oligomer custom cDNA microarray previously developed in our laboratories, which comprises 1070 probes targeting 640 transporters (including all of the known 48 ABC transporters) and ion channel genes, as well as 430 genes belonging to families of growth factors and receptors, cell adhesion molecules, and signal transduction factors (Anderle et al., 2004; Huang et al., 2004). For some genes of special interest (e.g., all ABC transporter genes), two different probes were designed and printed on the arrays. Total RNA was extracted from the parental and daughter cells individually with TRIzol (Invitrogen) and further purified using the RNeasy Mini column (QIAGEN, Valencia, CA). Eighteen micrograms of total RNA were used for cDNA synthesis, and the cDNA was labeled with Cy5 or Cy3 by amino-allyl coupling. The protocol is available at http://derisilab.ucsf.edu/pdfs/amino-allylprotocol.pdf. A paired and dye-swap design was applied in that the cDNA samples of parent cell lines were in one experimental group labeled with Cy3 dye and the samples of resistant daughter cells were labeled with Cy5 dye, and vice versa in another experimental group. The samples then were mixed, and the labeled cDNA was resuspended in 20 μl of HEPES buffer [25 mM (pH 7.0)] containing 1 μl of tRNA, 1.5 μl of polyA+, and 0.45 μl of 10% SDS. The mixture was hybridized to the slides for 16 h at 65°C. Slides were washed, dried, and scanned in an Affymetrix 428 scanner (Santa Clara, CA) to detect Cy3 (green) and Cy5 (red) fluorescence.
Background subtraction and calculation of medians of pixel measurements per spot were carried out using GenePix software 3.0 (Molecular Devices, Sunnyvale, CA). Spots were filtered out if they had red and green intensity <500 units after background subtraction or if they were flagged for any visual reason (e.g., odd shapes and background noise). Data normalization was carried out using the statistical software package R (www.r-project.org). This method is based on transformations: R/G →log2R/G - cj(A) = log2R/kj(A) × G →(1/aj) × log2R/kj(A) × G, where R and G represent the green fluorescence intensity of Cy5 and the green fluorescence intensity for Cy3. The parameter cj(A) is the lowest fit of the M versus A plot for spots on the jth grid of each slide (M = log2R/G and A = log2(R × G)1/2), and aj is the scale factor for the jth grid (to obtain equal variances along individual slides). To identify differentially expressed genes, we calculated the R/G ratios for each primer (four prints per primer) and each dye-swap group (two groups per primer). The cDNA level ratios between HCT15R and HCT15 cells were calculated based on the R/G ratios and then averaged, and the S.D. was calculated. In cases when two primers were used for one gene, the R/G values were individually calculated for each primer.
RT-PCR. Total RNA was extracted from HCT15 and HCT15R cells by TRIzol. The RNA was further purified using isopropyl alcohol precipitation and 70% ethanol washing. Single-strand cDNA was prepared from the extracted RNA using oligo-dT priming (Thermo-script RT Kit; Invitrogen). PCR was performed using primers for MDR1 (5′-CAGCAAAGGAGGCCAACATAC-3′ and 5′-TGAGGCTGTCTAACAAGGGCA-3′) and β-actin (5′-CCTGGCACCCAGCACAAT-3′ and 5′-GCCGATCCACACGGAGTACT-3′). The PCR conditions were as follows: 95°C for 10 min, 94°C for 45 s, 60°C for 45 s, 72°C for 45 s, and 72°C for 7 min (20 cycles).
Western Immunoblot Analysis. For Western blot analysis, cells were washed twice with phosphate-buffered saline and lysed in lysis buffer containing 950 mM Tris-HCl, 250 mM NaCl, 5 mM EDTA, 50 mM NaF, 0.15% Igepal CA-630, and 1.5 mM phenylmethylsulfonyl fluoride. Equal amounts of proteins (100 μg) were size-fractionated on 6% (Pgp) or 15% (acetylated histone proteins H3 and H4) SDS-polyacrylamide gel electrophoresis. Proteins then were transferred onto a nitrocellulose membrane. The membrane was blocked with blocking buffer (5% nonfat milk, 200 mM NaCl, 50 mM Tris, and 0.05% Tween 20) at room temperature for 2 h. The blocked membrane then was incubated with primary antibodies at 4°C overnight. After washing the membrane with Tris-buffered saline/Tween 20 buffer (20 mM Tris, 500 mM NaCl, and 0.05% Tween 20) for 3 × 15 min, the membrane was incubated with secondary antibody at room temperature for 1 h. The detection of specific protein binding was performed with the enhanced chemiluminescence Western blot detection reagents (Amersham Biosciences AB, Uppsala, Sweden). The antibodies used were mouse monoclonal JSB-1 anti-human Pgp antibody (1:50; Research Diagnostics Inc., Flanders, NJ), mouse monoclonal AC-15 anti-β-actin antibody (1:5000; Abcam Inc, Cambridge, MA), peroxidase-conjugated AffiniPure donkey anti-mouse IgG (1: 10,000; Research Diagnostics), rabbit polyclonal antiacetylated histone H3 antibody (1:500; Upstate Biotechnology, Lake Placid, NY), rabbit antiacetylated histone H4 antibody chromatin immunoprecipitation grade (1:500; Upstate Biotechnology), and peroxidase-conjugated donkey anti-rabbit IgG (1:2000; Upstate Biotechnology).
HAT and HDAC Activity Assays. Nuclear contents were extracted from the 8 × 106 HCT15 and HCT15R cells using a nuclear extract kit (Upstate Biotechnology). The resistant cells were cultured in FK228-free medium for 3 days before nuclear extraction. The HAT activity contained in the nuclear extracts was determined using a nonradioactive colorimetric kit (Upstate Biotechnology), and the HDAC activity was determined using a fluorescent kit (Upstate Biotechnology). The experiments were conducted according to the manufacturer's protocols.
Results
FK228 Is a Pgp Substrate. AP→BL and BL→AP transport of FK228 was investigated at concentrations ranging from 0.5 to 20 μM. Figure 2 shows the amount of FK228 transported across Caco-2 monolayers over time for AP→BL and BL→AP directions (Fig. 2a). For AP→BL transport at 0.5 μM FK228, only trace amounts of FK228 were found, and the levels were below the quantification limit and thus were not included in the calculation. FK228 transport was found to be linear with time for up to 180 min for both directions. The flux (J) was found to be proportional to the donor-side FK228 concentration (Fig. 2b). The calculated Papp values were 4.07 ± 0.74 × 10-6 cm/s (n = 18) and 1.27 ± 0.73 × 10-7 cm/s (n = 15) for BL→AP and AP→BL, respectively. The BL→AP transport was 32 times faster than that of AP→BL (p < 0.005). No apparent Pgp saturation was observed.

Transepithelial flux of FK228 across the Caco-2 cell monolayer. a, FK228 flux linear with time up to 180 min for BL→AP and AP→BL directions as measured at 5 μM (n = 3). b, flux proportional to the FK228 concentration throughout the concentration range with no apparent saturation for BL→AP and AP→BL directions (n = 3).
In the inhibition studies, pretreatment and coincubation with 5 μM CsA or 100 μM Ver caused dramatic decreases in the BL→AP transport rate (Fig. 3). Between the two inhibitors, CsA showed higher inhibition, causing a 13.6-fold decrease (p < 0.005) in Papp compared with 8.4 times by Ver (p < 0.005). For AP→BL direction, both inhibitors caused small but significant increases of Papp (p < 0.05). In the presence of the inhibitors, the Teff ratios decreased from 32 to 1.65 and 2.47 for CsA and Ver, respectively, suggesting essentially complete inhibition of efflux.
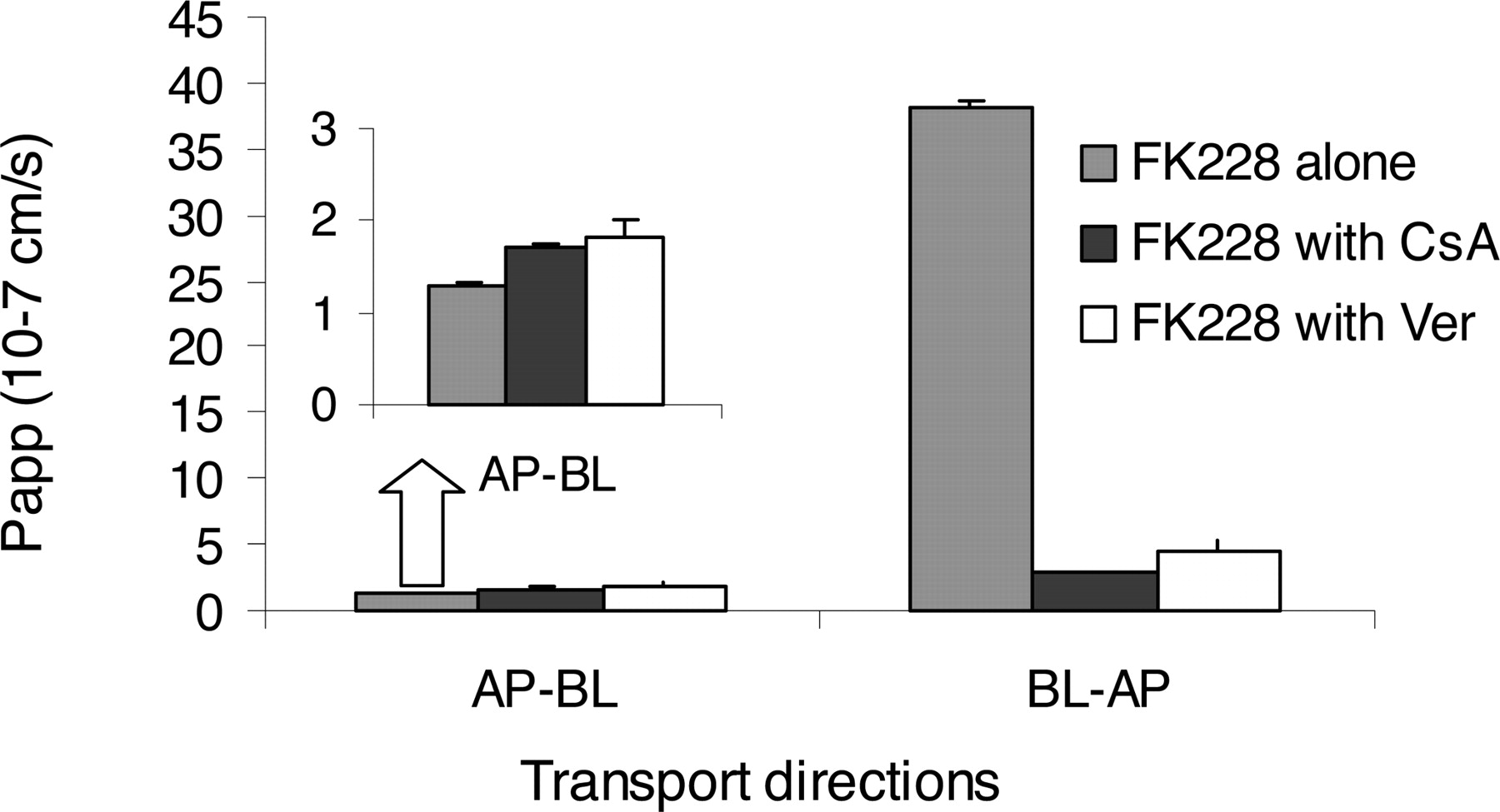
Effects of CsA and Ver on FK228 flux across the Caco-2 cell monolayer. Transport of FK228 was studied with pretreatment and coincubation of 5 μM CsA or 100 μM Ver. Transport of FK228 in the absence of CsA and Ver was used as a control. For AP→BL direction, inhibitors caused small but significant increase in Papp (p < 0.05); for BL→AP direction, the inhibitors caused dramatic decrease in Papp (p < 0.005).
FK228 Is an MRP1 Substrate. To investigate the contribution of MRP1 to FK228 transport and uptake, FK228 was incubated in human blood from eight healthy volunteers. FK228 showed a concentration-dependent uptake (Fig. 4) and was taken up by RBC more rapidly and extensively at the higher concentration (18 μM) than at the lower concentration (1.8 μM). MRP1 inhibition by 50 μM MK571 in the blood significantly increased the rate of FK228 removal from the plasma at 1.8 μM.

Human RBC uptake/metabolism of FK228. Blood samples from eight healthy volunteers were spiked with FK228 and incubated at 37°C, and the RBC uptake/metabolism was followed over time up to 60 min. RBCs seemed to uptake/metabolize FK228 more rapidly at higher FK228 concentrations. Treatment of blood with MRP inhibitor MK571 at 50 μM significantly increased the rate of FK228 removal from plasma.
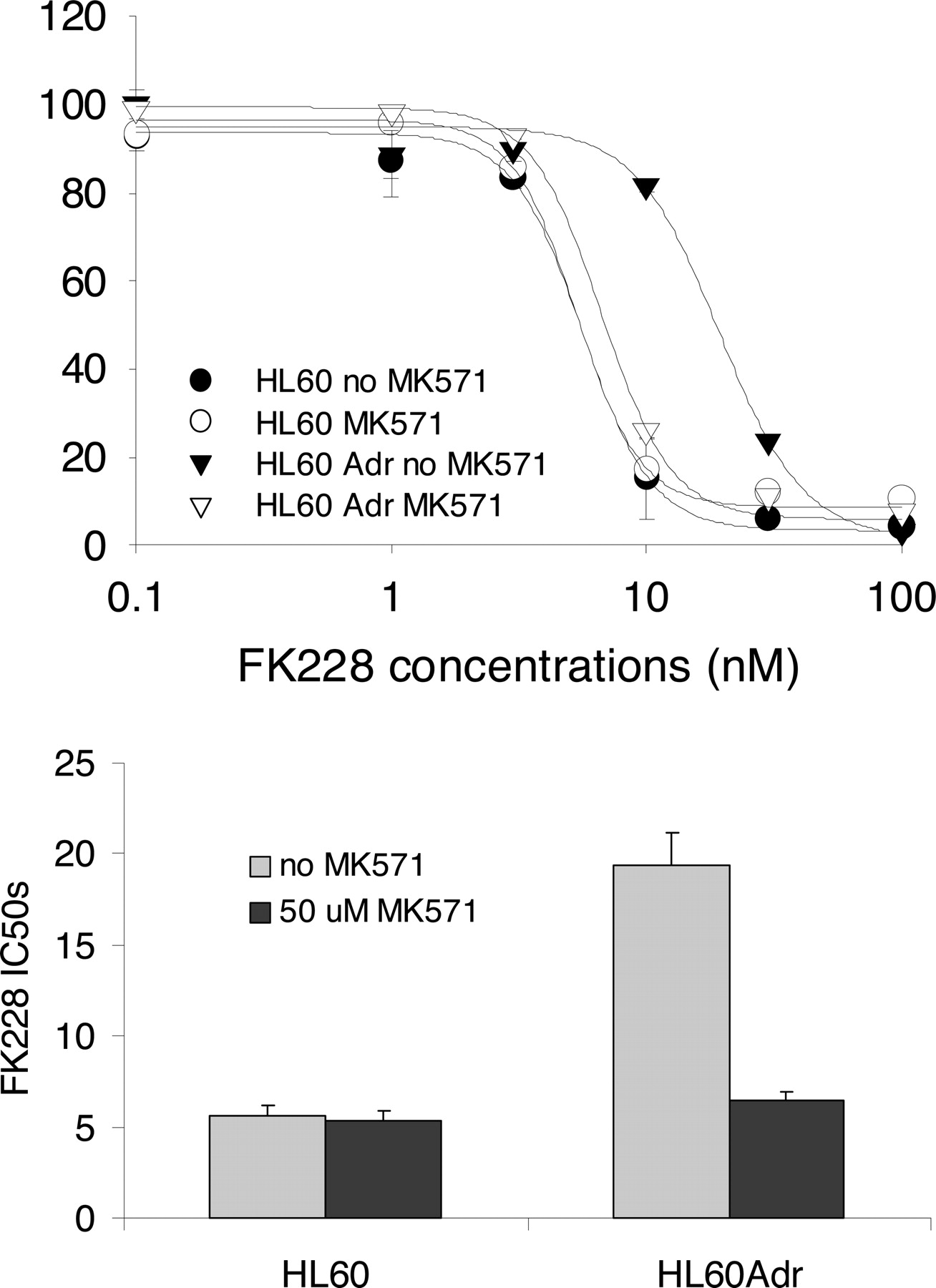
HL60 cells are Pgp(-)/MRP1(-), whereas the derivative HL60Adr cells are Pgp(-)/MRP1(+) (Bhalla et al., 1985; Gollapudi and Gupta, 1992). If FK228 is an MRP1 substrate, HL60Adr cells should be more resistant to FK228 than HL60 cells. For this reason, we conducted cytotoxicity assays in these two cell lines. Our results showed that in the absence of the MRP inhibitor MK571, the IC50 values of FK228 were 5.6 ± 0.56 and 19.4 ± 1.8 nM for HL60 and HL60Adr cells, respectively. In the presence of MK571, the IC50 did not change appreciably for HL60 cells (5.3 ± 0.59 nM), but for HL60Adr cells, it was reduced by >3-fold (6.5 ± 0.46 nM) (Fig. 5). This confirmed that FK228 is an MRP1 substrate.

Cytotoxicity assays of FK228 in Pgp(-)/MRP1(-) HL60 and Pgp(-)/MRP1(+) HL60Adr cells. In the absence of the specific MRP inhibitor MK571, the IC50 values of FK228 were 5.6 ± 0.56 and 19.4 ± 1.8 nM for HL60 and HL60Adr cells, respectively. The addition of 50 μM MK571 caused little change of the IC50 in HL60 cells (5.3 ± 0.59 nM) but totally reversed the IC50 value in HL60Adr cells (6.5 ± 0.46 nM; p < 0.05). Top, cytotoxicity curves; bottom, comparison of IC50 values of FK228 between HL60 and HL60Adr cells in the presence or absence of MK571.
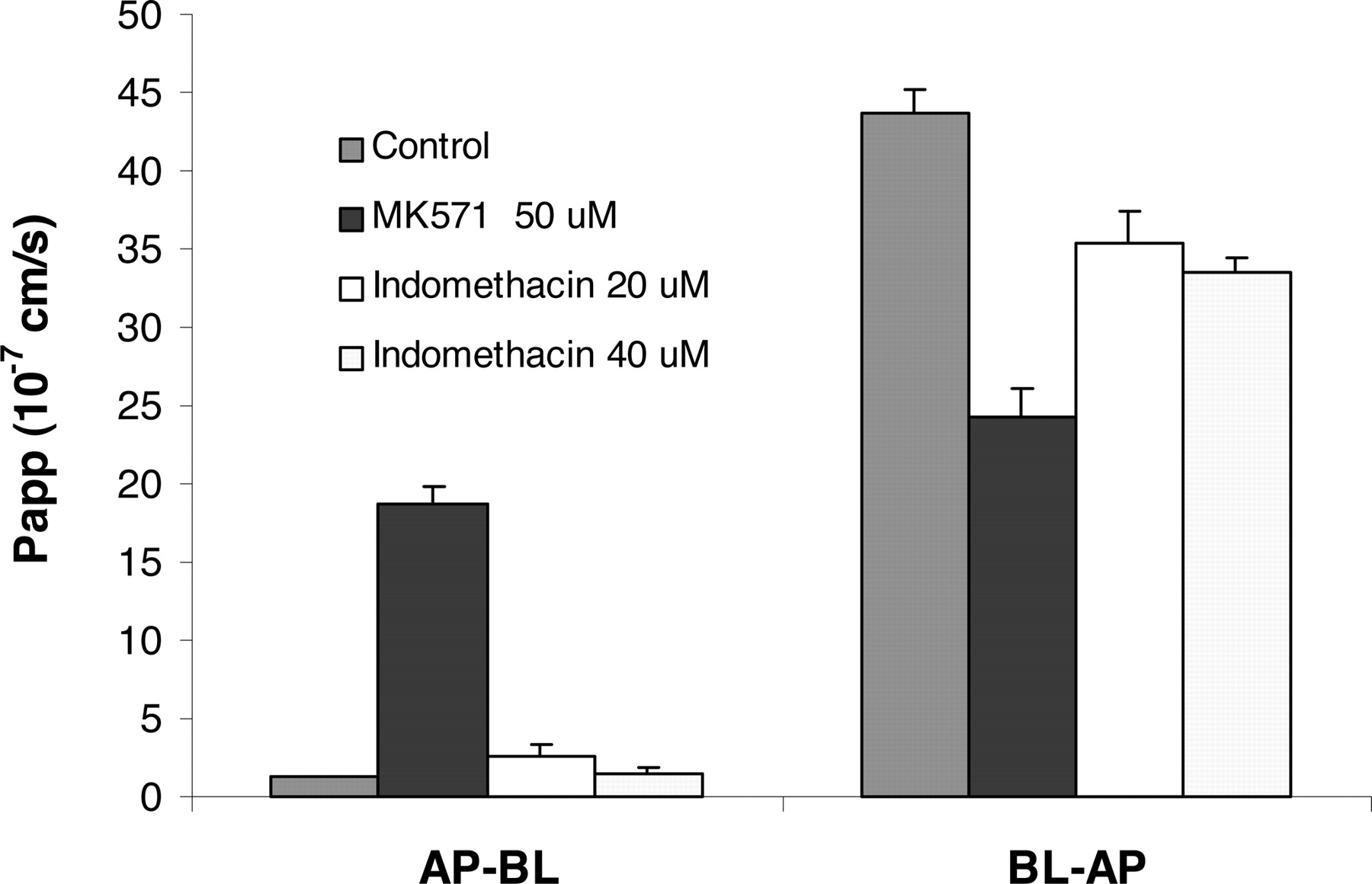
To further investigate whether FK228 also is a substrate of another major member of MRP family, MRP2, the effects of MRP inhibitors MK571 and indomethacin on FK228 transport across the Caco-2 cell monolayer were studied. MK571 at 50 μM significantly decreased BL→AP transport (p < 0.005) but increased AP→BL transport (p < 0.005) across the Caco-2 monolayer. Indomethacin at 20 or 40 μM showed no significant effect (Fig. 6) on FK228 transport of either direction (p > 0.05). The change of FK228 transport in the presence of MRP inhibitors was consistent with the apical localization of MRP2 in the differentiated Caco-2 cell monolayer (Hirohashi et al., 2000; Sun et al., 2002; Cooper et al., 2004).

Effects of MRP inhibition on FK228 transport across the Caco-2 cell monolayer in AP-BL and BL-AP directions. MRP inhibitor MK571 at 50 μM increased AP-BL transport (p < 0.005) and decreased BL-AP transport (p < 0.005) of FK228, whereas the other MRP inhibitor indomethacin had less influence on FK228 transport at either 20 or 40 μM (p > 0.05).
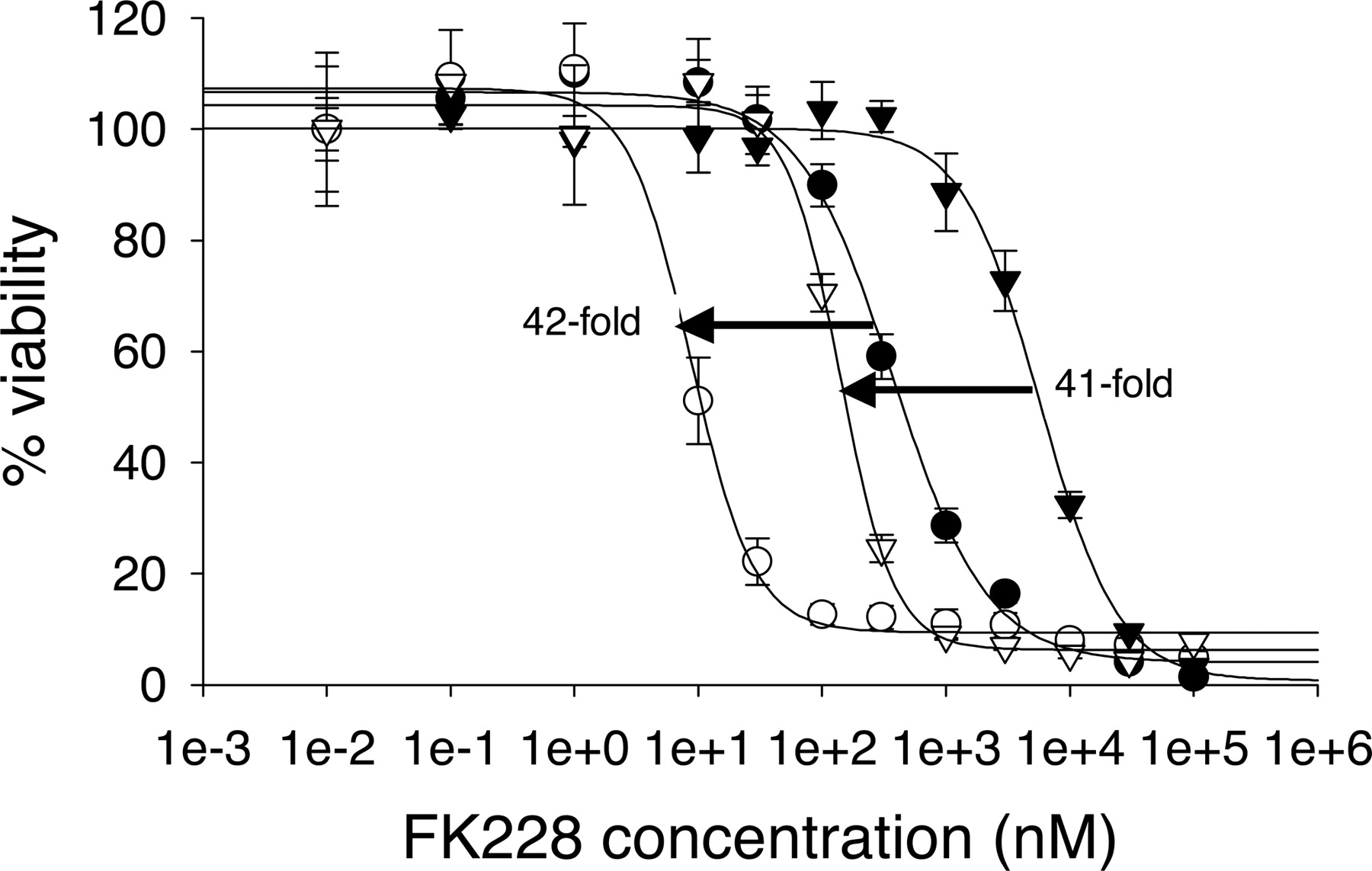
Development of FK228-Resistant HCT15R Cell Line. The HCT15 colon carcinoma cells acquired significant FK228 resistance fast (within 1 month) with no observable massive cell kills (Fig. 7a; Table 1). However, the HCT15R cells did not show apparent morphological change (data not shown). In the absence of CsA, the IC50 of FK228 on HCT15 cells was 378 ± 49.5 nM compared with 7139 ± 813 nM on HCT15R cells. This 19-fold difference in the IC50 was decreased by 5 μM CsA, which dramatically reduced IC50 values to 7.44 ± 1.1 and 11.4 ± 0.4 nM for HCT15 and HCT15R cells, respectively. HCT15R cells also showed cross-resistance to other two Pgp substrates, paclitaxel and doxorubicin (Fig. 7, b and c; Table 1). Similar to the case of FK228, CsA caused sensitization of HCT15 and HCT15R cells and resulted in essentially superimposable IC50 curves. These results suggest that Pgp may play an important role in the resistance and cross-resistance because paclitaxel and doxorubicin are well known Pgp substrates.

Cytotoxicity assays in HCT15 and HCT15R cells. (a) FK228, (b) doxorubicin, and (c) paclitaxel in HCT15 and HCT15R cells. IC50 values in HCT15 cells treated with the drug alone (•) or in combination with 5 μM CsA (○) were compared with those in HCT15R cells treated with the drug alone (▾) or in combination with 5 μM CsA (▿).
IC50 values of FK228, paclitaxel, and doxorubicin on HCT15 and HCT15R cells in the presence or absence of 5 μM CsA
In contrast to Pgp inhibition, MRP1 inhibition by 50 μM MK571 resulted in parallel IC50 shifts for HCT15 and HCT15R cells (Fig. 8), suggesting that the MRP1 expressions are essentially the same in HCT15 and HCT15R cells.
Cytotoxicity assays of FK228 in HCT15 and HCT15R cells. In the absence of MK571, the IC50 values of FK228 were 371 ± 4 and 5770 ± 549 nM for HCT15 and HCT15R cells, respectively. In the presence of 50 μM MK571, the IC50 values shifted to 8.7 ± 1.0 and 140 ± 10 nM for the two cell lines, respectively. The IC50 shifts for both cell lines were about 40-fold, suggesting the MRP1 levels in these two cell lines are essentially the same.
Further Characterization of FK228-Resistant HCT15R Cell Line. The resistance of HCT15R cells to FK228 was found to be reversed by CsA (Fig. 7a; Table 1). This suggests that Pgp up-regulation is a major mechanism for the acquired resistance. However, this resistance may also be derived from other factors such as up-regulation of MRPs. For this reason, we used a custom 70-oligomer cDNA microarray (the list of included genes is available upon request) to screen possible up-regulation or down-regulation of genes. The results showed that Pgp (gene name ABCB1), but not other ABC transporter genes, was predominantly up-regulated among all of the tested genes (Fig. 9). The Pgp up-regulation was further confirmed at the mRNA level by semiquantitative RT-PCR (Fig. 10a) and at the protein level by Western immunoblot analysis (Fig. 11, panels 0 and 7). The lack of MRP1 up-regulation (gene name ABCC1) (Fig. 9) suggests that expressions of Pgp and MRP1 may be controlled by different mechanisms.

The mRNA level comparison between HCT15R and HCT15 cell lines using a custom 70-oligomer cDNA microarray. ABCB1 (gene name for Pgp) was the only gene that was up-regulated among all genes included in the array. Two 70-mer probes were used for Pgp and showed similar results. No ABCC1 or ABCC2 (gene names for MRP1 and MRP2) up-regulation was observed.

Characterization of HCT15 and HCT15R cell lines by RT-PCR and Western blot analysis. a, RT-PCR showed Pgp up-regulation at the mRNA level in HCT15R cells using β-actin for normalization. b, Western blot analysis showed a global increase of acetylation of histone proteins H3 and H4 using β-actin as the loading control.
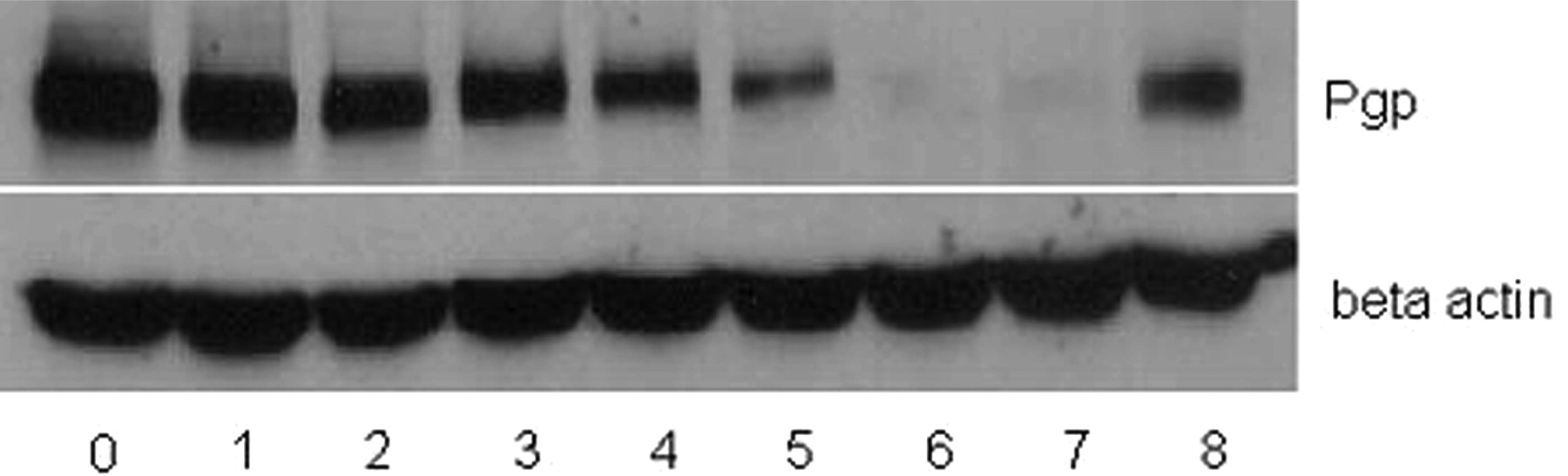
Reversible Pgp induction in HCT15R cells as measured by Western blot analysis. HCT15R cells were either cultured in medium containing 1000 nM FK228 (lane 0) or FK228-free medium for 1 to 6 weeks (lanes 1–6). The Pgp expression level decreased over time. HCT15 cells were used as a negative control (lane 7). Treating the resulting 6-week HCT15R cells with 500 nM FK228 readily restored the high Pgp expression (lane 8). β-Actin was used as the loading control.
FK228 is an HDAC inhibitor and up-regulates Pgp through histone hyperacetylation (Jin and Scotto, 1998; El-Osta et al., 2002). However, treatment with a Pgp substrate drug, such as paclitaxel (Schondorf et al., 2003) or doxorubicin (Marie et al., 1993; Schondorf et al., 2003), also can lead to rapid Pgp up-regulation. To find out whether FK228 induces Pgp through HDAC inhibition, we determined the histone acetylation status in HCT15 and HCT15R cells (Fig. 10B). Global increases of histone H3 and H4 acetylation suggested that Pgp induction is associated with HDAC inhibition by FK228.
FK228 has been reported to be a reversible inhibitor of class I HDACs (Marks et al., 2000). Consistent with this result, we found that FK228 reversibly up-regulates Pgp, and the sustained up-regulation depends on continuous FK228 exposure (Fig. 11). Pgp expression of HCT15R cells decreased over time in the absence of FK228 and decreased to the baseline Pgp level within 6 weeks. Retreatment of HCT15R cells after 6 weeks with 500 nM FK228 rapidly restored the Pgp expression (Fig. 11). The relatively slow decrease of Pgp expression following FK228 cessation of treatment was probably caused by a long turnover time of Pgp protein.
Histone acetylation status is controlled by HDAC and HAT enzymes, and deregulation of HAT and HDAC also may contribute to the acquired FK228 resistance. For this reason, we determined the HDAC and HAT activities in HCT15 and HCT15R cells (Fig. 12). However, no significant change of HAT or HDAC activity was detected.

HDAC and HAT activity assays of HCT15 and HCT15R cells. Nuclear extracts from HCT15 and HCT15R cells were determined for relative (a) HDAC and (b) HAT activities. The relative HDAC activities in HCT15 and HCT15R cells were 150.7 ± 5.2 and 150.0 ± 5.0%, respectively, and the relative HAT activities were 82.9 ± 37.5 and 64.5 ± 31.1%, respectively. Neither HDAC nor HAT activity was significantly changed in the two cell lines (n = 3; p > 0.05).
Discussion
FK228 Is a Substrate for Pgp and MRP1. Pgp and MRP1 are associated with resistance of many anticancer drugs (Lee, 2000). A National Cancer Institute screening project showed that FK228 is a Pgp substrate but not a Pgp inhibitor (Scala et al., 1997). However, no quantitative data regarding FK228 transport kinetics were available, and it was not clear whether other membrane transporters are associated with FK228 efflux.
Our current study confirmed that FK228 is a Pgp substrate, with BL→AP transport more than 30 times faster than AP→BL direction across the Caco-2 cell monolayer. The low AP→BL Papp values help to explain the low oral bioavailability of FK228 (Chan et al., 1997; Li and Chan, 2000). However, no apparent Pgp saturation was achieved throughout the rather wide FK228 concentration range from 0.5 to 20 μM. A possible explanation would be that FK228 is not a strong Pgp substrate and that its Km value for Pgp binding is >20 μM, the highest concentration we tested. This explanation is consistent with the data published by Scala et al. (1997), which showed that FK228 did not competitively inhibit cellular efflux of several model Pgp substrates by SW620 Ad300 cells. Conversely, 5 μM CsA significantly blocked FK228 BL→AP transport, and the resulting transport rates for both directions became similar, with a Teff ratio of 1.65 (Fig. 3). This could be readily explained if FK228 is not a strong Pgp substrate because CsA, as a moderate competitive Pgp inhibitor at the same concentration, seemed to bind Pgp more efficiently than FK228.
Paradoxically, being a relatively weak Pgp substrate, the cellular efflux of FK228 is expected to be minimal; however, our data showed an almost unidirectional FK228 transport across the Caco-2 monolayer. We hypothesize that FK228, being highly lipophilic (Chan et al., 1997), is likely to be trapped within the cell membrane. It has been reported that Pgp works as a flippase or a “hydrophobic vacuum cleaner” that pumps out substrate drugs from within the inner layer of cell membrane rather than from cytosol (Teodori et al., 2002). Thus, although FK228 is a relative weak Pgp substrate, its high concentration within the cell membrane may increase the efficiency of its Pgp-mediated efflux. This efflux is expected to decrease the FK228 concentration in the inner layer of cell membrane, as well as the FK228 concentration gradient between the inner membrane and cytosol. Because the concentration gradient is the driving force for FK228 to diffuse into the cell, Pgp-mediated efflux thus is expected to result in significantly decreased FK228 concentration in cytosol.
The aforementioned findings may have clinical significance. FK228 can be potentially used against Pgp-positive cancers in combination with a Pgp inhibitor because Pgp-related efflux can be readily reverted by Pgp inhibition.
The concept of FK228 functioning as an MRP1 substrate was first established by a human RBC uptake study (Fig. 4). Human RBC not only expresses high concentrations of MRP1 (Rychlik et al., 2000), but the membrane structure is also relatively simple and contains fewer interfering membrane transporters (e.g., Pgp). For these reasons, RBC is a good model to study MRP1-related uptakes (Zaman et al., 1996; Evers et al., 1997; Klokouzas et al., 2001). The saturable RBC uptake kinetics and the effect of MRP1 inhibition on the uptake were consistent with FK228 being an MRP1 substrate (Fig. 4). The Pgp(-)/MRP1(-) HL60 and Pgp(-)/MRP1(+) HL60Adr cell pair served as another good system to study MRP1-mediated cellular uptake and showed an inverse correlation between MRP1 expression and FK228 cytotoxicity (Fig. 5). Collectively, this evidence indicated that FK228 is an MRP1 substrate.
Caco-2 cell monolayer is widely used to study Pgp-related transport. However, its use to study MRP-mediated transport and uptake has been limited because of debates on whether differentiated Caco-2 cells express functional MRP proteins. It was recently reported that MRP2 is the major functional MRP transporter on the apical membrane of Caco-2 cell, whereas MRP1 expression is minimal (Hirohashi et al., 2000; Sun et al., 2002; Cooper et al., 2004). Using Caco-2 cell monolayer as a model, we found that 50 μM MK571 significantly increased FK228 AP→BL but decreased BL→AP transport (Fig. 6), suggesting that MRP2, or other related transporters that are expressed on the apical membrane, might play a role in FK228 transport across the Caco-2 monolayer. Another commonly used MRP inhibitor, indomethacin, at either 20 or 40 μM, showed little on FK228 transport profile across the Caco-2 cell monolayer, probably because of its weaker intrinsic inhibitory activity against MRP2 compared with MK571. The phenomenon that either Pgp or MRP1 inhibition reversed the net BL→AP flux of FK228 across the Caco-2 monolayer was rather difficult to explain, probably because of the nonspecificity of CsA and Ver as Pgp inhibitors. For this reason, we use a more specific Pgp inhibitor, PSC833 (Valspodar), at 1 μM to better determine the effect of Pgp on FK228 transport. Because of between-experiment variations, we were only able to determine the Papp values in the BL→AP direction of 75.4 ± 13 and 4.98 ± 1.2 × 10-7 cm/s in the absence and presence of PSC833, respectively (p < 0.005). This 15-fold decrease in Papp, BL→AP suggests Pgp plays a major role in FK228 transport. However, because of the lack of PSC833 inhibition data in the AP→BL direction, as well as the fact that Caco-2 is not a dedicated model for MRP transporters, the contribution of MRP2 or related transporters to FK228 transport cannot be quantitatively determined.
Pgp Induction Is the Major Mechanism for Acquired FK228 Resistance. HCT15 cells readily acquired FK228 resistance. As shown by our data, reversible Pgp up-regulation seemed to be a major resistance mechanism. This was confirmed by a series of cytotoxicity assays (Fig. 7; Table 1), microarray (Fig. 9), RT-PCR (Fig. 10A), and Western blot analysis (Fig. 11). However, there seemed to be no MRP1 up-regulation in HCT15R cells according to our cytotoxicity (Fig. 8) and microarray (Fig. 9) results. This suggests that expressions of Pgp and MRP1 may be controlled by different mechanisms. Moreover, we found the HAT/HDAC machinery is not deregulated in HCT15R cells.
To date, FK228 is the only known HDAC inhibitor being a Pgp substrate, and it is not clear whether Pgp induction is associated with the overall low response rate during several FK228 clinical trials (Marshall et al., 2002; Sandor et al., 2002). However, potential Pgp induction by FK228, and maybe by other HDAC inhibitors of clinical interest (e.g., suberoylanilide hydroxamic acid and valproic acid), should be considered when these drugs are used to pretreat patients receiving Pgp substrate drugs.
Footnotes
-
This work was supported by National Institutes of Health Grant 1R21CA 96323 and by BioMedical Mass Spectrometry Laboratory at The Ohio State University.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.104.072033.
-
ABBREVIATIONS: FK228, (E)-1S,4S,10S,21R)-7[(Z)-ethylideno]-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,20,23-tetraazabicyclo[8,7,6]-tricos-16-ene-3,6,9,22-pentanone; HDAC, histone deacetylase; Pgp, P-glycoprotein; MRP, multidrug resistance-associated protein; RBC, red blood cell; RT-PCR, reverse transcription-polymerase chain reaction; HAT, histone acetyl transferase; MK571, 3-[[3-[2-(7-chloroquinolin-2-yl)vinyl]phenyl]-(2-dimethylcarbamoylethylsulfanyl)methylsulfanyl] propionic acid; CsA, cyclosporin A; Ver, (±)-verapamil hydrochloride; HPLC, high-performance liquid chromatography; HBSS, Hanks' balanced salt solution; DPBS, Dulbecco's phosphate-buffered saline; FBS, fetal bovine serum; AP, apical; BL, basolateral; ABC, ATP-binding cassette; PCR, polymerase chain reaction.
-
↵1 Current address: Department of Pharmaceutical Sciences, University of Maryland, Baltimore, MD.
- Received May 28, 2004.
- Accepted January 4, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}