


Abstract
Previous studies demonstrated that analogs of benztropine (BZT) possess high affinity for the dopamine transporter, inhibit dopamine uptake, but generally have behavioral effects different from those of cocaine. One hypothesis is that muscarinic-M1 receptor actions interfere with cocaine-like effects. Several tropane-nitrogen substitutions of 4′,4′′-diF-BZT have reduced M1 affinity compared with the CH3-analog (AHN 1-055; 3α-[bis-(4-fluorophenyl)methoxy]tropane). All of the compounds displaced [3H]WIN 35,428 (2β-carbomethoxy-3β-(4-fluorophenyl)tropane) binding with affinities ranging from 11 to 108 nM. Affinities at norepinephrine ([3H]nisoxetine) and serotonin ([3H]citalopram) transporters ranged from 457 to 4810 and 376 to 3260 nM, respectively, and at muscarinic M1 receptors ([3H]pirenzepine) from 11.6 (AHN 1-055) to higher values, reaching 1030 nM for the other BZT-analogs. Cocaine and AHN 1-055 produced dose-related increases in locomotor activity in mice, with AHN 1-055 less effective than cocaine. The other compounds were ineffective in stimulating activity. In rats discriminating cocaine (29 μmol/kg i.p.) from saline, WIN 35,428 fully substituted for cocaine, whereas AHN 1-055 produced a maximal substitution of 79%. None of the other analogs fully substituted for cocaine. WIN 35,428 produced dose-related leftward shifts in the cocaine dose-effect curve, whereas selected BZT analogs produced minimal changes in the effects of cocaine. The results suggest that reducing M1 affinity of 4′,4′′-diF-BZT with N-substitutions reduces effectiveness in potentiating the effects of cocaine. Furthermore, although the BZT-analogs bind with high affinity at the dopamine transporter, their behavioral effects differ from those of cocaine. These compounds have reduced efficacy compared with cocaine, a long duration of action, and may serve as leads for the development of medications to treat cocaine abuse.
Novel analogs of benztropine [BZT; 3α-(diphenylmethoxy)-1H,5H-tropane] have been developed that have high affinity for the dopamine transporter and are selective for the dopamine transporter over the other monoamine transporters (Newman et al., 1994, 1995; Kline et al., 1997; Newman and Kulkarni, 2002). Although these compounds bind to the dopamine transporter and inhibit the uptake of dopamine in vitro, their behavioral effects are generally different from those of the typical dopamine uptake inhibitors, for which cocaine is a prototype (Katz et al., 1999). Many of these compounds have relatively high affinity for muscarinic M1 receptors as well as the dopamine transporter, and it has been suggested that these other actions may interfere with the cocaine-like behavioral effects of BZT analogs (Katz et al., 1999). For example, 4′-chloro-3α-(diphenylmethoxy)tropane (4′-Cl-BZT) has a 30 nM affinity for the dopamine transporter, which is comparable with the high-affinity binding of cocaine. However, 4′-Cl-BZT is only marginally efficacious as a stimulant of locomotor activity and does not produce cocaine-like discriminative-stimulus effects (Katz et al., 1999; Tolliver et al., 1999). Furthermore, this compound does not maintain rates of responding as high as those maintained by cocaine in a self-administration paradigm (Woolverton et al., 2000) or break points as high as those for cocaine in a self-administration progressive ratio procedure (Woolverton et al., 2001). As such, this and similar compounds suggest a challenge to the dopamine transporter hypothesis of the behavioral effects of cocaine. According to that hypothesis, compounds that bind to the dopamine transporter and inhibit dopamine uptake will have behavioral effects like those of cocaine (Kuhar et al., 1991). In addition, an understanding of the differences in pharmacological mechanisms of cocaine and the BZT analogs may provide insight into the neurobiological substrates that underlie the abuse liability of cocaine and help to understand the functioning of the dopamine transporter.
The present studies further examined the pharmacology of 4′,4′′-diF-BZT analogs (Fig. 1), both alone and in combination with cocaine. The present report focuses on analogs substituted on the nitrogen, which are among the BZT analogs with the highest affinities for the dopamine transporter (Newman and Kulkarni, 2002). These N-substituted analogs retained high affinity for the dopamine transporter and had a reduced affinity for muscarinic M1 receptors (Agoston et al., 1997). As such, studies of these compounds will serve as additional assessments of the role of M1 muscarinic activity in the behavioral effects of BZT analogs.

Basic structures of cocaine and BZT analogs with substitutions on the nitrogen.
Materials and Methods
Subjects. Studies in rats and mice were conducted, respectively, with male Sprague-Dawley and Swiss-Webster strains (Taconic Farms, Germantown, NY). All animals were housed in a temperature- and humidity-controlled vivarium with a 12-h light/dark cycle (lights on 7:00 AM). All experiments were conducted during the light phase of the light/dark cycle, between 8:00 AM and 3:00 PM. With the exception of the behavioral studies in rats, the subjects were group housed with unrestricted access to food and water.
[3H]Nisoxetine Binding Assay. Membranes from frozen frontal cortex dissected from male Sprague-Dawley rats (Taconic Farms) were homogenized in 20 volumes (w/v) of 50 mM Tris containing 120 mM NaCl and 5 mM KCl (pH 7.4 at 25°C), using a Brinkman Polytron (at setting 6 for 20 s). The tissue was centrifuged at 50,000g for 10 min at 4°C. The resulting pellet was resuspended in buffer and recentrifuged. The final pellet was resuspended in cold buffer to a concentration of 80 mg/ml (original wet weight).
Ligand binding experiments were conducted in assay tubes containing 0.5 ml of buffer, 0.5 nM [3H]nisoxetine (PerkinElmer Life Sciences, Boston, MA), and 8 mg of frontal cortex tissue. The reaction was started with the addition of the tissue, and the tubes were incubated for 60 min at 0–4°C. The incubation was terminated by rapid filtration through Whatman GF/B filters, presoaked in 0.05% polyethylenimine, using a cell harvester (Brandel Inc., Gaithersburg, MD). The filters were washed twice with 5 ml of cold buffer and transferred to scintillation vials to which Beckman Ready Safe was added. Nonspecific binding was determined using 1 μM desipramine. Data were analyzed using GraphPad Prism software (GraphPad Software Inc., San Diego, CA).
[3H]Citalopram Binding Assay. Membranes from frozen rat midbrain were homogenized in 20 volumes (w/v) of 50 mM Tris containing 120 mM NaCl and 5 mM KCl (pH 7.4 at 251°C), using a Brinkman Polytron (at setting 6 for 20 s). The tissue was centrifuged at 20,000g for 10 min at 41°C. The resulting pellet was resuspended in buffer and recentrifuged. The final pellet was resuspended in cold buffer to a concentration of 15 mg/ml (original wet weight).
Ligand binding experiments were conducted in assay tubes containing 0.5 ml of buffer, 1.4 nM [3H]citalopram (PerkinElmer Life Sciences), and 1.5 mg of midbrain tissue. The reaction was started with the addition of the tissue, and the tubes were incubated for 60 min at 251°C (room temperature). The incubation was terminated by rapid filtration through Whatman GF/B filters (presoaked in 0.3% polyethylenimine in water) using a cell harvester (Brandel Inc.). The filters were washed twice with 5 ml of cold buffer and transferred to scintillation vials to which Beckman Ready Safe was added. Nonspecific binding was determined using 10 μM fluoxetine (Sigma/RBI, Natick, MA). Data were analyzed using GraphPad Prism software (GraphPad Software Inc.).
[3H]Pirenzepine Binding Assay. Membranes from frozen rat brains, excluding cerebellum, were thawed in ice-cold buffer (10 mM Tris-HCl and 320 mM sucrose, pH 7.4) and homogenized with a Brinkman Polytron in a volume of 10 ml/g of tissue. The homogenate was centrifuged at 1000g for 10 min at 41°C. The resulting supernatant was then centrifuged at 10,000g for 20 min at 41°C. The resulting pellet was resuspended in a volume of 200 mg/ml in 10 mM Tris buffer (pH 7.4).
Ligand binding assays were conducted in tubes containing 0.5 ml of buffer (10 mM Tris-HCl and 5 mM MgCl2), 3 nM [3H]pirenzepine (PerkinElmer Life Sciences), and 20 mg of brain tissue. The reaction was started with the addition of the tissue, and the tubes were incubated for 60 min in a 371°C water bath. The incubation was terminated by the addition of 5 ml of ice-cold buffer (10 mM Tris-HCl, pH 7.4) and rapid filtration through Whatman GF/B glass fiber filter paper (presoaked in 0.5% polyethylenimine) using a cell harvester (Brandel Inc.). The filters were washed twice with 5 ml of cold buffer and transferred to scintillation vials to which absolute ethanol and Beckman Ready Safe was added. Quinuclidinyl benzilate, 100 μM final concentration, was used to determine nonspecific binding. Data were analyzed by using GraphPad Prism software (GraphPad Software Inc.).
Receptor Screen. Three of the compounds, AHN 2-005, GA-I-103, and JHW 007, were screened for their activity at various receptor sites by examining their competition with the appropriate radioligands (ProfilingScreen procured from MDS Panlabs Pharmacology Services, Bothell, WA). The screen consisted of assays designed to assess the activity of the compounds at 31 mammalian receptors. Each compound was tested in duplicate in each assay at a concentration of 10 μM. If at this concentration there was greater than 50% displacement of specific binding of ligand, the test was repeated in duplicate at the original concentration and at 10-, 100-, and 1000- fold lower concentrations to obtain an approximation of affinity for the site. Concurrent vehicle and reference standards were conducted with each assay, and the sites listed in Table 1 were targeted. Significant details of the procedures are also provided in the table. For other details, see the MDS Panlabs catalog (MDS Panlabs Pharmacology Services, 2000).
Assay conditions for activity at various receptor sites by examining competition with the appropriate radioligands (ProfilingScreen; MDS Panlabs Pharmacology Services)
For sites at which activity was identified, IC50 values and inhibition constants (Ki) were computed from the displacement data using a nonlinear, least-squares regression analysis (GraphPad Prism; GraphPad Softwae Inc.). The Ki values were calculated using the equation of Cheng and Prusoff (1973) using the obtained IC50 value of the tested compound, the concentration of radioligand used in the assay, and the MDS Panlabs historical value for the Kd of the ligand. Because IC50 were determined from four concentrations of cold compound, the derived binding constants should be interpreted as estimates.
Locomotor Activity. Mice were placed one at a time in clear acrylic chambers (40 cm3) for the assessment locomotor activity on a horizontal plane. The acrylic chambers fit within monitors (Omnitech Electronics, Columbus, OH), which were equipped with light-sensitive detectors, spaced 2.5 cm apart along two perpendicular walls. Mounted on the opposing walls and directed at the detectors were infrared light sources. One activity count was registered each time the subject interrupted a single light beam. Mice were injected (i.p. in volumes of 1 ml/100 g) and immediately placed in the apparatus for 1 h, with activity counts totaled each 10 min. Each drug dose was studied in eight mice, and mice were used only once. In experiments on the time course of effects, mice were injected, immediately placed in the apparatus, and data were collected for 8 h. All other aspects of these experiments were identical to those in which activity was assessed for 60 min.
Cocaine Discrimination. Rats weighing 320 to 350 g served as subjects. They were fed daily about 15 g of standard lab chow at least 30 min after testing that maintained them at their individual weights throughout the study. Subjects were tested daily in two-lever operant-conditioning chambers (model ENV 007; Med Associates, St. Albans, VT) that were housed within light- and sound-attenuating enclosures. White noise was present throughout testing to mask extraneous sounds. Ambient illumination was by a lamp in the top center of the front panel (house light). Levers were set 17 cm apart, with pairs of lamps (light-emitting diodes; LEDs) above each of the levers, also on the front panel. A downward force on either lever of 0.4 N through about 1 mm was defined as a response and produced an audible click. Reinforced responses dispensed one 45-mg pellet (BioServe, Frenchtown, NJ) into a food tray centered between the levers on the front panel of the chamber. On-line experimental control and data collection were by PC MS-DOS computers with Med Associates' interfacing equipment and operating software (Med Associates).
Subjects were initially trained to press both levers under a 20-response fixed ratio (FR 20) schedule of food reinforcement and to discriminate i.p. injections of 29 μmol/kg cocaine (10 mg/kg) from i.p. injections of saline. After cocaine injection, responses on only one lever were reinforced; after saline injection, responses on the other lever were reinforced. The assignment of cocaine- and saline-appropriate levers was counterbalanced across rats. Immediately after injection, rats were placed inside the experimental chambers. A 5-min time-out period, during which the house light and LEDs were extinguished and responding had no scheduled consequences preceded the illumination of the house light and the LEDs. Only responses on the appropriate lever were reinforced, and responses on the inappropriate lever reset the FR response requirement. Each food presentation was followed by a 20-s time-out period during which all lights were off, and responding had no scheduled consequences. Sessions ended after 20 food presentations or 15 min, whichever occurred first. Training sessions with cocaine (C) and saline (S) injections were conducted daily 5 days per week and ordered in a double alternation sequence (e.g., SCCS).
Testing was initiated when performances reached criteria of at least 85% appropriate responding overall and during the first FR 20 of the session over four consecutive sessions. Tests were conducted with different doses of cocaine, doses of the novel compounds, or combinations of doses administered before sessions. Selected doses of the test compounds were administered at different times up to 120 min after injection to examine the time course of the discriminative-stimulus effects. After a test session, a subject was required to meet the above-mentioned performance criteria over two consecutive (cocaine and saline) training sessions to be tested again. Repeated test sessions were conducted, with at least two training sessions between tests, until entire dose-effects were determined in each subject. Test sessions were identical to training sessions, with the exception that 20 consecutive responses on either lever were reinforced.
For each of the rats studied in the cocaine-discrimination procedure, the overall response rate and the percentage of responses occurring on the cocaine-appropriate lever were calculated. The mean values were calculated for each measure at each drug dose tested. If less than one-half of the rats responded at a particular dose, no mean value was calculated for percentage of cocaine-appropriate responding at that dose. At least 20% cocaine-appropriate responding was adopted as a conservative criterion at which to assume a significant difference from saline; 80% or higher cocaine-appropriate responding was taken as similar to the training dose of cocaine, and intermediate levels of cocaine-appropriate responding were considered partial substitution.
Data Analyses. Each dose-effect curve was analyzed using standard analysis of variance (ANOVA) and linear regression techniques. Locomotor activity in mice was assessed with counts collected during each successive 10-min epoch; counts during the first and last three epochs of the 1-h assessments were cumulated for separate analyses of the first and last 30 min. Effects of individual doses were determined significant by ANOVA and subsequent planned comparisons (Stevens, 1990). Results of cocaine discrimination studies were assessed with data collected during the entire session, which lasted a maximum of 15 min. In general, the effects obtained in the first and second 30-min periods were comparable, although there were some quantitative differences. Therefore, only the analyses of data from the 30-min period in which maximal stimulation was obtained are described. When locomotor stimulant effects were obtained, maximal effects were in the first 30 min after injection, and those data are shown in the figure.
Half-maximum stimulation of locomotor activity was calculated by adding the number of horizontal locomotor activity counts at the dose that produced the largest increase in activity to the number of counts after vehicle injection, and the sum was divided by two. The dose that produced this half-maximal stimulation (ED50 value) was determined by linear regression. For cocaine discrimination, the dose producing a half-maximal effect (50% cocaine-appropriate responding) was calculated. For these analyses, points on the linear part of the ascending portions of the dose-effect curves were used (Snedecor and Cochran, 1967). Pairs of ED50 values were considered to be significantly different if their 95% confidence limits did not overlap. To assess the degree of change in the cocaine dose-effect curve produced by coadministration of the BZT analogs, data were also analyzed by standard parallel-line bioassay techniques as described by Finney (1964). This analysis consists of a one-way ANOVA that determines whether the slopes of the two dose-response curves are significantly different from parallel and fits a common slope to the two dose-response curves. It then compares the ratio of doses for a 50% effect to provide a value for relative potency as a measure of the degree of shift in the cocaine dose-effect curve. The relative potency value represents the dose of cocaine, in subjects coadministered one of the BZT analogs, equal to 1 μmol/kg cocaine alone (i.e., a relative potency value of 0.5 indicates a 2-fold shift to the left of the cocaine dose-effect curve in the presence of the BZT analog). A significant shift in the cocaine dose-effect curve is indicated when the 95% confidence limits for the relative potency ratio do not include the value 1.0. Differences in the effectiveness of selected pairs of drugs were assessed by comparing maximal effects with a Student's t test.
Drugs. The drugs studied were (-)-cocaine HCl (Sigma-Aldrich) and N-substituted analogs of 3α-diphenylmethoxytropane analogs. The synthesis of these analogs was conducted in the Medicinal Chemistry Section of the national Institute on Drug Abuse Intramural Research Program and has been described previously (Agoston et al., 1997). Substitutions examined in the present study were exclusively on the nitrogen and are shown in Fig. 1. All drugs were dissolved in 0.9% NaCl or water, with heat and sonication, as necessary. The drugs were administered IP on the basis of body weight at 1 ml/kg (rats) or 10 ml/kg (mice).
Results
Radioligand Binding Assays. The Ki values determined for the BZT analogs at norepinephrine and serotonin transporters ranged from 457 to 4810 and 376 to 3260 nM, respectively (Table 2). These values were considerably higher than those determined previously (Agoston et al., 1997) for displacement of [3H]WIN 35,428 from the dopamine transporter, which ranged from 8.5 to 108 nM (Table 2). Consistent with the Agoston et al. (1997) results, N-substitutions for CH3 decreased binding affinity at M1 muscarinic receptors: the 12 nM affinity of the parent compound AHN 1-055 was reduced to values ranging from 177 to 1030 nM (Table 1). In general, the N-substituted compounds retained selectivity for the dopamine transporter over the other monoamine transporters and had increased selectivity for the dopamine transporter over muscarinic sites.
Potencies of N-substituted BZT analogs in binding to the dopamine, norepinephrine, and serotonin transporters and M1 muscarinic receptors Numbers are Ki values (S.E.M.).
The sites at which 10 μM AHN 2-005, GA-I-103, or JHW 007 displaced ligand to less than 50% specific binding are shown in Table 3, along with approximate Ki values. Among the 31 sites evaluated, all three compounds had activity in the less than 10 nM range at central histamine H1 receptors (Ki values ranged from 2.77 to 5.97 nM). Two of the compounds also had low nanomolar affinity at σ binding sites, with affinity at this site reduced for AHN 2-005 compared with the other compounds. Affinity in the range of 0.01 to 0.1 μM was obtained with two of the three compounds at α1 adrenergic and dopamine D2L receptors. In general, activity at the remaining sites was within the 0.1 to 10 μM range. Occasionally, lower values were obtained for one of the three compounds (e.g., 5-HT2 and M2 sites; Table 3).
Estimated Ki values (nanomolar) of selected N-substituted BZT analogs in binding to sites examined in the receptor screen
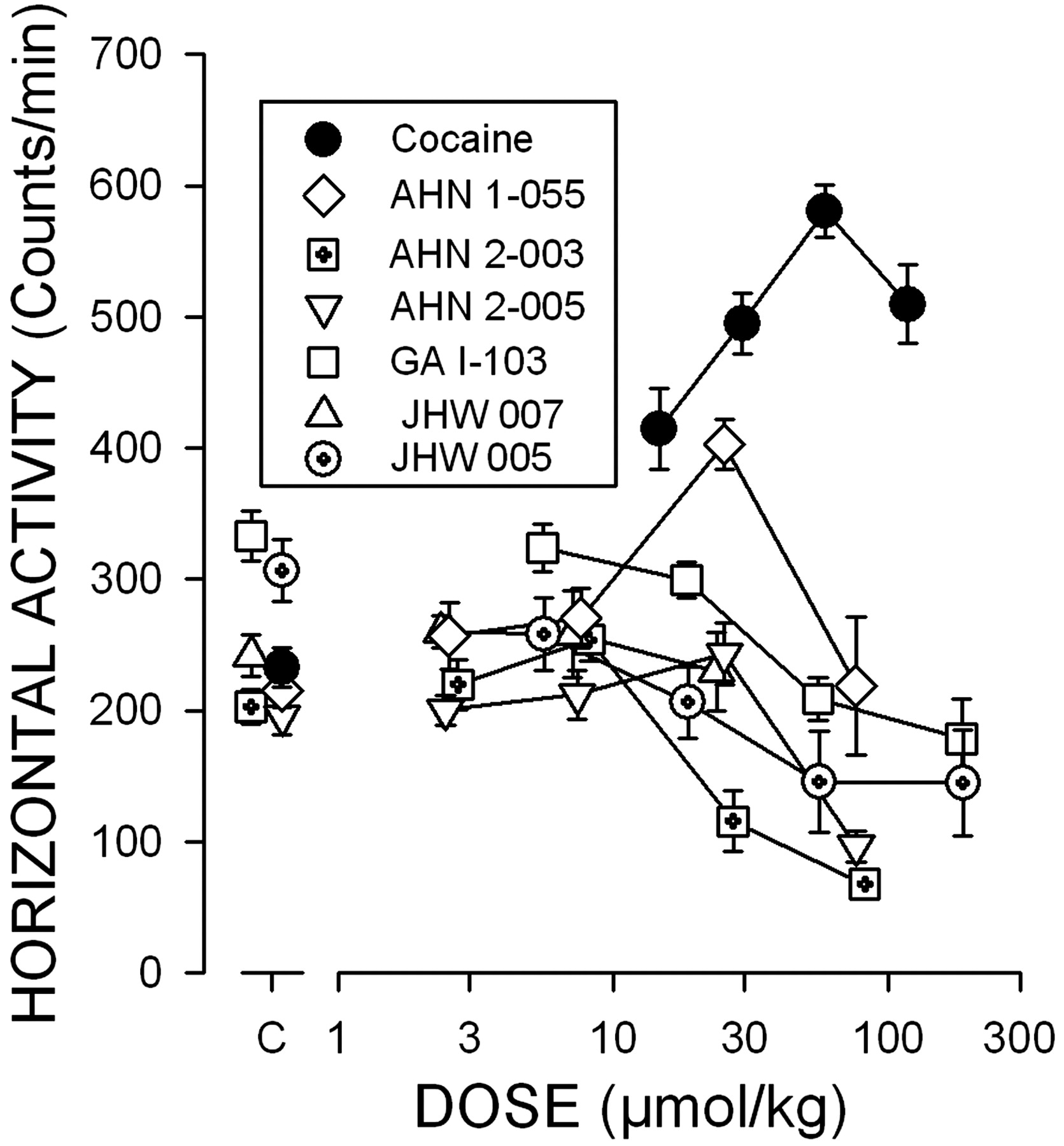
Locomotor Activity. Cocaine, as has been demonstrated previously, increased ambulatory activity with a maximum of 580 counts/min during the first 30 min of the session at 59 μmol/kg (Fig. 2, filled circles; Table 4), which seemed to be the maximum stimulation as a higher dose had a reduced effect (Fig. 2). In addition, AHN 1-055 at a dose of 25.4 μmol/kg also produced a significant stimulation of activity (Fig. 2; diamonds), although the maximal stimulation of 403 counts/min was significantly less (t22 = 5.676; p < 0.0001) than that produced by cocaine (Table 4). In contrast, none of the other 4′,4′′-diF-BZT analogs produced significant stimulation of activity that was greater than that observed after vehicle injection (Fig. 2). Each of the drugs was examined at doses ranging from those having no effects on activity to those that decreased locomotor activity. Each of the compounds was active across a comparable range of doses, with the exceptions of GA-I-103 and JHW 005, which minimally decreased activity and did so to a degree that was not statistically significant.

Dose-dependent effects of N-substituted BZT analogs on locomotor activity in mice. Ordinates, horizontal locomotor activity counts after drug administration in counts per second. Abscissae, dose of drug in micromoles per kilogram, log scale. Each point represents the average effect determined in eight mice. The data are from the 30-min period immediately after drug administration. Vehicle values averaged 247 ± 16.2 counts/min. Note that none of the BZT analogs produced a stimulation of activity that was equivalent to that of cocaine and that among these only the AHN 1-055 significantly stimulated activity.
Comparisons of potencies and maximal stimulant effects on locomotor activity of N-substituted BZT analogs
Cocaine Discrimination. As has been shown previously, there was a dose-related increase in the percentage of cocaine-appropriate responses in subjects trained to discriminate cocaine (29 μmol/kg) from saline (Fig. 3, A–F, filled symbols). In addition, the phenyltropane analog of cocaine, WIN 35,428, also fully substituted for cocaine (Fig. 3A, open circles). Several of the other N-substituted BZT analogs (AHN 1-055, AHN 2-003, AHN 2-005, and JHW 005) also produced a level of substitution greater than that produced by saline, although none of these compounds fully substituted for cocaine (Fig. 3A, triangles; and B–D, respectively). In contrast, JHW 007, GA-I-103, and JHW 025 did not produce substitution significantly greater than saline levels (Fig. 3, E and F). The maximal substitution produced by any of the BZT analogs, other than AHN 1-055, was 54% at 15.3 μmol/kg AHN 2-003 (Table 5). Each of the compounds that did not fully substitute for cocaine was examined from doses having no effects to those producing pronounced decreases in response rates (Fig. 3, G–L). An exception to this was JHW 025, a quaternary derivative, which was studied at doses as high as possible with restrictions imposed by its solubility.

Effects of N-substituted BZT analogs in rats trained to discriminate injections of cocaine from saline. Top, ordinates, percentage of responses on the cocaine-appropriate key. Bottom, ordinates, rates at which responses were emitted (as a percentage of response rate after saline administration). Abscissae, drug dose in micromoles per kilogram (log scale). Each point represents the effect in 4 to 16 rats. Filled circles in each panel of the figure represent the effects of cocaine, replotted for reference. The percentage of responses emitted on the cocaine-appropriate key was considered unreliable, and not plotted, if fewer than one-half of the subjects responded at that dose. Note that only the cocaine analog, WIN 35,428, fully substituted for cocaine, and among the N-substituted BZT analogs only AHN 1-055 approached the effects of cocaine.
Comparisons of potencies and maximal effects of N-substituted BZT analogs in substituting for cocaine and affecting rates of responding
The lack of full substitution for cocaine produced by the BZT analogs does not depend on the measure of substitution used. Other measures that have been used previously to assess the substitution of the test drug for the training drug were also examined. These alternate measures were 1) the percentage of responses on the cocaine-appropriate lever before the first reinforcer (Table 6, column B); 2) the percentage of subjects selecting the cocaine-appropriate lever (Table 6, column C); and 3) the percentage of subjects selecting the cocaine-appropriate lever on the first trial of the session (Table 6, column D). There was a very close correspondence among all of these alternative measures of maximal effect with the percentage of responses on the cocaine-appropriate lever tabulated over the entire session (Table 6, column A). The R2 values for the correlations among all four measures of maximal effect were all above 0.980. All four of these measures indicated that, other than the N-methyl substituted analog, none of the compounds had maximal effects comparable with those of cocaine or its analog WIN 35,428.
Efficacy of N-substituted BZT analogs in substituting for cocaine in rats: comparisons of different methods of assessing efficacy Correlations between columns: B with A: R2 = 0.999 (F1,8 = 5267.2; p < 0.001); C with A: R2 = 0.989 (F1,8 = 659.6; p < 0.001); D with A: R2 = 0.986 (F1,8 = 479.2; p < 0.001); C with B: R2 = 0.987 (F1,8 = 547.1; p < 0.001); D with B: R2 = 0.987 (F1,8 = 519.3; p < 0.001); and D with C: R2 = 0.992 (F1,8 = 862.0; p < 0.001).
Increasing the time between injection and testing did not significantly change the efficacy of AHN 2-003 and AHN 2-005, the two drugs for which the time courses of effects were assessed (Fig. 4). With AHN 2-003, there was a trend toward an increase in the efficacy in producing cocaine-like discriminative stimulus effects; however, that effect was not significant (F3,43 = 2.642; p = 0.061). Additionally, over the same time there was a trend toward a decrease in the efficacy of AHN 2-003 in reducing response rates (F3,48 = 2.640; p = 0.060; Fig. 4). Similarly, with AHN 2-005 there were no significant changes in efficacy in producing cocaine-like discriminative stimulus effects (F4,113 = 0.417; p = 0.796) over the course of 120 min. As with AHN 2-003, there was a trend toward a reduction in the effects of the drug on response rates (F4,116 = 2.431; p = 0.051; Fig. 4).

Effects of N-substituted BZT analogs in rats trained to discriminate injections of cocaine from saline at various times after injection. Top, ordinates, percentage of responses on the cocaine-appropriate key. Bottom, ordinates, rates at which responses were emitted (as a percentage of response rate after saline administration). Abscissae, drug dose in micromoles per kilogram (log scale). Each point represents the effect in four to six rats. Filled circles in each panel of the figure represent the effects of cocaine, redetermined during the course of assessing the effects of pretreatment time. Note that with pretreatments of up to 2 h the effects of the drugs in substituting for cocaine were not significantly increased, and the effects on response rates tended to be diminished though this effect was not statistically significant.
Drug Interactions. In general, treatment with the BZT analogs did not produce significant changes in the effects of cocaine, whereas WIN 35,428 produced a significant leftward shift in the cocaine dose-effect curve (Fig. 5). The change in the effects of cocaine produced by WIN 35,428 was significant; the relative potency estimate (Table 7) of 574 had 95% CL values exclusive of 1.0 at a WIN 35,428 dose of 1.0 mg/kg. In contrast to the effects obtained with WIN 35,428, among the N-substituted analogs of BZT only AHN 1-055 (at 14.2 μmol/kg) and JHW 005 (at 18.7 μmol/kg) produced significant changes in the discriminative-stimulus dose effects of cocaine (Table 7; Fig. 5).

Changes in the cocaine dose-effect curve for discriminative stimulus effects produced by pretreatments with WIN 35,428 or the N-substituted BZT analogs. Ordinates, percentage of responses on the cocaine-appropriate key. Abscissae, cocaine dose in micromoles per kilogram (log scale). Each point represents the effect in four to six rats. The percentage of responses emitted on the cocaine-appropriate key was considered unreliable, and not plotted, if fewer than one-half of the subjects responded at that dose. Note that WIN 35,428 shifted the cocaine dose-effect curve to the left, whereas the BZT analogs were either inactive or only marginally shifted the cocaine dose-effect curve to the left.
Alterations in the discriminative-stimulus potency of cocaine produced by treatment with N-substituted BZT analogs ED50 values and potencies relative to cocaine alone (with 95% CL) are shown.
Table 7 shows the ED50 values for cocaine, either alone or in combination with the BZT analogs. Relative potency estimates with 95% CLs are also shown. The cocaine ED50 value was not significantly changed by the coadministration of any of the BZT analogs (note that 95% CLs overlapped). In addition, the relative potency estimates generally showed no significant change induced by administering a BZT analog with cocaine (95% CLs include the value 1.0). The exceptions were AHN 1-055 and JHW 005, which at the highest doses studied, produced an approximate 2-fold leftward shift in the cocaine dose-effect curve for the discriminative-stimulus effects of cocaine. As indicated in Fig. 5, AHN 2-005 produced a trend toward a shift to the left in the cocaine dose effect curve; however, this trend did not approach statistical significance as indicated by the relative potency value (Table 7). For JHW 007 and AHN 2-005, the highest dose studied in combination with cocaine produced an increased substitution when these drugs were examined in combination with the lowest dose (2.9 μmol/kg) of cocaine. This change resulted in a decrease in the slopes of the dose-effect curves, and a lack of significance of the linear regression (Table 7).
To ensure a comparability of doses of BZT analogs studied in combination with cocaine, the doses that were studied were selected based on their potency for decreasing response rates (Fig. 3, G–L). Each compound was examined in combination with cocaine at doses that bracketed either their ED50 values, or the minimally effective dose, for decreasing response rates. The pharmacological equivalence of the doses examined in combination with cocaine can be seen in Fig. 5, bottom. Each of the drugs produced a decrease in the slope of the dose effects of cocaine on response rates, as well as a generalized decrease in the rates of responding across the range of cocaine doses.
Discussion
In the present study, the pharmacology of a selected series of 4′,4′′-diF, N-substituted analogs of BZT was evaluated. Each of these compounds had relatively high affinity for the dopamine transporter (Agoston et al., 1997). In addition to this action, the compounds were relatively selective among the monoamine transporters, having affinities at the norepinephrine and serotonin transporters that were relatively low compared with those for the dopamine transporter. The present results are consistent with previous studies that have demonstrated that N-methyl analogs of BZT can have high affinity and selectivity for the dopamine transporter compared with the norepinephrine and serotonin transporters (Newman et al., 1995). Among the previously studied N-methyl-BZT analogs was the parent compound of the present series, AHN 1-055, which was previously reported to have relatively high affinity for M1 muscarinic receptors (Newman et al., 1995; Katz et al., 1999). In contrast, the present N-substituted BZT analogs have reduced affinity for M1 receptors. For example, AHN 2-005 had approximately 100-fold lower affinity for M1 muscarinic receptors and a 4-fold higher affinity for the dopamine transporter than does the parent compound BZT. Comparably increased selectivities were obtained with the other N-substituted analogs.
Activity at the dopamine transporter is generally thought to be sufficient for cocaine-like behavioral effects, and consequently actions at the dopamine transporter should result in compounds having cocaine-like behavioral effects. For example, phenyltropane analogs of cocaine generally have high affinity for the dopamine transporter, inhibit dopamine uptake in vitro, and produce cocaine-like behavioral effects, often with significantly greater potency than cocaine (Spealman et al., 1977, 1979; Carroll et al., 1995; Fleckenstein et al., 1996). Moreover, cocaine-like activity typically is produced by cocaine analogs with relatively high selectivity for the dopamine transporter (Kimmel et al., 2001). Thus, the selectivity of the BZT analogs for the dopamine transporter compared with the other monoamine transporters, would not by itself be expected to produce behavioral effects different from those of cocaine. The original observation that N-methyl analogs of BZT were generally not as effective as other dopamine uptake inhibitors in producing cocaine-like behavioral effects was surprising (Newman et al., 1995) and has led to investigations into the mechanisms for the differences (Katz et al., 1999).
One explanation for the differences in behavioral effects between BZT analogs and cocaine-like stimulants is that other actions of the BZT analogs may interfere with the expression of cocaine-like effects. The high affinity for muscarinic M1 receptors of the N-methyl analogs (Newman et al., 1995; Katz et al., 1999) was the focus of previous studies. In general, anticholinergics, such as atropine and scopolamine, have generally been reported to potentiate the behavioral effects of CNS stimulants (Carlton and Didamo, 1961; Scheckel and Boff, 1964). This type of interaction was also found for cocaine and either atropine or scopolamine using behavioral procedures identical to those used in the present study (Katz et al., 1999). Those results suggested it unlikely that antimuscarinic effects of BZT analogs could interfere with their effectiveness in producing the presently examined cocaine-like effects.
In the present study, N-substituted BZT analogs were examined for which modifications in structure resulted in a relatively low affinity for muscarinic M1 receptors compared with BZT. These compounds retained their relatively high affinity and selectivity for the dopamine transporter and were used to further examine the potential of M1 activity to influence the behavioral effects of BZT analogs. Despite a relatively decreased muscarinic affinity, the N-substituted BZT analogs did not produce cocaine-like locomotor stimulant or discriminative-stimulus effects. These results are consistent with those reported previously (Katz et al., 1999, 2001) for the N-methyl analogs of BZT, which had relatively high affinity for M1 muscarinic receptors. AHN 1-055, which has 12 nM affinity for M1 receptors, as in the previous study (Katz et al., 1999), produced some stimulation of locomotor activity and substituted partially (79%) for cocaine, effects that approached those of cocaine.
In a previous study AHN 1-055 fully substituted for cocaine when administered 90 min before testing its cocaine-like discriminative-stimulus effects. Along those lines, recent pharmacokinetic studies (Raje et al., 2003) have indicated that the elimination of several BZT analogs (including AHN 1-055, AHN 2-003, AHN 2-005, and JHW 007) from plasma and brain is substantially slower than that for cocaine. Thus, it is possible that providing greater amounts of time for uptake and distribution may have rendered the present compounds more effective in producing cocaine-like discriminative stimulus effects. In the present study, AHN 2-003 and AHN 2-005 were examined at time points up to 2 h after injection without a significant increase in their cocaine-like discriminative-stimulus effects. In addition, we generally examined a range of doses of the compounds, from those having no effects to those virtually eliminating response rates. The decreases in response rates suggest that doses examined were more than adequate for a centrally mediated effect. The study by Raje et al. (2003) demonstrated that AHN 1-055, AHN 2-003, AHN 2-005, and JHW 007 readily and rapidly crossed the blood-brain barrier, having brain to plasma ratios either equal to, or greater than 2-fold higher than that of cocaine. Together, the present studies on time course and the pharmacokinetic studies indicate that, whereas these compounds have a long duration of action, their uptake and distribution in brain is sufficient to rule out a limited uptake and distribution of the drugs as an explanation for their lack of substitution for cocaine.
It is currently not clear what differences in the pharmacology of AHN 1-055 and the N-substituted compounds account for their differences in behavioral effects. However, one possibility that is consistent with the literature (Carlton and Didamo, 1961; Scheckel and Boff, 1964) is that the antimuscarinic effects, rather than interfering with, contribute to the cocaine-like pharmacology of the BZT analogs with effects most closely resembling those of cocaine (also see Katz et al., 2001). Consistent with this hypothesis, the N-substituted BZT analogs that had reduced M1 receptor affinity were uniformly less active as stimulant-like drugs than cocaine, its phenyltropane analog WIN 35,428, and AHN 1-055. However, BZT itself has high affinity for M1 receptors, yet was relatively inactive as a cocaine-like agent (Katz et al., 1999). In contrast to the other compounds, BZT has relatively low affinity for the dopamine transporter. It seems reasonable to suggest that some minimal activity at the dopamine transporter is necessary for the potentiation of cocaine-like effects by antimuscarinic activity.
A recent study of cocaine self-administration in rhesus monkeys was designed to address the role of antimuscarinic actions in the reinforcing effects of BZT analogs. In that study, the addition of the anticholinergic, scopolamine, to the cocaine solution being self-administered decreased the effectiveness of cocaine and did not shift the cocaine dose-effect curve leftward (Ranaldi and Woolverton, 2002), differentiating reinforcing effects from other effects of cocaine. The lack of a potentiation of the reinforcing effects of cocaine by scopolamine suggests further that the anticholinergic component of action will not potentiate whatever reinforcing effects are exhibited by BZT analogs. As such, that finding is consistent with previous findings indicating a lesser effectiveness of BZT analogs in self-administration compared with cocaine (Woolverton et al., 2000, 2001). Ranaldi and Woolverton (2002) further speculated that the decreased effectiveness of scopolamine-cocaine combinations compared with cocaine alone, could be due to an antagonism of the reinforcing effects of cocaine by scopolamine or an added punishment of cocaine-maintained behavior by the response-dependent administration of scopolamine. In a previous study (Wilson and Schuster, 1973) the effects of the anticholinergic, atropine, were examined in rhesus monkeys self-administering cocaine. Atropine was administered before experimental sessions independently of responding, and therefore could not function as a punishing stimulus. In that study, the effects of atropine were similar to the effects of lowering the injected dose of cocaine. Thus, the most parsimonious interpretation of the results together suggests a physiological antagonism of the reinforcing effects of cocaine by the anticholinergic agents. However, as Ranaldi and Woolverton note, a more thorough study of alternative hypotheses is in order. Certainly, studies of self-administration of the present N-substituted BZT analogs that have reduced affinity for muscarinic receptors will add important information in assessing the role of antimuscarinic actions in the reinforcing effects of BZT analogs. Such studies are ongoing.
Activity at sites other than the monoamine transporter and muscarinic M1 receptors was investigated for several of the compounds. Among the sites evaluated, activity in the less than 10 nM range was demonstrated for central histamine H1 receptors. This activity was not surprising because the parent compound BZT has antihistaminic activity (Richelson, 1981). Nonetheless, it is possible that actions at histamine H1 receptors contributed to the present effects, and the influence of activity at histamine H1,H2, and H3 receptors on the effects of cocaine has been examined (Campbell et al., 2002). In general, antagonists at each of these receptors were inactive in significantly modifying the dose-effect curves for the discriminative-stimulus effects of cocaine. In addition, a large number of BZT analogs were assessed for their affinities at each of these histamine receptors and compared with their affinities for the dopamine transporter. Those compounds with higher affinity for the dopamine transporter than histamine receptors were no more likely to produce increases in locomotor activity than were compounds for which the affinity at a histamine receptor was greater than its affinity at the dopamine transporter. From these results together, it seems unlikely that antihistaminic effects of BZT analogs could interfere with their effectiveness in producing the presently examined cocaine-like effects.
Two of the compounds (JHW 007 and GA-I-103) also had low nanomolar affinity at sigma binding sites, which was greater than their affinity for the dopamine transporter. The other compound, AHN 2-005, had lower σ receptor affinity than dopamine transporter affinity. Actions at σ receptors have been noted previously to influence the behavioral effects of cocaine. For example, Menkel et al. (1991) found that relatively low doses of the σ antagonists rimcazole and BMY 14802 could block the locomotor stimulant effects of cocaine. In addition, other sigma antagonists have been demonstrated to alter locomotor stimulant and acute toxic effects of cocaine (Matsumoto et al., 2001). Moreover, subjects treated with sigma1 receptor antisense oligodeoxynucleotides show diminished effects of cocaine (Romieu et al., 2000). Using the present procedures, several analogs of the σ antagonist rimcazole were found to diminish the behavioral effects of cocaine, although the effects were most pronounced in only one of the compounds (Katz et al., 2003). Together, these results indicate that the possibility that the reduced cocaine-like effectiveness of the present N-substituted BZT analogs is due to their actions at sigma receptors cannot be discounted and deserves further study.
Because of the reduced effectiveness among the present BZT analogs compared with cocaine, it was of interest to examine the interactions of these compounds with cocaine. Previous studies of interactions have found that BZT analogs shift the cocaine dose-effect curve for discriminative-stimulus effects to the left (Tolliver et al., 1999; Katz et al., 2001). That leftward shift was not expected for compounds that bind to the same site as cocaine but have reduced effectiveness. In the present study, WIN 35,428 produced a pronounced left-ward shift in the cocaine dose-effect curve similar to those produced by other dopamine transport inhibitors (Holtzman, 2001; Katz et al., 2003). In contrast, the BZT analogs examined in this study failed to produce large or significant shifts to the left in the cocaine dose-effect curve. To ensure an assessment of doses that would be behaviorally active, the doses of BZT analogs that were studied in combination with cocaine were selected for the pharmacological equivalence of their effects on response rates. Thus, the present N-substituted analogs of BZT seemed to be qualitatively different from other dopamine uptake inhibitors as indicated by their lack of substitution for cocaine and a lack of potentiation of the behavioral effects of cocaine.
The lack of a significant shift leftward in the cocaine dose-effect curve in the present study with N-substituted BZT analogs distinguishes these compounds from other BZT analogs. As mentioned above, previous studies (Tolliver et al., 1999; Katz et al., 2001) have shown that BZT analogs shift the cocaine dose-effect curve leftward. It is possible that the antimuscarinic effects of the previously studied compounds contributed significantly to the leftward shift in the cocaine dose-effect curve, as was found with atropine and scopolamine (Katz et al., 1999). The general absence of a significant alteration in the effects of cocaine by the present N-substituted BZT analogs is being further investigated.
By analogy to treatments for heroin dependence, such as methadone and buprenorphine, the present results suggest that analogs of BZT may be useful as treatments for cocaine abuse, in situations in which an agonist treatment is indicated. Moreover, these compounds may possess a preclinical profile indicative of therapeutic advantage over other dopamine uptake inhibitors in that they have significantly reduced effectiveness as stimulants compared with other ligands for the dopamine transporter. In addition, as mentioned above, previous studies have indicated that BZT analogs are less effective in maintaining self-administration than are either cocaine or another dopamine uptake inhibitor GBR 12909 (Woolverton et al., 2000, 2001). These findings together suggest that BZT analogs may have potential as leads in the development of medications to treat cocaine abuse and that these drugs may have reduced liability for their abuse. The reduced affinity for muscarinic M1 receptors, as well as the sites examined in the broad screen of activity that was obtained with the present N-substituted analogs of BZT, suggests further that these particular compounds may be efficacious with a reduced liability for side effects.
Acknowledgments
We thank Bettye Campbell, Jianjing Cao, and Dawn French for technical support; Patty Ballerstadt for administrative and clerical support; and Dawn French for expert data analysis.
Footnotes
-
Portions of this research were presented at the 1999 Annual Meeting of the College on Problems of Drug Dependence.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
DOI: 10.1124/jpet.103.060525.
-
ABBREVIATIONS: BZT, benztropine; 4′-Cl-BZT, 4′-chloro-3α-(diphenylmethoxy)tropane; LED, light-emitting diode; FR, fixed ratio; C, cocaine; S, saline; ANOVA, analysis of variance; 5-HT, 5-hydroxytryptamine; CL, confidence limit; AHN 1-055, 3α-[bis-(4-fluorophenyl)methoxy]tropane; AHN 2-005, N-allyl-3α-[bis-(4-fluorophenyl)methoxy]tropane; GA-I-103, N-(phenyl-n-butyl)-3α-[bis-(4-fluorophenyl)methoxy]tropane; JHW 005, N-benzyl-3α-[bis-(4-fluorophenyl)methoxy]tropane; JHW 007, N-(n-butyl)-3α-[bis-(4-fluorophenyl)methoxy]tropane; GBR 12909, 1-{2-[bis-(4-fluorophenyl)methoxy]ethyl}-4-(3-phenylpropyl)piperazine; WIN 35,428, 2β-carbomethoxy-3β-(4-fluorophenyl)tropane; BMY 14802, α-(4-fluorophenyl)-4-(5-fluoro-2-pyrimidinyl)-1-piperazine.
-
↵1 Current address: EntreMed, Inc., 9640 Medical Center Dr., Rockville, MD 20850.
- Received September 25, 2003.
- Accepted December 3, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}