


Abstract
S32504 [(+)-trans-3,4,4a,5,6,10b-hexahydro-9-carbamoyl-4-propyl-2H-naphth[1,2-b]-1,4-oxazine] displayed marked affinity for cloned, human (h)D3 receptors (pKi, 8.1) at which, in total G-protein ([35S]GTPγS binding, guanosine-5′-O-(3-[35S]thio)-triphosphate), Gαi3 (antibody capture/scintillation proximity), and mitogen-activated protein kinase (immunoblot) activation procedures, it behaved as an agonist: pEC50 values, 8.7, 8.6, and 8.5, respectively. These actions were blocked by haloperidol and the selective D3 receptor antagonist S33084 [(3aR,9bS)-N-[4-(8-cyano-1,3a,4,9b-tetrahydro-3H-benzopyrano[3,4-c]pyrrole-2-yl)-butyl]-(4-phenyl) benzamide)]. S32504 showed lower potency at hD2S and hD2L receptors in [35S]GTPγS binding (pEC50 values, 6.4 and 6.7) and antibody capture/scintillation proximity (hD2L, pEC50, 6.6) procedures. However, reflecting signal amplification, it potently stimulated hD2L receptor-coupled mitogen-activated protein kinase (pEC50, 8.6). These actions were blocked by haloperidol and the selective D2 receptor antagonist L741,626 [4-(4-chlorophenyl)-1-(1H-indol-3-ylmethyl)piperidin-4-ol]. The affinity of S32504 for hD4 receptors was low (5.3) and negligible for hD1 and hD5 receptors (pKi, <5.0). S32504 showed weak agonist properties at serotonin1A ([35S]GTPγS binding, pEC50, 5.0) and serotonin2A (Gq, pEC50, 5.2) receptors and low affinity for other (>50) sites. In anesthetized rats, S32504 (0.0025-0.01 mg/kg, i.v.) suppressed electrical activity of ventrotegmental dopaminergic neurons. Correspondingly, S32504 (0.0025-0.63 mg/kg, s.c.) potently reduced dialysis levels (and synthesis) of dopamine in striatum, nucleus accumbens, and frontal cortex of freely moving rats, actions blocked by haloperidol and L741,626 but not by S33084. In contrast, S32504 only weakly inhibited serotonergic transmission and failed to affect noradrenergic transmission. Actions of S32504 were expressed stereospecifically versus its less active enantiomer S32601 [(-)-trans-3,4,4a,5,6,10b-hexahydro-9-carbomoyl-4-propyl-2H-naphth[1,2-b]-1,4-oxazine]. Although the D3/D2 agonist and antiparkinsonian agent ropinirole mimicked the profile of S32504, it was less potent. In conclusion, S32504 is a potent and selective agonist at dopamine D3 and D2 receptors.
Dopaminergic mechanisms are broadly implicated in the etiology of psychiatric and neurological disorders such as depression, Parkinson's disease (PD), and schizophrenia. Accordingly, dopaminergic ligands are of substantial interest as therapeutic agents, and elucidation of the physiological significance of individual classes (D1-D5) of dopamine (DA) receptor is a task of considerable importance.
Particular attention has been devoted to D3 receptors that display patterns of ligand recognition and intracellular coupling similar to those of closely related D2 receptors (Levant, 1997; Vallone et al., 2000), of which short (D2S) and long (D2L) isoforms are predominantly localized pre- and postsynaptically, respectively, relative to dopaminergic pathways (Usiello et al., 2000; Centonze et al., 2002). Although D2 receptors appear to be of broad functional significance, the precise role of D3 receptors has proven difficult to identify inasmuch as conventional dopaminergic ligands such as the antagonist haloperidol and the agonist apomorphine interact indiscriminately with both D2 and D3 sites (Levant, 1997; Vallone et al., 2000; Joyce, 2001; Millan et al., 2002). Nevertheless, crucial insights into the functional significance of D3 versus D2 receptors have recently been provided by: first, use of the preferential D2 versus D3 receptor antagonist L741,626, together with highly selective D3 versus D2 receptor antagonists, such as GR218,231, S33084, and SB277,011 (Millan et al., 2000a,b; Crider and Scheideler, 2001); second, characterization of mice genetically deprived of either D2 and/or D3 receptors (Sibley, 1999); and third, use of antisense probes directed against D3 or D2 receptors (Ekman et al., 1998).
Accordingly, a consensus has emerged that inhibitory D2 autoreceptors predominate over their D3 counterparts on dopaminergic pathways (Joyce, 2001), although postsynaptic populations of D3 receptor may modulate the activity of ascending dopaminergic pathways via a feedback loop (Millan et al., 2000a,b; Zapata et al., 2001; Joseph et al., 2002). At the postsynaptic level, broadly distributed, cerebral populations of D2 receptors fulfill diverse roles in, for example, the control of mood and motor function (Picetti et al., 1997; Vallone et al., 2000). Dopamine D3 receptors show a more restricted distribution, being concentrated in limbic regions (Stanwood et al., 2000; Joyce, 2001). These populations are involved in the control of emotion and reward, actions of relevance to depressive and psychotic states as well as drug abuse (Picetti et al., 1997; Joyce, 2001). Of particular interest is the contrasting influence of postsynaptic D2 and D3 receptors upon motor behavior. There is, thus, unequivocal evidence for a facilitatory influence of striatal and limbic populations of D2 receptors upon motor function (Sibley, 1999; Joyce, 2001). On the other hand, D3 sites, which are primarily localized on different classes of striatal neurons compared with their D2 counterparts (Joyce, 2001), may exert an opposite, tonic, inhibitory influence upon locomotion (Waters et al., 1993). However, this remains disputed and the significance of D3 receptors to the beneficial (restoration of motor function) and deleterious (induction of dyskinesia) actions of antiparkinsonian agents remains controversial (Ekman et al., 1998; Bézard et al., 2003; Millan et al., 2004c). The role of D3 compared with D2 receptors in the neuroprotective properties of dopaminergic agonists in Parkinson's patients (Whone et al., 2003) also remains unclear. Nevertheless, as discussed in the accompanying paper (Millan et al., 2004b), D3 sites participate in the ability of dopamine D3/D2 receptor agonists to protect dopaminergic neurons from neurotoxic damage in rodents (Joyce et al., 2003; Ramirez et al., 2003).
In light of the above comments, there is considerable interest in novel ligands at D3 and/or D2 receptors for the improved treatment of psychiatric and neurological disorders (Crider and Scheideler, 2001). Although many agonists at D2/D3 sites have been described, the majority potently interact with other classes of dopaminergic, adrenergic, and serotonergic receptors. Such actions modify their functional profiles and limit their utility as pharmacological tools for exploration of the significance of D3 and D2 sites (Joyce, 2001; Millan et al., 2002; Newman-Tancredi et al., 2002a,b). Indeed, the indolinone, ropinirole (Fig. 1), remains one of the few genuinely selective D2/D3 agonists (Millan et al., 2002) to be therapeutically employed in the management of PD (Coldwell et al., 1999; Matheson and Spencer, 2000; Rascol et al., 2000).
Chemical structures of S32504 and ropinirole.
The chemically novel naphtoxazine derivative, S32504 (Fig. 1), is of particular interest since, as described in this series of papers (Millan et al., 2004a,2004b), it behaves as a highly selective agonist at D3/D2 receptors and shows pronounced activity in experimental models of potential antiparkinsonian, neuroprotective, and antidepressant properties. Employing a complementary cellular, neurochemical, and electrophysiological approach, the present study characterizes: 1) the binding profile of S32504; 2) its influence upon transduction mechanisms controlled by hD3, hD2L, hD2S, and hD4 receptors; 3) its modulation of the electrical, synthetic, and DA-releasing activity of ascending dopaminergic pathways; and 4) its (comparatively weak) actions at serotonin (5-HT)1A and 5-HT2A receptors, which are implicated in the influence of many antiparkinsonian agents upon motor function, mood, and cognition (Gresch and Walker, 1999; Bibbiani et al., 2001; Millan et al., 2002; Newman-Tancredi et al., 2002a,b). To underpin the specificity of actions of S32504, its effects were compared with those of its less active enantiomer S32601. Furthermore, its actions were systematically compared with those of ropinirole. Finally, employing haloperidol, S33084, and L741,626 (see above citations; Millan et al., 2000a), we examined the role of D3 compared with D2 receptors in the actions of S32504.
Materials and Methods
Determination of Binding Affinities. Drug affinities at multiple classes of dopaminergic receptor and other sites were determined by use of conventional procedures, which we have extensively employed and described previously (Millan et al., 2002). The protocols for dopaminergic receptor subtypes and other receptors for which S32504 showed significant affinity (pKi values, > 5.0) are summarized in Tables 1 and 4. For all sites, IC50 values were derived from isotherms by nonlinear regression analysis using the program PRISM (GraphPad Software Inc., San Diego, CA). IC50 values were transformed into Ki values according to the Cheng-Prusoff equation: Ki = IC50/(1 + L/Kd), where L corresponds to the radioligand concentration and Kd to its dissociation constant.
Affinities of (+)S32504 compared with its enantiomer (-)S32601, racemic (±)S31411, and ropinirole at multiple classes of dopamine receptor Data are means ± S.E.M. of ≥3 determinations, each performed in triplicate.
Affinities of S32504 at multiple classes of 5-HT receptor and α-adrenoceptor Data are means ± S.E.M. of two to four determinations, each performed in triplicate. For additional, procedural details, see Millan et al. (2002).
Determination of Drug Efficacies at hD3, hD2S, hD2L, and hD4 Receptors by [35S]GTPγS Binding. The protocols employed for determination of drug efficacies at Chinese hamster ovary (CHO)-expressed, recombinant hD3, hD2S, hD2L, and hD4 (hD4.4 isoform) receptors by total [35S]GTPγS binding have been described in detail previously (Millan et al., 2000a; Newman-Tancredi et al., 2002a). In brief, the concentration of [35S]GTPγS was 1.0 nM (hD3) or 0.1 nM (hD2S, hD2L, and hD4); the pH was 7.4 in each case, and the temperature 22°C. The incubation period was 40 min for hD3, hD2S, and hD2L sites and 20 min for hD4 sites. The buffer contained Hepes (20 mM), NaCl (150 mM for hD3 and l00 mM for hD2S, hD2L, and hD4 receptors), guanosine diphosphate (3 μM), and MgCl2 (3 mM for D3 and 10 mM for other receptors). Membranes were incubated with drug for 15 min before the addition of [35S]GTPγS. Agonist efficacies are expressed as a percentage of the effect observed with maximally effective concentrations of DA (3 μM). Experiments were terminated by rapid filtration through Unifilter-96 GF/B filters (PerkinElmer Life and Analytical Sciences, Boston, MA) using a filtermate harvester (PerkinElmer Life and Analytical Sciences). Radioactivity retained on the filters was determined by liquid scintillation counting using a Top Count microplate scintillation counter (PerkinElmer Life and Analytical Sciences). Data are expressed as means ± S.E.M. of at least three independent determinations performed in triplicate.
Determination of Drug Efficacies at hD3 and hD2L Receptors Coupled to Gαi3 by Antibody Capture Assay/Scintillation Proximity Assay (SPA). Gαi3 subunit stimulation by CHO-transfected hD3 and hD2L receptors was quantified using SPA procedures essentially as previously described for coupling of 5-HT2C receptors (Cussac et al., 2002b). In brief, [35S]GTPγS binding was carried out as described above for conventional filtration protocols but in 96-well optiplates (PerkinElmer Life and Analytical Sciences). At the end of the incubation period, 20 μl of NP40 (0.27% final concentration) was added to each well, and the plates were incubated with gentle agitation for 30 min. Thereafter, 10 μl of anti-Gαi3 (0.87 μg/ml final dilution) was added and incubation continued for a further 30 min. SPA beads coated with secondary anti-mouse antibodies (Amersham Biosciences UK, Ltd., Little Chalfont, Buckinghamshire, UK) were added in a volume of 50 μl and plates incubated for 3 h with gentle agitation. The plates were then centrifuged (10 min at 1300g) and radioactivity quantitated on a Top Count microplate scintillation counter. Membranes were incubated with agonists alone or with S32504 plus antagonist for 15 min before the addition of [35S]GTPγS. The efficacies of S32504 and ropinirole are expressed relative to those of DA, which was tested at a maximally effective concentration (1 μM for hD3 and 10 μM for hD2L) in each experiment. Antagonist KB values for inhibition of S32504-stimulated Gαi3 subunit stimulation were calculated according to the Cheng-Prusoff equation: KB = IC50/(1 + (agonist/EC50)), where IC50 is IC50 of antagonist, agonist is concentration of S32504, and EC50 is EC50 of S32504 alone. All data are expressed as means ± S.E.M. of at least three independent determinations, each performed in triplicate.
Determination of Drug Efficacies at hD3 and hD2L Receptors by Induction of Mitogen-Activated Protein (MAP) Kinase Phosphorylation. CHO cells expressing hD3 or hD2L receptors were grown in 24-well plates until 90% confluent, and MAP kinase phosphorylation was determined as previously described (Cussac et al., 1999). In brief, the cells were washed once with serum-free medium and incubated overnight in this medium. Drugs were diluted in the serum-free medium and added to cells to obtain the appropriate final concentration. For antagonist studies, cells were preincubated for 20 min with the antagonist at concentrations indicated and then exposed to S32504 for a further 5 min. At the end of the incubation period, 0.25 ml per well of Laemmli sample buffer containing 200 mM of dithiothreitol was added. Whole-cell lysates were boiled for 3 min at 95°C. Cell extracts (14 μl) were loaded onto 15-well 10% polyacrylamide gels, and “fully” activated MAP kinase was revealed using a monoclonal antibody specifically raised against the phosphorylated pp42MAP-KINASE (ERK 2) and pp44MAP-KINASE (ERK 1) forms on both threonine and tyrosine residues (NanoTools, Denzlingen, Germany), followed by chemiluminescence detection with horseradish peroxidase as a secondary antibody (Amersham, Les Ulis, France). Immunoblots shown are from representative experiments repeated at least three times with comparable results. Autoradiograms were analyzed by computerized densitometry using AIS software (Imaging Research, St. Catherines, ON, Canada), and phosphorylated MAP kinase was quantified. Isotherms were analyzed by nonlinear regression using PRISM (Graphpad Software Inc.). The efficacies of S32504 and ropinirole are expressed relative to those of DA, which was tested at a maximally effective concentration (0.1 μM for hD3 and 1 μM for hD2L) in each experiment. KB values of antagonists for inhibition of S32504-stimulated MAP kinase phosphorylation were calculated according to the Cheng-Prusoff equation (see above).
Determination of Drug Efficacies at h5-HT1A Receptors and at h5-HT2A Receptors. The efficacy of S32504 at CHO-expressed h5-HT1A receptors was determined by a total [35S]-GTPγS binding procedure as described previously (Newman-Tancredi et al., 2002b). The efficacy of S32504 at CHO-transfected h5-HT2A receptors was determined as described in detail elsewhere (Cussac et al., 2002c) by depletion of membrane-bound [3H]phosphatidylinositol ([3H]PI), a measure of phospholipase C activation. Efficacy at h5-HT2A receptors was also determined by Gq activation employing the same SPA protocol used for activation of Gq-coupled h5-HT2C receptors (Cussac et al., 2002b). The efficacies of S32504 and ropinirole are expressed relative to those of 5-HT, which was tested at a maximally effective concentration (10 μM) in each experiment. KB values of antagonists for inhibition of agonist actions of S32504 were calculated according to the Cheng-Prusoff equation (see above). All data are expressed as means ± S.E.M. of at least three independent determinations performed in triplicate.
Animals. Unless otherwise specified below, studies employed male Wistar rats of 180 to 250 g (Iffa Credo, L'Arbresele, France) housed in sawdust-lined cages with unrestricted access to standard chow and water. There was a 12-h light/dark cycle with lights on at 7:30 AM. Laboratory temperature and humidity were 21 ± 0.5°C and 60 ± 5%, respectively. Animals were adapted to laboratory conditions for at least 1 week prior to testing. All animals use procedures conformed to international European ethical standards (86/609-EEC) and the French National Committee (décret 87/848) for the care and use of laboratory animals.
Influence of Drugs upon the Electrical Activity of Dopaminergic Compared with Serotonergic and Adrenergic Cell Bodies. The influence of drugs upon the firing rate of ventrotegmental area (VTA)-localized dopaminergic cell bodies compared with dorsal raphe nucleus (DRN)-localized serotonergic and locus coeruleus-localized adrenergic perikarya was determined in anesthetized rats as described previously (Millan et al., 2000a). In brief, following anesthesia with chloral hydrate (400 mg/kg, i.p.), rats were placed in a stereotaxic apparatus, and a tungsten microelectrode was lowered into the VTA, DRN, or locus coeruleus. Coordinates were as follows: VTA, AP = -5.5 from bregma, L = 0.8, and H = -7/-8.5 from dura; DRN, AP = -7.8 from bregma, L = 0.0, and H = -5.5/-6.5 from dura; and locus coeruleus, AP = -1.0 from zero, L = 1.2, and H = -5.5/6.0 from dura. Neurons in each structure were characterized by their distinctive wave form and their discharge rhythm (Millan et al., 2000b). Following baseline recording (≥5 min), in studies of the VTA, vehicle, S32504, S31411 (the racemic form), S32601 (its less active enantiomer), or ropinirole were administered i.v. (in a volume of 0.5 ml/kg) in cumulative doses every 2 to 3 min. Subsequent to vehicle or drug administration, a further injection of haloperidol (16 μg/kg, i.v.) was made. For studies of the DRN, S32504 or ropinirole were administered (in a volume of 0.5 ml/kg) in cumulative doses every 2 to 3 min. Subsequent to vehicle or S32504 administration, a further injection of the selective 5-HT1A receptor antagonist WAY100,635 (100 μg/kg, i.v.) was made. For the locus coeruleus, the influence of S32504 was likewise examined. Drug effects were quantified over the 60-s bin corresponding to their time of peak action. Spike2 software (CED, Cambridge, UK) was employed for data acquisition and analysis. Data are expressed as percent change from baseline firing rate (defined as 0%). Data were analyzed by two-way analysis of variance (ANOVA) followed by Newman-Keuls test for paired data, and ID50 values were calculated.
Influence of Drugs upon Extracellular Levels of DA Compared with 5-HT and Noradrenaline (NA) in the Frontal Cortex (FCX), Nucleus Accumbens, and Striatum of Freely Moving Rats. Quantification of extracellular levels of DA, 5-HT, and NA in single dialysate samples of the FCX, nucleus accumbens (DA and 5-HT), and striatum (DA and 5-HT) was performed as previously described (Millan et al., 2000a). Guide cannulae were implanted under pentobarbital anesthesia (60 mg/kg, i.p.) 1 week before experimentation at the following coordinates: FCX, AP = +2.2 from bregma, L = ±0.6, and H = -0.2 from dura; nucleus accumbens, AP = +0.8 from bregma, L = +0.6, and H = -4.5 from dura; and striatum, AP = +0.5 from bregma, L = -2.8, and H = -3.0 from dura. On the test day, a cuprophane CMA/11 probe (4 mm in length for the FCX and striatum, 2 mm in length for nucleus accumbens, and, in each case, 0.24 mm of outer diameter) was lowered into position. Three basal samples of 20 min each were taken. Vehicle, S32504, or ropinirole were administered s.c., and samples were taken for an additional 3 h. In the antagonist experiments of DA release (nucleus accumbens and striatum), haloperidol, L741,626, S33084, or vehicle were injected, followed, 20 min later, by S32504 (0.63 mg/kg, s.c.) or vehicle. In the antagonist experiments of 5-HT release (striatum), the selective 5-HT1A receptor antagonist WAY100,635 (0.63 mg/kg, s.c.) or vehicle were injected before S32504 (10.0 mg/kg, s.c.). DA, 5-HT, and NA levels were quantified by high-performance liquid chromatography followed by coulometric detection. The assay limit of sensitivity was 0.1 to 0.2 pg/sample for DA, 5-HT, and NA in each case. Data were analyzed by ANOVA with sampling time as the repeated within-subject factor.
Influence of Drugs upon the Turnover of Dopamine Compared with 5-HT. As previously described (Millan et al., 2000a), the ratio of levels of the DA metabolite, dihydroxyphenylacetic acid (DOPAC), to those of DA were determined in projection targets of the mesocortical pathway (FCX), the mesolimbic pathway (nucleus accumbens and olfactory tubercles), and the nigro-striatal pathway (striatum). The ratio of levels of the 5-HT metabolite, 5-hydroxyindole-acetic acid (5-HIAA), to those of 5-HT were also determined in these structures. The influence of S32504, ropinirole, and vehicle were evaluated 30 min following their administration. Tissue levels of DOPAC, DA, 5-HIAA, and 5-HT were determined by high-performance liquid chromatography and electrochemical detection. DOPAC/DA and 5-HIAA/5-HT ratios were expressed relative to those of vehicle values (defined as 100%). Data were analyzed by ANOVA followed by Dunnett's test.
Drugs. In general, full dose (concentration)-response curves were generated for S32504 and ropinirole. Dose (concentration)-response relationships were also established for antagonists, the dose ranges of which were based upon our previous characterization of selective actions at their respective targets (Millan et al., 1998, 2000a,b, 2004c; Silverdale et al., 2002). All drug doses are in terms of the base. Drugs were dissolved in sterile water, if necessary, plus a few drops of lactic acid, and pH adjusted to as close to normality (>5.0) as possible. Unless otherwise specified, drugs were injected s.c. in an injection volume of 1 ml/kg. Drug structures, sources, and salts were as follows. Haloperidol (Sigma, St. Quentin Fallevier, France) and L741,626 (Tocris Cookson Inc., Bristol, UK). S33084 and MDL100,907 were synthesized by G. Lavielle (Institut de Recherches Servier, Paris, France). Ropinirole HCl, WAY100,635 HCl, S31411, its (+)enantiomer S32504, and its (-)enantiomer S32601 were synthesized by J.-L. Peglion (Institut de Recherches Servier).
Results
Binding Profile of S32504 Compared with S32601, S31411, and Ropinirole at hD3, hD2L, and hD2S Receptors. In an initial series of experiments, the binding profile of (+)S32504 was compared with that of its enantiomer (-)S32601 and with that of their racemic form, (+)S31411, at cloned, recombinant, CHO-transfected hD3, hD2S, and hD2L receptors. Racemic S31411 concentration-dependently occupied hD3 receptors and, at higher concentrations, hD2S and hD2L receptors. This profile was potently mimicked by S32504, whereas S32601 displayed only low affinity for hD3, hD2S, and hD2L sites. On this basis, S32504 was selected as the isomer for intensive study, whereas S32601 served as a reference ligand for confirmation of the enantioselectivity (stereoselectivity) and specificity of its functional actions in vitro and in vivo. As documented previously (Millan et al., 2002), ropinirole likewise behaves as a preferential ligand of hD3 compared with hD2L and hD2S receptors, although it is less potent than S32504 (Fig. 2; Table 1).

Binding profile of S32504 compared with S31411, S32601, and ropinirole at multiple classes of dopamine receptor. Panels A-C, interaction of (+)S32504 compared with racemic (±)S31411 and its enantiomer (-)S32601 with cloned, human hD3, hD2L, and hD2S receptors, respectively. Panel D, interaction of S32504 with hD3 compared with hD2S and hD2L receptors. E, interaction of ropinirole with hD3 compared with hD2S and hD2L receptors. Isotherms (means ± S.E.M.) are from representative experiments, each of which was performed in triplicate at least three times. See Table 1 for analyses. The data in panel E have been documented elsewhere in tabular form (Millan et al., 2002) and are reproduced here graphically to illustrate the comparative binding profiles of ropinirole and S32504 at hD3 versus hD2L and hD2S receptors.
Binding Profile of S32504 at Other Dopaminergic Receptor Subtypes. Compared with hD3 receptors, the affinity of S32504 for cloned hD4 receptors was very weak (>500-fold lower). Indeed, in contrast to hD3,hD2L, and hD2S receptors (see above), the affinity of S32504 for hD4 receptors was lower than that of ropinirole (Millan et al., 2002). S32504 revealed (not shown) negligible affinity (pKi values of <5.0) for cloned hD1 receptors expressed in L cells and labeled by [3H]SCH23390 (0.3 nM) and for cloned hD5 receptors expressed in CHO cells and likewise labeled by [3H]SCH23390 (0.3 nM). Ropinirole also shows negligible affinity for these sites (Millan et al., 2002). At native, rat, striatal D2 receptors, the affinity of S32504 was modest and slightly superior to that of ropinirole. Neither S32504 nor ropinirole displayed (not shown) significant affinity (pKi values of <5.0) for native, rat, striatal D1 receptors labeled with [3H]SCH23390 (0.2 nM). S32504 and ropinirole had negligible affinity (pKi values of <5.0) for native rat and cloned human DA transporters labeled with [3H]GBR12935 (1 nM) (not shown) (Table 1).
Agonist Properties of S32504 at hD3 Receptors: Total [35S]GTPγS Binding. Corresponding with previous studies (Newman-Tancredi et al., 2002a), DA (pEC50, 8.00 ± 0.07) elicited a robust 1.6-fold elevation in [35S]GTPγS binding at hD3 receptors expressed in CHO cells (Bmax, 15,000 fmol/mg protein): its effect was defined as “100”. S32504 showed high potency in likewise activating [35S]GTPγS binding at hD3 receptors (pEC50, 8.66 ± 0.20), although with somewhat less than maximal efficacy (74 ± 3%) compared with DA. Racemic S31411 also potently and markedly elevated [35S]GTPγS binding at hD3 receptors with a pEC50 of 7.71 ± 0.04 (not shown). In contrast, paralleling its low affinity for hD3 receptors in binding studies, the less potent enantiomer S32601 only weakly activated [35S]GTPγS binding at hD3 receptors with a pEC50 and a percent maximal effect (Emax) of 5.67 ± 0.10% and 18 ± 7%, respectively (not shown). At hD3 receptors, then, in a model of total [35S]GTPγS binding, S32504 is both a more potent and more efficacious agonist than ropinirole (Fig. 3; Table 2).
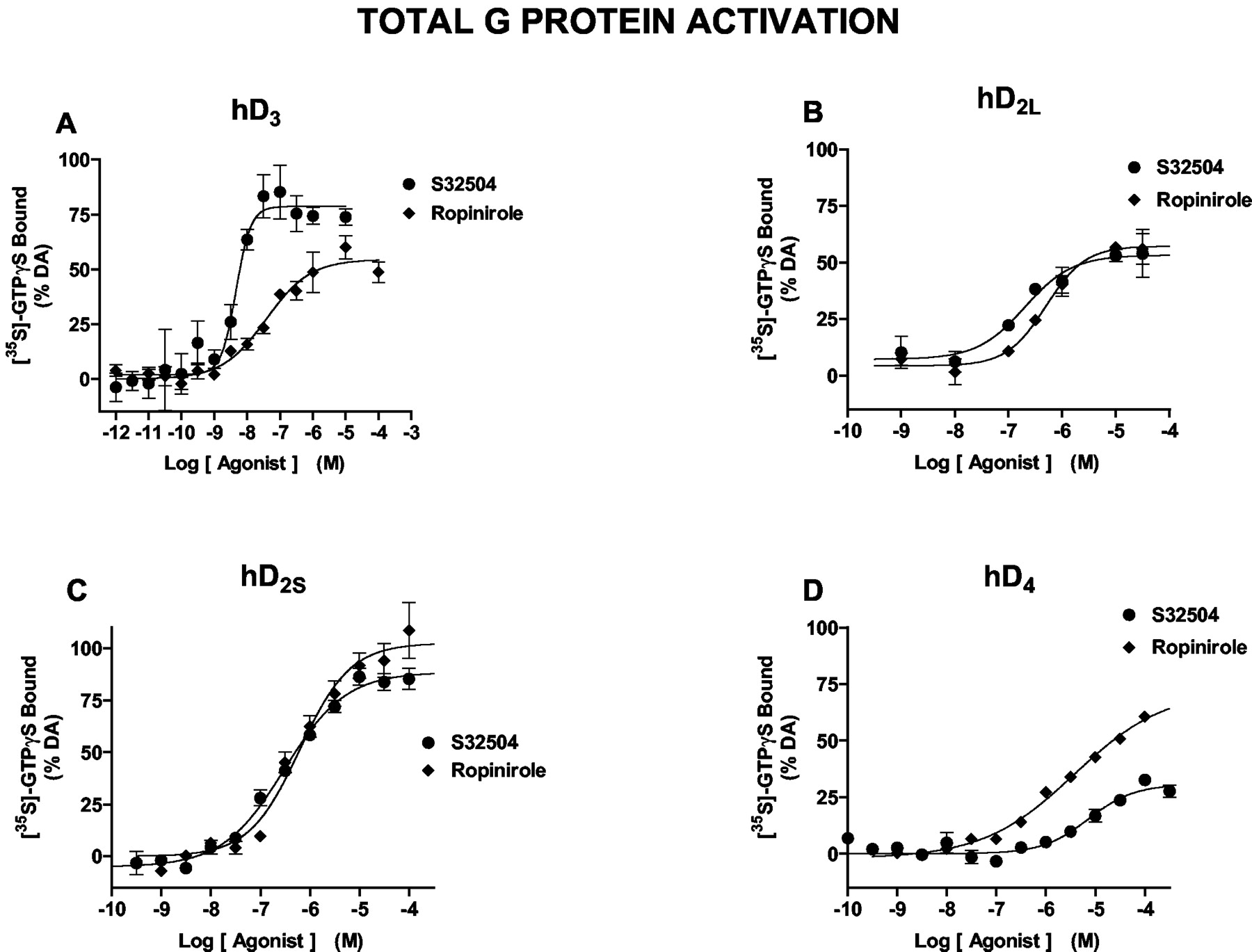
Induction of [35S]GTPγS binding by S32504 compared with ropinirole binding at hD3, hD2S, hD2, and hD4 receptors. Panel A, induction of [35S]GTPγS binding at hD3 receptors; panel B, induction of [35S]GTPγS binding at hD2L receptors; panel C, induction of [35S]GTPγS binding at hD2S receptors; panel D, induction of [35S]GTPγS binding at hD4 receptors. See Table 2 for analyses. Data (means ± S.E.M.) are from representative experiments, each of which was performed in triplicate at least three times. The data for ropinirole in panel D were presented previously in tabular form (Newman-Tancredi et al., 2002a) and are reproduced here graphically to facilitate comparisons with S32504.
Activation of hD3, hD2L, and hD2S receptors by S32504 and ropinirole as determined by measures of total G-protein activation ([35S]GTPγS binding), Gαi3 activation (immunoprecipitation coupled to SPA detection), and MAP kinase phosphorylation (immunoblot) Data are means ± S.E.M. of ≥3 determinations, each performed in triplicate.
Agonist Properties of S32504 at hD2S and hD2L Receptors: Total [35S]GTPγS Binding. In line with previous studies (Newman-Tancredi et al., 2002a), DA (pEC50, 6.45 ± 0.03) elicited a pronounced, 2.5-fold increase in [35S]GTPγS binding at hD2S receptors expressed in CHO cells (Bmax, 1600 fmol/mg of protein). Compared with DA, S32504 likewise produced a robust and concentration-dependent elevation in [35S]GTPγS binding at hD2S receptors displaying an Emax of 87 ± 1% with a pEC50 of 6.39 ± 0.07. Racemic S31411 similarly elicited a robust increase in [35S]GTPγS binding at hD2S receptors with a pEC50 of 6.13 ± 0.08 and an Emax of 96 ± 5% (not shown), whereas S32601 was inactive (pEC50 < 5.0). At CHO cell-transfected hD2L sites (Bmax, 2200 fmol/mg of protein), in agreement with previous studies (Newman-Tancredi et al., 2002a), DA evoked a 1.9-fold increase in [35S]GTPγS binding with a pEC50 of 6.48 ± 0.05. S32504 behaved as a partial agonist at hD2L sites in stimulating [35S]GTPγS binding less markedly (50 ± 3%) than at their hD2S counterparts, although showing slightly higher potency (pEC50, 6.71 ± 0.02). In procedures of [35S]GTPγS binding, thus, S32504 shows similar potency and intrinsic activity to ropinirole (Table 2; Millan et al., 2000a) both at hD2S receptors (high efficacy) and at hD2L receptors (modest efficacy) (Fig. 3; Table 2).
Weak Agonist Properties of S32504 at hD4 Receptors: Total [35S]GTPγS Binding. At hD4 sites transfected into CHO cells (Bmax, 1400 fmol/mg protein), corresponding with previous studies (Newman-Tancredi et al., 2002a), DA evoked a 2.2-fold increase in [35S]GTPγS binding with a pEC50 of 6.97 ± 0.07. The potency (pEC50, 5.22 ± 0.10) and efficacy (28 ± 4%) of S32504 at these sites was low. This low activity was confirmed by studies of racemic S31411, which yielded a pEC50 of 5.26 ± 0.14 and an Emax of 7.5% (not shown). S32601 was inactive (pEC50 < 5.0). S32504 is, thus, a less efficacious ligand than ropinirole at hD4 receptors (Newman-Tancredi et al., 2002a): pEC50 = 5.54 ± 0.06 and Emax = 74 ± 2% (Fig. 3; Table 2).
Stimulation of Gαi3 Coupled to hD3 and hD2L Receptors by S32504: Scintillation Proximity Assays. At hD3 receptors, DA potently induced [35S]GTPγS binding to Gαi3 by 2-fold with a pEC50 of 7.90 ± 0.06 (Fig. 4; Tables 2 and 3): its Emax was defined as 100%. Its actions were potently mimicked by S32504 with a pEC50 of 8.65 ± 0.07 and an efficacy of 68 ± 2% (Table 2). Ropinirole likewise, albeit less potently, enhanced [35S]GTPγS binding to Gαi3 with a pEC50 of 8.09 ± 0.04 and a similar Emax of 60 ± 3% (Table 2). The induction of [35S]GTPγS binding by S32504 was concentration dependently and potently suppressed by haloperidol and S33084 with pKB values of 8.68 ± 0.03 and 9.44 ± 0.10, respectively (Table 3), whereas L741,626 (pKB, 7.66 ± 0.1) was less active. These pKB values correlate well with their pKi values for hD3 sites (Table 3) (Millan et al., 2000a). Dopamine (though less potently) also elicited a marked and concentration-dependent 2-fold elevation in [35S]GTPγS binding at hD2L receptor-coupled Gαi3 with a pEC50 of 6.22 ± 0.04. Its actions were mimicked by S32504 and ropinirole with pEC50 values/percent efficacies of 6.63 ± 0.10/51 ± 1% and 6.59 ± 0.06/54 ± 5%, respectively (Table 2). Haloperidol and L741,626, which did not influence basal [35S]GTPγS binding (not shown), concentration dependently and potently blocked the action of S32504 at hD2L sites with pKB values of 9.44 ± 0.09 and 8.42 ± 0.07, respectively. S33084, which was likewise inactive alone, less potently (pKB, 7.7 ± 0.03) blocked the action of S32504 at hD2L receptors (Table 3). These pKB values for haloperidol, L741,626, and S33084 correlated well with their affinities for hD2L receptors (Millan et al., 2000a) (Table 3).

Activation of Gαi3 coupled to hD3 and hD2L receptors by S32504 compared with ropinirole as determined by antibody capture/scintillation proximity assays. Panel A, activation of hD3 receptor-coupled Gαi3 by S32504 compared with ropinirole; panel B, activation of hD2L receptor-coupled Gαi3 by S32504 compared with ropinirole; panel C, blockade of the activation of hD3 receptor-coupled Gαi3 by S32504 (0.1 μM) with haloperidol, the selective dopamine D3 receptor antagonist S33084, and the preferential dopamine D2L receptor antagonist L741,626; panel D, blockade of the activation of hD2L receptor-coupled Gαi3 by S32504 (10 μM) with haloperidol, the preferential dopamine D2L receptor antagonist L741,626, and the selective dopamine D3 receptor antagonist S33084. Data (means ± S.E.M.) are from representative experiments, each of which was performed in triplicate at least three times.
Blockade by D2 and/or D3 receptor antagonists (pKB values) of S32504-induced Gαi3 activation and MAP kinase phosphorylation at hD3 and hD2L receptors Data (pKB values for antagonist properties) are means ± S.E.M. of three determinations, each performed in triplicate.
Activation of MAP Kinase Coupled to hD3 and hD2L Receptors. At hD3 receptors, corroborating our previous study (Cussac et al., 1999), DA evoked a marked increase in MAP kinase phosphorylation with a pEC50 of 7.51 ± 0.09 (Fig. 5; Tables 2 and 3). S32504 and ropinirole mimicked this action of DA with pEC50 values of 8.45 ± 0.15 and 8.51 ± 0.11, respectively, and Emax values of 90 ± 6% and 80 ± 8%, respectively (Table 2). The induction of MAP kinase phosphorylation by S32504 was concentration dependently suppressed by haloperidol and S33084 with pKB values of 9.83 ± 0.07 and 9.27 ± 0.2, respectively (Table 3). At hD2L receptors, DA elicited a pronounced induction of MAP kinase phosphorylation with a pEC50 of 8.08 ± 0.03. S32504 likewise potently activated MAP kinase with a similar Emax (100 ± 6%) and a pEC50 of 8.59 ± 0.15 (Table 2). This effect of S32504 was reproduced by ropinirole with a pEC50 of 8.21 ± 0.07 and an Emax of 98 ± 2% (Table 2). Haloperidol and L741,626 concentration dependently blocked the action of S32504 with pKB values of 9.48 ± 0.14 and 8.61 ± 0.08, respectively (Table 3).

Activation of MAP kinase coupled to hD3 and hD2L receptors by S32504 compared with ropinirole as determined by immunoblot assays. Panel A, activation of hD3 receptor-coupled MAP kinase by S32504 compared with ropinirole; panel B, activation of hD2L receptor-coupled MAP kinase by S32504 compared with ropinirole; panel C, blockade of the activation of hD3 receptor-coupled MAP kinase by S32504 with haloperidol and the selective dopamine D3 receptor antagonist S33084; panel D, blockade of the activation of hD2L receptor-coupled MAP kinase by S32504 with haloperidol and the preferential dopamine D2L receptor antagonist L741,626. The immunoblots are depicted below the respective panels of quantified data. Data (means ± S.E.M.) are from representative experiments, each of which was performed in triplicate at least three times.
Binding Profile of S32504 to Nondopaminergic Receptors. Compared with hD3 receptors, S32504 revealed modest affinity for cloned h5-HT1A receptors, and it also showed modest affinity for native (rat) hippocampal 5-HT1A receptors: pKi, 5.98 ± 0.16. The affinity of S32504 for h5-HT1B sites was low, but it showed modest affinity for h5-HT1D and h5-HT7 sites (Table 4). For h5-HT2A and h5-HT2B receptors, the affinity of S32504 was weak, whereas it had low affinity for h5-HT2C receptors. For all other classes of 5-HT receptor examined (5-HT3, 5-HT4, 5-HT5A, and 5-HT6), S32504 and ropinirole revealed negligible (pKi, < 5.0) affinity. S32504 displayed low affinity for native, rat, cortical α2D-adrenoceptors and cloned, hα2A-, hα2B-, and hα2C-adrenoceptors. At native, rat, cortical α1-adrenoceptors and cloned hα1A-, hα1B-, and hα1D-adrenoceptors, as well as cloned hβ1- and β2-adrenoceptors, the affinity of S32504 was negligible (pKi values, <5.0). Neither S32504 nor ropinirole recognized (pKi values, <5.0) cloned hNA or h5-HT transporters labeled with [3H]nisoxetine (2.0 nM) and [3H]citalopram (2.0 nM), respectively (not shown). S32504 revealed low affinities—in all cases, pKi values of <5.0 —in a screen of >50 binding sites including monoamine oxidases A and B, multiple classes of histaminergic (hH1-hH4) and muscarinic (hM1-hM5) receptors, acetylcholinesterase, GABAA and GABAB receptors, central and peripheral benzodiazepine receptors, N-methyl-d-aspartate, glycineB and glycineA receptors, α-amino-2,3-dihydro-5-methyl-3-oxo-4-isoxazolepropionic acid receptors, adenosine1 and adenosine2A receptors, corticotrophin-releasing factor1 receptors, neurokinin1-3 receptors, melanin-concentrating hormone1 receptors, neuropeptide Y1 receptors, cannabinoid receptors, and σ1 and σ2 sites.
Stimulation of h5-HT1A Receptors by S32504: [35S]GTPγS Binding. At h5-HT1A receptors expressed in CHO cells (Bmax, 3600 fmol/mg), in line with previous work (Newman-Tancredi et al., 2002b), 5-HT elicited (pEC50, 7.7) a 1.5-fold increase in [35S]GTPγS binding (Fig. 6). Compared with 5-HT, S32504 only weakly and partially enhanced [35S]GTPγS binding with a pEC50 of 5.0 ± 0.14 and an Emax of 74 ± 2%. Ropinirole displays a similar profile showing a pEC50 and Emax of 5.30% and 73%, respectively (Newman-Tancredi et al., 2002b). The selective 5-HT1A receptor antagonist WAY100,635, which did not modify [35S]GTPγS binding alone (not shown), concentration-dependently abolished the action of S32504 with a pKB of 9.00 ± 0.12.

Activation of cloned, human 5-HT1A and h5-HT2A receptors by S32504. Panel A, induction of [35S]GTPγS binding at h5-HT1A receptors by S32504; panel B, blockade of the action of S32504 at h5-HT1A receptors by the selective 5-HT1A receptor antagonist WAY100,635; panel C, stimulation of h5-HT2A receptor-coupled Gαq by S32504 as determined in an SPA procedure; panel D, blockade of the action of S32504 at h5-HT2A receptors by the selective 5-HT2A receptor antagonist MDL100,907. Data (means ± S.E.M.) are from representative experiments, each of which was performed in triplicate at least three times.
Stimulation of h5-HT2A Receptors by S32504: [3H]PI Depletion and Activation of Gαq. At h5-HT2A receptors expressed in CHO cells (Bmax, 2000 fmol/mg), in line with previous studies (Newman-Tancredi et al., 2002b), 5-HT elicited (pEC50, 7.51) a robust decrease in [3H]PI levels (Fig. 6). This action was weakly mimicked by S32504 with a pEC50 of 5.5 and an Emax of 70% (data not shown), indicating that it behaves as a low potency partial agonist at these sites. At h5-HT2A receptors, 5-HT also potently (pEC50, 7.56 ± 0.06) and markedly (1.5-fold) enhanced [35S]GTPγS binding to Gαq as determined by a SPA procedure. This effect was weakly (pEC50, 5.23 ± 0.04) mimicked by S32504 with an Emax of 79 ± 2%, confirming its weak partial agonist properties at h5-HT2A sites. This action of S32504 was concentration-dependently abolished by the selective 5-HT2A receptor antagonist, MDL100,907, with a pKB of 9.47 ± 0.11. MDL100,907 did not itself modify [35S]GTPγS binding to Gαq over a comparable range of concentrations (not shown).
Modulation of the Electrical Activity of Dopaminergic Compared with Serotonergic and Noradrenergic Cell Bodies. By analogy to racemic S31411 and in distinction to its enantiomer S32601, S32504 dose-dependently and completely abolished the electrical activity of ventrotegmental dopaminergic perikarya in anesthetized rats (Fig. 7). The ID50 values were 0.57 and 1.15 μg/kg, i.v. for S32504 and S31411, respectively; S32601 did not attain 50% inhibition at the highest dose (64 μg/kg, i.v.) evaluated, so no ID50 was determined. The inhibitory action of S32504 was associated with a reduction in the number of bursts emitted. Haloperidol completely reversed these actions of S32504. Only doses far in excess of those required to markedly suppress the firing rate of dopaminergic neurons reduced the electrical activity of serotonergic neurons of the DRN, an action blocked by the selective 5-HT1A antagonist WAY100,635. S32504 failed to significantly (P > 0.05) affect the firing rate of noradrenergic neurons of the locus coeruleus (not shown): percent firing rate versus basal (defined as 100%): 2.0 μg/kg, i.v. (88.7 ± 7.2%); 8.0 (84.4 ± 11.3%); 31 (85.1 ± 11.6); and 125 (94.4 ± 13.0%). Ropinirole mimicked the inhibitory influence of S32504 upon the electrical discharge of ventrotegmental dopaminergic neurons with an ID50 = 0.87 μg/kg, i.v. It likewise exerted little influence upon serotonergic perikarya even at doses markedly higher than those inhibiting dopaminergic neurons.
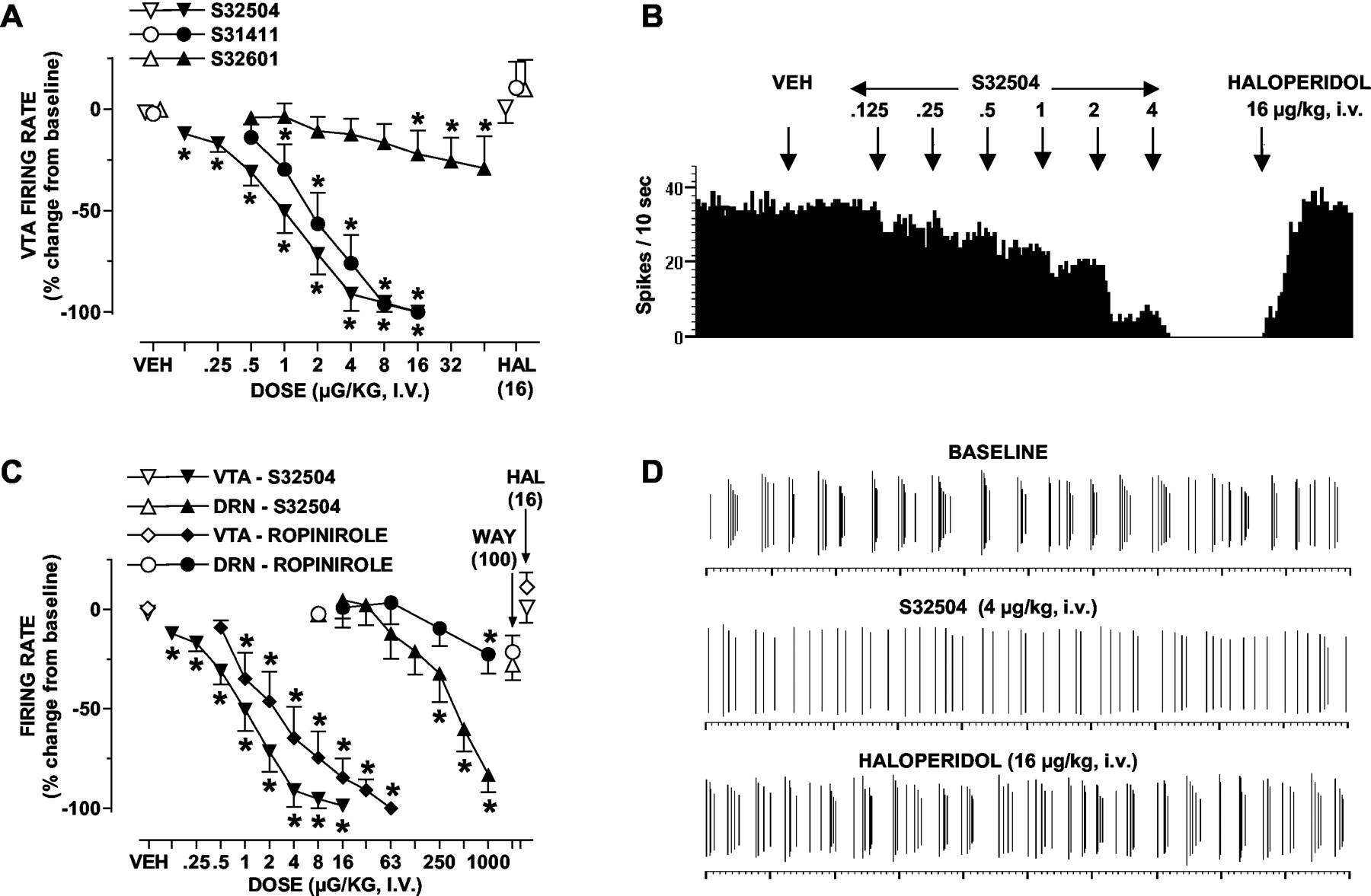
Influence of S32504 compared with S31411, S32601, and ropinirole upon the electrical activity of dopaminergic and serotonergic neurons. Panel A, dose-dependent suppression of the firing rate of ventrotegmental dopaminergic neurons by (+)S32504 compared with racemic (±)S31411 and its enantiomer (-)S32601; panel B, influence of S32504 upon the electrical activity of a representative dopaminergic neuron; panel C, preferential suppression of the electrical activity of ventrotegmental dopaminergic compared with raphe serotonergic neurons by S32504 and ropinirole; panel D, a 20-s recording of a representative bursting dopaminergic neuron showing sequential suppression of bursts by S32504 and reversal of its actions by subsequent administration of haloperidol. Data (panels A and C) are means ± S.E.M. n ≥ 5 per value. ANOVA as follows: panel A, S32504, F(8,40) = 85.6, P < 0.001; S31411, F(6,24) = 31.2, P < 0.001; and S32601, F(8,3) = 5.3, P < 0.001. Panel C, S32504, ventrotegmental area (as for panel A); S32504, DRN, F(7,28) = 29.0, P < 0.001; ropinirole, VTA, F(8,32) = 28.2, P < 0.001; and dorsal raphe nucleus, F(5,20) = 3.0, P < 0.05. For dopaminergic neurons, the influence of haloperidol (HAL) upon agonist actions was significant (P < 0.05) in each case in paired Student's t tests. For serotonergic neurons, the influence of WAY100,635 (WAY) upon the action of S32504 was significant (P < 0.05) in a paired Student's t test. Asterisks indicate significance of drug differences to vehicle values in Newman-Keuls test following ANOVA. *, P < 0.05.
Modulation of Extracellular Levels of DA Compared with 5-HT and NA. In the FCX of freely moving rats, S32504 elicited a pronounced, sustained, and dose-dependent diminution in dialysis levels of DA, whereas levels of 5-HT and NA were not significantly modified at equivalent doses (Fig. 8). Ropinirole exerted a similar pattern of selective influence upon levels of DA compared with 5-HT and NA in this structure (Fig. 8). S32504 and ropinirole also potently and dose dependently suppressed extracellular levels of DA in the nucleus accumbens and striatum (Fig. 9). In the presence of haloperidol or the preferential D2 receptor antagonist L741,626, which elevated levels of DA in the nucleus accumbens and striatum, the influence of S32504 upon DA levels was abolished (Figs. 10 and 11). In contrast, the selective D3 receptor antagonist S33084, which was ineffective alone, did not significantly modify the action of S32504. In the nucleus accumbens, levels of 5-HT were only affected by the highest dose of S32504 (10.0 mg/kg, s.c.) (not shown). Area under the curve analysis was as follows: vehicle (n = 8), +1.2 ± 2.2% versus S32504 (n = 5), -40.3% ± 4.2, F(1,11) = 20.2, P < 0.01. Likewise, in the nucleus accumbens, 5-HT levels were only modified by the highest dose of ropinirole (10.0 mg/kg, s.c.) (not shown). Area under the curve analysis was as follows: vehicle (n = 8), +1.2 ± 2.2 versus ropinirole (n = 6), -16.0 ± 3.6, F(1,12) = 5.9, P < 0.05. In the striatum, the minimal dose of S32504 required to significantly diminish levels of 5-HT (2.5 mg/kg, s.c.) was 64-fold higher than the minimum dose needed to significantly suppress DA levels (0.04 mg/kg, s.c.) (Fig. 9). The inhibitory influence of S32504 (10.0 mg/kg, s.c.) upon striatal levels of 5-HT was blocked in the presence of the selective 5-HT1A receptor antagonist WAY100,635 (0.63 mg/kg, s.c.), which did not modify levels of 5-HT (not shown). Area under the curve analysis was as follows: vehicle/vehicle (n = 7), - 4.0 ± 2.4; vehicle/S32504 (n = 5), - 42.7 ± 2.2; WAY100,635/vehicle (n = 5) +1.6 ± 2.9; and WAY100,635/S32504 (n = 5) - 14.5 ± 1.8; influence of WAY100,635, F(1,10) = 1.1, P > 0.05; influence of S32504, F(1,10) = 51.2, P < 0.01; and interaction, F(1,8) = 66.9, P < 0.01. The difference between WAY100,635/S32504 and vehicle/S32504 values was significant (P < 0.05) in Dunnett's test. The reduction by S32504 (10.0 mg/kg, s.c.) of striatal DA levels was not modified in the presence of WAY100,635 (0.63 mg/kg, s.c.) (not shown). By analogy to S32504, in the striatum, only a dose of ropinirole (10.0 mg/kg, s.c.) far higher than the minimal effective dose required to suppress DA levels significantly reduced levels of 5-HT (Fig. 9).

Influence of S32504 and ropinirole upon dialysis levels of DA compared with 5-HT and NA in dialysates of the frontal cortex of freely moving rats. Left panels, influence of S32504; right panels, influence of ropinirole. Data are means ± S.E.M. n ≥ 5 per value. Absolute basal levels of monoamines in pg/20 μl of dialysate were DA, 1.11 ± 0.14; 5-HT, 0.68 ± 0.06; and NA, 1.25 ± 0.12. ANOVA as follows: DA, S32504 (0.0025), F(1,9) = 0.4, P > 0.05; S32504 (0.01), F(1,8) = 9.6, P < 0.05; S32504 (0.04), F(1,9) = 22.8, P < 0.01; S32504 (0.63), F(1,9) = 39.3, P < 0.01; ropinirole (0.0025), F(1,8) = 1.4, P > 0.05; ropinirole (0.04), F(1.8) = 15.8, P < 0.01; ropinirole (0.63), F(1,8) = 47.3, P < 0.01. 5-HT, S32504 (0.0025), F(1,8) = 0.1, P > 0.05; S32504 (0.01), F(1,8) = 0.1, P > 0.05; S32504 (0.04), F(1,8) = 0.1, P > 0.05; S32504 (0.63), F(1,8) = 0.1, P > 0.05; ropinirole (0.0025), F(1,8) = 0.4, P > 0.05; ropinirole (0.04), F(1.8) = 0.3, P > 0.05; ropinirole (0.63), F(1,8) = 0.9, P > 0.05. NA, S32504 (0.0025), F(1,9) = 0.1, P > 0.05; S32504 (0.01), F(1,7) = 0.1, P > 0.05; S32504 (0.04), F(1,9) = 0.1, P > 0.05; S32504 (0.63), F(1,9) = 0.5, P > 0.05; ropinirole (0.0025), F(1,8) = 1.7, P > 0.05; ropinirole (0.04), F(1.8) = 0.1, P > 0.05; ropinirole (0.63), F(1,8) = 0.1, P > 0.05. Asterisks indicate significance of drug-treated versus vehicle-treated values. *, P < 0.05.

Influence of S32504 compared with ropinirole upon dialysis levels of DA compared with 5-HT in the striatum and nucleus accumbens of freely moving rats. Left panels, influence of S32504; right panels, influence of ropinirole. Data are means ± S.E.M. n ≥ 5 per value. Absolute basal levels of monoamines in pg/20 μl dialysate were DA, striatum, 13.9 ± 1.4; 5-HT, striatum, 0.55 ± 0.04; and DA, nucleus accumbens, 7.1 ± 1.0. ANOVA as follows: nucleus accumbens, DA, S32504 (0.0025), F(1,12) = 0.2, P > 0.05; S32504 (0.01), F(1,12) = 6.3, P < 0.05; S32504 (0.04), F(1,12) = 14.6, P < 0.01; S32504 (0.16), F(1,12) = 21.9, P < 0.01; S32504 (0.63), F(1,12) = 48.7, P < 0.01; S32504 (2.5), F(1,13) = 63.2, P < 0.01; S32504 (10.0), F(1,12) = 131.5, P < 0.01; ropinirole (0.01), F(1,11) = 0.7, P > 0.05; ropinirole (0.04), F(1.11) = 13.0, P < 0.01; ropinirole (0.63), F(1,11) = 44.5, P < 0.01; ropinirole (10.0), F(1,12) = 115.9, P < 0.01. Striatum, DA, S32504 (0.0025), F(1,11) = 1.2, P > 0.05; S32504 (0.01), F(1,12) = 2.4, P > 0.05; S32504 (0.04), F(1,11) = 11.9, P < 0.01; S32504 (0.16), F(1,12) = 26.8, P < 0.01; S32504 (0.63), F(1,12) = 20.1, P < 0.01; S32504 (2.5), F(1,13) = 117.5, P < 0.01; S32504 (10.0), F(1,12) = 222.0, P < 0.01; ropinirole (0.01), F(1,11) = 0.7, P > 0.05; ropinirole (0.04), F(1,11) = 20.1, P < 0.01; ropinirole (0.63), F(1,11) = 91.2, P < 0.01; ropinirole (10.0), F(1,12) = 245.0, P < 0.01. Striatum, 5-HT, S32504 (0.0025), F(1,11) = 4.3, P > 0.05; S32504 (0.01), F(1,12) = 1.4, P > 0.05; S32504 (0.04), F(1,11) = 2.9, P > 0.05; S32504 (0.16), F(1,11) = 2.0, P > 0.05; S32504 (0.63), F(1,12) = 2.9, P > 0.05; S32504 (2.5), F(1,13) = 67.5, P < 0.01; S32504 (10.0), F(1,12) = 116.6, P < 0.01; ropinirole (0.01), F(1,11) = 0.1, P > 0.05; ropinirole (0.04), F(1,11) = 1.3, P > 0.05; ropinirole (0.63), F(1,11) = 3.3, P > 0.05; ropinirole (10.0), F(1,12) = 41.5, P < 0.01. Asterisks indicate significance of drug-treated versus vehicle-treated values. *, P < 0.05.
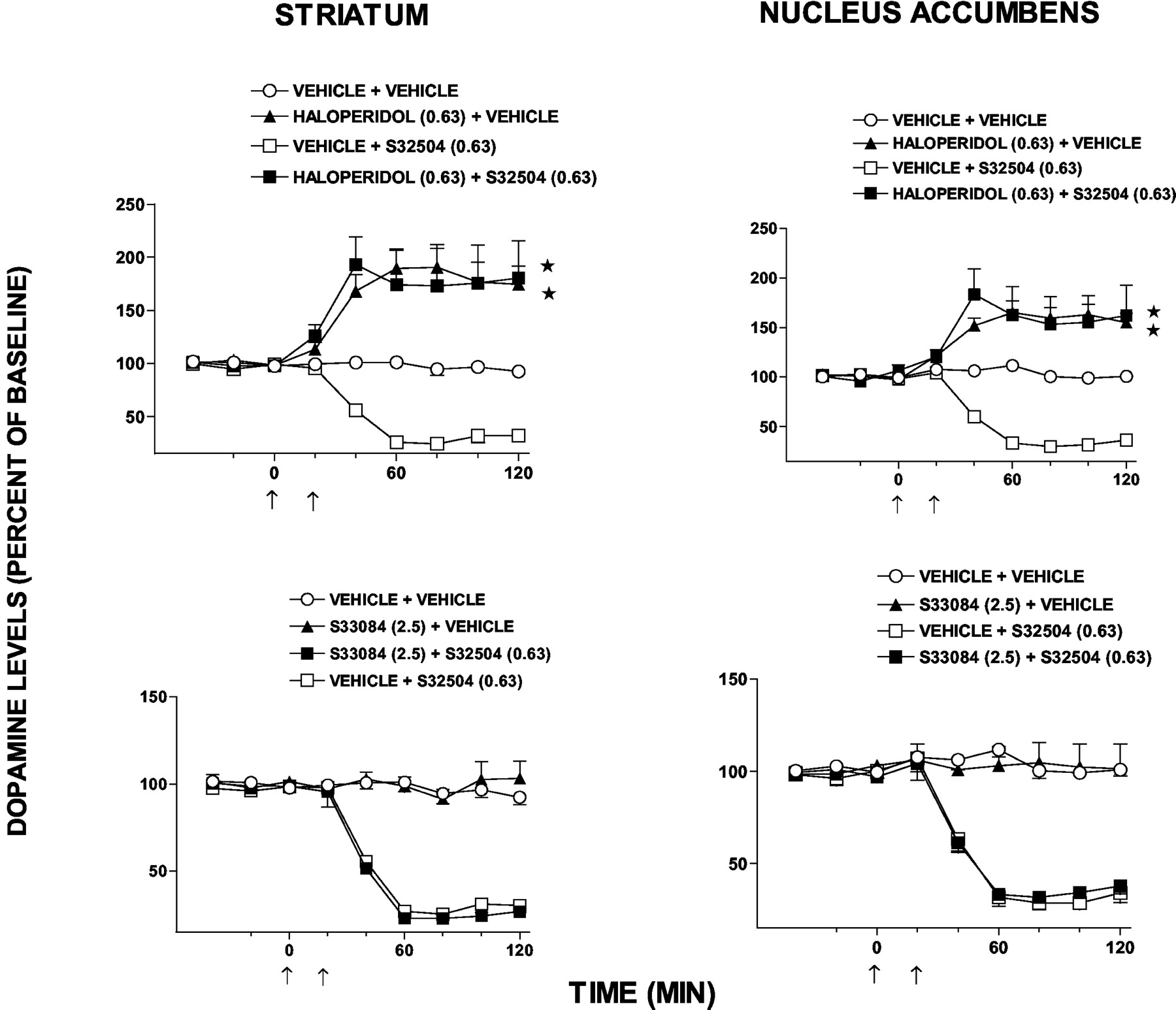
Influence of haloperidol and the selective D3 receptor antagonist S33084 upon the reduction by S32504 of dialysis levels of DA in the striatum and nucleus accumbens of freely moving rats. Left panels, striatum; right panels, nucleus accumbens. Data are means ± S.E.M. n ≥ 5 per value. Absolute basal levels of monoamines in pg/20 μl dialysate were DA, striatum, 14.9 ± 1.1; and DA, nucleus accumbens, 5.2 ± 0.4. ANOVA as follows: haloperidol, striatum: influence of haloperidol, F(1,15) = 29.0, P < 0.05; influence of S32504, F(1,16) = 109.4, P < 0.05; interaction, F(1,10) = 209, P < 0.05. Haloperidol, nucleus accumbens: influence of haloperidol, F(1,16) = 55.3, P < 0.05; influence of S32504, F(1,17) = 160.9, P < 0.05; interaction, F(1,9) = 26.5, P < 0.05. S33084, striatum: influence of S33084, F(1,15) = 0.7, P > 0.05; influence of S32504, F(1,14) = 148.7, P < 0.05; interaction, F(1,10) = 1.8, P > 0.05. S33084, nucleus accumbens: influence of S33084, F(1,16) = 0.1, P > 0.05; influence of S32504, F(1,17) = 158.9, P < 0.05; interaction, F(1,10) = 2.6, P > 0.05. Asterisks indicate significance of haloperidol + S32504-treated values versus vehicle + S32504-treated values and of haloperidol + vehicle versus vehicle + vehicle values. *, P < 0.05.

Influence of the preferential D2 receptor antagonist L741,626 upon the reduction by S32504 of dialysis levels of DA in the striatum and nucleus accumbens of freely moving rats. Left panels, striatum; right panels, nucleus accumbens. Data are means ± S.E.M. n ≥ 5 per value. Absolute basal levels of monoamines in pg/20 μl dialysate were DA, striatum, 14.9 ± 1.1; and DA, nucleus accumbens, 5.2 ± 0.4. ANOVA as follows: L741,626 (0.63), striatum: influence of L741,626, F(1,16) = 13.2, P < 0.05; influence of S32504, F(1,16) = 144.7, P < 0.05; interaction, F(1,10) = 18.4, P < 0.05. L741,626 (0.63), nucleus accumbens: influence of L741,626, F(1,16) = 15.7, P < 0.05; influence of S32504, F(1,17) = 174.1, P < 0.05; interaction, F(1,9) = 4.5, P > 0.05. L741,626 (2.5), striatum: influence of L741,626, F(1,13) = 90.9, P < 0.05; influence of S32504, F(1,16) = 120.3, P < 0.05; interaction, F(1,10) = 38.6, P < 0.05. L741,626 (2.5), nucleus accumbens: influence of L741,626, F(1,14) = 77.0, P < 0.05; influence of S32504, F(1,17) = 162.4, P < 0.05; interaction, F(1,10) = 33.6, P < 0.05. L741,626 (10.0), striatum: influence of L741,626, F(1,15) = 134.5, P < 0.05; influence of S32504, F(1,15) = 117.6, P < 0.05; interaction, F(1,12) = 50.0, P < 0.05. L741,626 (10.0), nucleus accumbens: influence of L741,626, F(1,17) = 34.0, P < 0.05; influence of S32504, F(1,16) = 137.9, P < 0.05; interaction, F(1,11) = 69.5, P < 0.05. Asterisks indicate significance of L741,626 + S32504-treated values versus vehicle + S32504-treated value and of L741,626 + vehicle versus vehicle + vehicle values. *, P < 0.05.
Modulation of Cerebral Turnover of DA Compared with 5-HT. In terminal regions of mesocortical dopaminergic pathways (the FCX), mesolimbic dopaminergic pathways (the nucleus accumbens and olfactory tubercles), and the nigrostriatal dopaminergic pathway (the striatum), S32504 elicited a marked and dose-dependent reduction in the ratio of DA to DOPAC, indicative of a suppression in DA turnover. There was no marked or significant difference among the various structures concerning the potency and the magnitude of the suppressive influence of S32504, suggesting that its inhibitory influence upon mesocortical, mesolimbic, and nigrostriatal dopaminergic pathways is expressed to a similar degree. Ropinirole mimicked this inhibitory influence of S32504 upon DA to DOPAC ratios. S32504 and ropinirole exerted only a modest influence upon 5-HT turnover (as estimated by the ratio of 5-HT to its metabolite, 5-HIAA) even at doses higher than these reducing DA turnover. These observations underpin electrophysiological and dialysis studies outlined above, indicating that S32504 and ropinirole affect serotonergic pathways only at doses markedly higher than those influencing dopaminergic transmission (Fig. 12).

Influence of S32504 compared with ropinirole upon the turnover of dopamine (DA) and 5-HT in projection targets of frontocortical, mesolimbic, and nigrostriatal dopaminergic pathways. Data are means ± S.E.M. n > 5 per value. Absolute levels of DOPAC/DA and ANOVA as follows: frontal cortex, DA = 0.9 ± 0.2 ng/mg protein and DOPAC = 0.2 ± 0.1 ng/mg protein; S32504, F(4,15) = 13.5, P < 0.05; and ropinirole, F(3,12) = 34.8, P < 0.05; nucleus accumbens, DA = 46.8 ± 3.2 ng/mg protein and DOPAC = 4.6 ± 0.4 ng/mg protein; S32504, F(4,15) = 14.7, P < 0.05; and ropinirole, F(3,12) = 178, P < 0.05; olfactory tubercles, DA = 28.2 ± 3.5 ng/mg protein and DOPAC = 3.5 ± 0.2 ng/mg protein; S32504, F(4,15) = 44.1, P < 0.05; ropinirole, F(3,12) = 19.6, P < 0.05; striatum, DA = 83.9 ± 5.6 ng/mg protein and DOPAC = 5.5 ± 0.2 ng/mg protein; S32504, F(4,15) = 20.9, P < 0.05; and ropinirole, F(3,12) = 168, P < 0.05. Absolute levels of 5-HIAA/5-HT and ANOVA as follows: frontal cortex, 5-HT = 2.6 ± 0.8 ng/mg protein and 5-HIAA = 1.3 ± 0.3 ng/mg protein; S32504, F(4,15) = 0.8, P > 0.05; and ropinirole, F(3,12) = 1.8, P > 0.05; nucleus accumbens, 5-HT = 2.6 ± 0.8 ng/mg protein and 5-HIAA = 1.3 ± 0.3 ng/mg protein; S32504, F(4,15) = 2.0, P > 0.05; and ropinirole, F(3,12) = 1.8, P > 0.05; olfactory tubercles, 5-HT = 3.1 ± 0.1 ng/mg protein and 5-HIAA = 2.6 ± 0.2 ng/mg protein; S32504, F(4,15) = 1.5, P > 0.05 and ropinirole, F(3,12) = 7.0, P < 0.05 and striatum, 5-HT = 3.1 ± 0.1 ng/mg protein and 5-HIAA = 2.6 ± 0.2 ng/mg protein; S32504, F(4,15) = 5.0, P < 0.05 and ropinirole, F(3,12) = 5.5, P < 0.05. Asterisks indicate significance of drug versus vehicle differences in Dunnett's test following ANOVA. *, P < 0.05.
Discussion
Receptor-Binding Profile of S32504: Marked Preference for hD3 Receptors. S32504, the optically pure, (+)isomer of racemic S31411, interacted stereospecifically and with high affinity at hD3 receptors compared with its enantiomer S32601. This stereospecific pattern of recognition was also apparent at hD2S and hD2L receptors for which, however, the affinity of S32504 was substantially lower. Notably, S32504 was a more potent ligand of hD3 receptors than ropinirole (Millan et al., 2002) from which it could also be distinguished by its high selectivity for hD3 versus hD4 receptors. Indeed, the weak stimulation of hD4 receptors by S32504 differentiates it from numerous antiparkinsonian agents (Millan et al., 2002) and is of significance inasmuch their activation does not contribute to improvement of motor function by antiparkinsonian agents and may even unfavorably influence mood and cognition (Oak et al., 2000; Newman-Tancredi et al., 2002a). More generally, the high selectivity of S32504 for hD3 receptors differentiates it from the majority of antiparkinsonian drugs that potently interact with various classes of α-adrenoceptor, 5-HT receptor, and/or histaminergic receptor (Millan et al., 2002; Newman-Tancredi et al., 2002a,b) (see below).
Agonist Properties of S32504 at hD3 Receptors: G-Protein Recruitment. In line with its high affinity, in a [35S]GTPγS model of G-protein activation (Newman-Tancredi et al., 1999, 2002a), S32504 manifested potent and robust agonist properties at hD3 receptors compared with ropinirole (Table 2). The efficacy of S32504 (74%) bears comparison to the constrained range of efficacies (39-94%) of 14 antiparkinsonian agents examined under identical conditions (Newman-Tancredi et al., 2002a). Employing an antibody neutralization strategy to uncouple hD3 receptors from their respective G-proteins, we previously showed that CHO-transfected hD3 receptors primarily couple to pertussis-sensitive Gi/o proteins (Newman-Tancredi et al., 1999). Of this family, exploiting an innovative SPA/antibody-based strategy, we found herein that S32504 and ropinirole activate hD3 receptor-coupled Gαi3, and the specificity of its engagement by S32504 was confirmed by blockade with haloperidol and the selective D3 receptor antagonist S33084 (Millan et al., 2000a). Employing other antibodies, S32504 (and DA) were found not to activate Go, Gq, or Gs in this hD3 receptortransfected CHO cell line (not shown). However, hD3 receptors couple in a cell-specific fashion to various G-protein subtypes, including Go and Gz (Liu et al., 1999; Obadiah et al., 1999; Zaworski et al., 1999), so it would be of interest to further evaluate G-protein recruitment by S32504 at hD3 receptors in other systems.
Agonist Properties of S32504 at hD3 Receptors: MAP Kinase Phosphorylation. Pertussis-sensitive Gi/o-proteins transduce MAP kinase phosphorylation by hD3 receptors (Cussac et al., 1999), which was accordingly powerfully stimulated by S32504. Moreover, mimicking the SPA study, S33084 potently blocked S32504-induced MAP kinase phosphorylation with a potency close to its affinity for hD3 receptors (Millan et al., 2000a). Although less potent than S32504, ropinirole was also an efficacious agonist, complementing studies of its agonist properties employing measures of adenylyl cyclase, mitogenesis, and extracellular acidification (Coldwell et al., 1999; Perachon et al., 1999). The precise physiological significance of S32504-induced MAP kinase via hD3 receptors remains to be clarified. However, this signal is implicated in the neuroprotective influence of S32504 and other dopaminergic agonists upon dopaminergic neurons that, as discussed in the accompanying paper (Millan et al., 2004b) and elsewhere (Joyce et al., 2003; Ramirez et al., 2003), appear to involve the activation of dopamine D3 receptors.
Agonist Properties of S32504 at hD2S and hD2L Receptors: G-Protein Recruitment. Exploiting a [35S]GTPγS binding paradigm, S32504 behaved as an agonist at hD2L and hD2S receptors with, respectively, high and modest efficacy. Inasmuch as these observations are comparable to ropinirole and many other antiparkinsonian drugs (Newman-Tancredi et al., 2002a), differential efficacy at hD2L versus hD2S sites may be an intrinsic feature of the cellular expression systems employed. Indeed, contrasting patterns of G-protein activation by hD2L versus hD2S receptors have been reported in certain other cell lines (Gardner et al., 1996; Picetti et al., 1997; Newman-Tancredi et al., 2002a). Filtration assays do not permit identification of individual G-proteins, but in a novel SPA procedure, S32504 and ropinirole potently activated Gαi3 via hD2L sites. Employing a complementary strategy— cotransfection with mutant G-proteins resistant to pertussis toxin—O'Hara et al. (1996) also showed that hD2L receptors recruit Gαi3. Using an antisense knockdown strategy, activation of Gαi3 by hD2L sites was likewise reported by Wolfe and Morris (1999). However, it would be interesting to examine the influence of S32504 upon other species of G-protein known to couple to hD2L and hD2S receptors: notably, Go in neuronal tissue (Wolfe and Morris, 1999; Alberts et al., 2000; Cordeaux et al., 2001; Jiang et al., 2001; Watts et al., 2001).
Agonist Properties of S32504 at hD2L Receptors: MAP Kinase Phosphorylation. Pertussis-sensitive Gi/o-proteins are implicated in the induction of MAP kinase via hD2L receptors (Choi et al., 1999; Oak et al., 2001), and both S32504 and ropinirole robustly increased MAP kinase phosphorylation. These findings correspond to the high efficacy of S32504 and ropinirole for suppression of forskolin-stimulated adenylyl cyclase activity at hD2L receptors (Maggio et al., 2003). Furthermore, they complement observations of agonist properties of ropinirole at hD2L (and hD2S) sites employing parameters of extracellular acidification and mitogenesis (Coldwell et al., 1999; Perachon et al., 1999). Relative to G-protein coupling, there was a substantial potency shift for both S32504 and ropinirole in eliciting MAP kinase phosphorylation. This amplification of signaling is specific since: 1) identical shifts in potency are seen for other agonists at hD2L sites (D. Cussac, unpublished observations); 2) S32601 did not stimulate MAP kinase phosphorylation (not shown); 3) the action of S32504 was abolished by haloperidol and L741,626 at doses similar to those blocking Gi/o activation via hD2L receptors; and 4) studies of other G-protein-coupled receptors, such as α2-adrenoceptors and h5-HT1A receptors, revealed comparable amplification between the G-protein and downstream effectors (Lopez-Ilasaca, 1998; Umland et al., 2001; Cussac et al., 2002a). This marked amplification between Gi/o- and MAP kinase at hD2L but not hD3 receptors likely reflects contrasting intracellular pathways for engagement of MAP kinase at hD2L versus hD3 sites (Luo et al., 1998; Welsh et al., 1998; Cussac et al., 1999; Oak et al., 2001). Notably, in addition to Gα-subunits, hD2L receptors may stimulate MAP kinase via Gβγ subunit release and other components of the MAP kinase cascade such as Grb2 and Shc (Hawes et al., 1995). Irrespective of mechanisms underlying amplification, the potent and efficacious activation of hD2L receptor-coupled MAP kinase by S32504, ropinirole, and other agonists provides a substrate for their pronounced actions at D2 receptors in vivo (Levant, 1997; Picetti et al., 1997; Joyce, 2001; Millan et al., 2004a,b). Furthermore, by analogy to other cellular signals downstream of G-proteins, the apparently pronounced preference of S32504 for hD3 versus hD2 receptors in binding and [35S]GTPγS-binding studies is eclipsed at the level of MAP kinase (Coldwell et al., 1999; Cussac et al., 1999; Millan et al., 2000a). Although the precise significance of MAP kinase to the actions of D2L agonists is unclear, it may transduce their antiparkinsonian properties in the striatum (Welsh et al., 1998; Cai et al., 2002).
Activation by S32504 of D2/D3 Autoreceptors. In line with other dopaminergic agonists such as ropinirole (Eden et al., 1991), S32504 stereospecifically suppressed the activity of frontocortical, mesolimbic, and nigrostriatal dopaminergic pathways. Anatomical (Stanwood et al., 2000), pharmacological (Millan et al., 2000a,b), gene knockout (Sibley, 1999), and antisense (Ekman et al., 1998) studies support a major role of D2 sites in the modulation of dopaminergic transmission (Levant, 1997; Millan et al., 2000b), and the D2S isoform is principally involved (Centonze et al., 2002). Indeed, the inhibitory influence of S32504 upon DA release in nucleus accumbens and striatum was resistant to S33084 yet abolished by haloperidol and L741,626, consistent with a key role of D2 sites. Nevertheless, a possible role of D3 receptors should not be entirely excluded. First, the facilitation by L741,626 and haloperidol of dopaminergic transmission complicates interpretation of their actions. Second, although we acquired an identical pattern of data employing a further D3 antagonist, PD128,907, in interaction with L741,626 (blocked) and S33084 (refractory) (Millan et al., 2004c), Taylor et al. (1999) observed an abrogation of the PD128,907-induced reduction of DA release in the nucleus accumbens, although not striatum, employing a further D3 antagonist, SB269,652. Third, likewise using PD128,907, we have obtained evidence that D3 receptors control DA release in the FCX (Millan et al., 2000a). Fourth, studies in genetically modified mice suggest that, under certain conditions, terminal D3 receptors modulate DA release (Zapata et al., 2001; Joseph et al., 2002). Finally, via a long-loop feedback mechanism, postsynaptic D3 receptors may moderate subcortical release of DA (see Joyce, 2001).
Weak Agonist Actions of S32504 at h5-HT1A and h5-HT2A Receptors. It has been proposed that 5-HT1A agonist properties are favorable for antiparkinsonian agents in improving mood and alleviating dyskinesias (Bibbiani et al., 2001). Although this remains to be clinically proven, virtually all antiparkinsonian agents possess at least moderate partial agonist properties at 5-HT1A receptors (Newman-Tancredi et al., 2002b). Indeed, like ropinirole, S32504 similarly showed mild potency and efficacy at h5-HT1A sites in vitro. However, despite the high sensitivity of presynaptic 5-HT1A receptors (Millan et al., 2000b), S32504 and ropinirole reduced cerebral 5-HT release only at doses markedly higher than those that suppressed DA release. It is unlikely, then, that 5-HT1A receptors make a major contribution to the functional actions of S32504 in vivo, an assertion underpinned by the lack of influence of 5-HT1A receptor antagonists upon its behavioral actions (Millan et al., 2004b). It has been suggested that engagement of 5-HT2A receptors facilitates motor function in PD but, less favorably, exacerbates psychiatric side effects (Gresch and Walker, 1999; Newman-Tancredi et al., 2002b). Compared with the potent agonist properties at h5-HT2A receptors of, for example, pergolide (Newman-Tancredi et al., 2002b), S32504 behaved as a very weak agonist at Gq and phospholipase C, prototypical transduction mechanisms coupled to 5-HT2A receptors. It is unlikely, then, that they are involved in its actions.
General Comments. First, this study focuses on actions of S32504 at D2(D3) autoreceptors, which likely participate in its anxiolytic and neuroprotective properties (Millan et al., 2004a,b). S32504 also shows robust and potent agonist actions at postsynaptic D2/D3 receptors mediating, for example, prolactin secretion and yawning (M. J. Millan, unpublished observations). Activation of postsynaptic populations of D2(D3) sites is implicated in the antiparkinsonian (motor) and antidepressant actions of S32504 (Millan et al., 2004a,b). Second, D3 and D2 receptors are colocalized on dopaminergic and certain other classes of neuron (Stanwood et al., 2000; Joyce, 2001). Inasmuch as S32504 behaves as a potent and efficacious agonist at D3/D2 heterodimers in vitro (Maggio et al., 2003), certain functional actions attributed to D3 or D2 monomers might actually involve recruitment of D2/D3 heterodimers. Third, as for all G-protein-coupled receptors, ligand efficacy at D3 and D2 receptors depends upon many factors, including the intracellular signal and cell line under study (Cordeaux et al., 2001; Cussac et al., 2002b). Additional work will be required to more fully explore the influence of S32504 upon multiple coupling cascades at diverse populations of D3 and D2 receptors. Finally, for other dopaminergic ligands, the precise relationship between its cellular actions and its therapeutic properties awaits further clarification.
Conclusions. The novel naphtoxazine S32504 is a potent and highly selective agonist at hD3 receptors and, reflecting signal amplification downstream of G-proteins, hD2S and hD2L receptors, whereas it is devoid of activity at hD4 sites. This distinctive profile is associated with a potent inhibitory influence upon dopaminergic pathways. These observations provide a framework for the following papers (Millan et al., 2004a,2004b) demonstrating marked activity of S32504 in models of potential antiparkinsonian, neuroprotective, antidepressant, and anxiolytic properties.
Acknowledgments
We thank M. Soubeyran for secretarial assistance and V. Pasteau, C. Chaput, L. Verrièle, M. Touzard, R. Billiras, and L. Cistarelli for technical assistance.
Footnotes
-
DOI: 10.1124/jpet.103.062398.
-
ABBREVIATIONS: PD, Parkinson's disease; DA, dopamine; L741,626, 4-(4-chlorophenyl)-1-(1H-indol-3-ylmethyl)piperidin-4-ol; S32504, (+)-trans-3,4,4a,5,6,10b-hexahydro-9-carbamoyl-4-propyl-2H-naphth[1,2-b]-1,4-oxazine; 5-HT, 5-hydroxytryptamine (serotonin); S33084, (3aR,9bS)-N-[4-(8-cyano-1,3a,4,9b-tetrahydro-3H-benzopyrano[3,4-c]pyrrole-2-yl)-butyl]-(4-phenyl) benzamide; [35S]GTPγS, guanosine-5′-O-(3-[35S]thio)-triphosphate; CHO, Chinese hamster ovary; SPA, scintillation proximity assay; MAP kinase, mitogen-activated protein kinase; PI, phosphatidylinositol; VTA, ventrotegmental area; DRN, dorsal raphe nucleus; S31411, (±)-trans-3,4,4a,5,6,10b-hexahydro-9-carbamoyl-4-propyl-2H-naphth[1,2-b]-1,4-oxazine; ANOVA, analysis of variance; NA, noradrenaline; FCX, frontal cortex; DOPAC, dihydroxyphenyl acetic acid; 5-HIAA, 5-hydroxyindole acetic acid; MDL100,907, R(+)-α-(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenylethyl)]-4-piperidinemethanol; WAY100,635 HCl, N-{2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl}-N-(2-pyridinyl) cyclohexanecarboxamide 3HCl; Emax, maximal effect; S32601, (-)-trans-3,4,4a,5,6,10b-hexahydro-9-carbomoyl-4-propyl-2H-naphth[1,2-b]-1,4-oxazine; GR218,231, 2(R,S)-di-n-propylamino)-6-(4-methoxyphenylsulphonyl-methyl)-1,2,3,4-tetrahydronaphthalene; SB277,077, trans-N-[4-[2-(6-cyano-1,2,3,4-tetrahydroisoquinolin-2yl)ethyl]cyclohexyl]-4-quinolinecarboxymide.
- Received November 5, 2003.
- Accepted February 12, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}