


Abstract
The rostral ventromedial medulla (RVM) is a major locus for the descending control of nociception and opioid analgesia. However, it is not clear how opioids affect synaptic inputs to RVM neurons. In this study, we determined the effect of μ-opioid receptor activation on excitatory and inhibitory synaptic transmission in spinally projecting RVM neurons. RVM neurons were retrogradely labeled with a fluorescent tracer injected into the dorsal horn of the spinal cord in rats. Whole-cell voltage-clamp recordings were performed on labeled RVM neurons in brain slices in vitro. The μ-receptor agonist [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO, 1 μM) significantly decreased the amplitude of evoked excitatory postsynaptic currents (EPSCs) in 52% (9 of 17) of labeled cells. DAMGO also significantly reduced the amplitude of evoked inhibitory postsynaptic currents (IPSCs) in 69% (11 of 16) of cells examined. Furthermore, DAMGO significantly decreased the frequency of miniature EPSCs in 55% (15 of 27) of cells and significantly decreased the frequency of miniature IPSCs in all 12 cells studied. Although most EPSCs and IPSCs were mediated by glutamate and GABA, the nicotinic and glycine receptor antagonists attenuated EPSCs and IPSCs, respectively, in some labeled RVM neurons. Immunocytochemical labeling revealed that only 35% of recorded RVM neurons were tryptophan hydroxylase-positive, and 15% cells had GABA immunoreactivity. Thus, this study provides important functional evidence that activation of μ-opioid receptors decreases the release of both excitatory and inhibitory neurotransmitters onto most spinally projecting RVM neurons.
The rostral ventromedial medulla (RVM) plays an important role in the descending modulation of nociceptive information and opioid analgesia (Fields et al., 1983a; Jones and Gebhart, 1988; Urban and Smith, 1994). The RVM receives synaptic inputs from the periaqueductal gray (PAG) and relays information to the dorsal horn of the spinal cord through the dorsolateral funiculus. The analgesia produced by microinjection of morphine into the PAG is dependent upon the connection to the RVM (Urban and Smith, 1994; Pan and Fields, 1996). Direct stimulation of the RVM also can produce potent analgesia. For example, medullary electrical stimulation or microinjection of glutamate and morphine into the RVM results in either an increase in the tail-flick latency or a decrease in spinal dorsal horn neuronal activity (Llewelyn et al., 1986; Jones and Gebhart, 1988; McGowan and Hammond, 1993; Urban and Smith, 1994). However, the cellular mechanisms of the action of opioids in the RVM remain unclear.
Three classes of RVM neurons have been identified, including ON-, OFF-, and NEUTRAL-cells, that show, respectively, increases, decreases, and no changes in the firing activity in response to noxious stimuli (Fields et al., 1983b, 1995). Furthermore, activation of μ-opioid receptors inhibits ON-cells, but stimulates OFF-cells during the application of noxious stimulation (Fields et al., 1983b). Although activation of OFF-cells by μ-opioids is considered an important mechanism of opioid analgesia, the mechanisms and the phenotype of RVM neurons are not fully known. The RVM contains a number of nuclei, including the nucleus raphe magnus (NRM). The NRM is a major serotonergic nucleus that supplies the spinal cord with the majority of its 5-hydroxytryptamine (serotonin) (5-HT) (Oliveras et al., 1977). It has been hypothesized that μ-opioids stimulate RVM serotonergic neurons, leading to inhibition of spinal dorsal horn neurons through 5-HT release. Recent studies, however, have shown that 5-HT is only contained in a few spinally projecting RVM cells and is not necessarily involved in opioid analgesia (Kalyuzhny et al., 1996; Gao et al., 1998; Marinelli et al., 2002).
The disinhibitory mechanism has been proposed for the analgesic action of opioids in the PAG. Several lines of evidence suggest that both inhibitory and excitatory neurotransmitters may be involved in opioid-induced analgesia in the RVM. In this regard, GABA-positive nerve terminals appose both ON- and OFF-cells, whereas another population of RVM neurons is double-labeled for GABA and the μ-opioid receptor (Skinner et al., 1997; Kalyuzhny and Wessendorf, 1998). Glutamate may also mediate the opioid action in the RVM because microinjection of both NMDA and non-NMDA receptor antagonists in the RVM attenuates morphine-induced analgesia in the PAG (Spinella et al., 1996). Furthermore, NMDA receptor blockade prevents morphine-induced disinhibition of OFF-cells (Heinricher et al., 2001). The RVM is a heterogeneous region that contains interneurons and different types of output neurons. Although some of the ON- and OFF-cells have projections to the spinal cord (Fields et al., 1995), very few studies have focused specifically on RVM neurons that project to the spinal cord. The μ-opioid receptor agonist DAMGO produces an outward current in some spinally projecting RVM neurons (Marinelli et al., 2002). However, the effect of μ-opioids on glutamatergic or GABAergic synaptic inputs to spinally projecting RVM neurons has not been studied previously. Therefore, in the present study, we used in vitro whole-cell recordings combined with the retrograde labeling technique to determine the effect of μ-receptor stimulation on excitatory and inhibitory synaptic inputs to spinally projecting RVM neurons. Furthermore, immunohistochemistry labeling was used to determine the presence of GABA and 5-HT immununoreactivity of recorded RVM cells.
Materials and Methods
Retrograde Labeling of Spinally Projecting RVM Neurons. Sprague-Dawley rats (8–14 days of age; Harlan, Indianapolis, IN) of either sex were used in this study. The surgical preparations and experimental protocols were approved by the Animal Care and Use Committee of The Pennsylvania State University College of Medicine and conformed to the National Institutes of Health guidelines on the ethical use of animals. The spinal cord was exposed at the T1-T3 level through dorsal laminectomy under halothane anesthesia. A rhodamine-labeled fluorescent microsphere suspension (Fluo-Spheres, 0.04 μm; Molecular Probes, Eugene, OR) was pressure ejected (Picospritzer II; General Valve, Fairfield, NJ) bilaterally into the region of the dorsal horn of the spinal cord in three or four separate 50-nl injections using a glass micropipette (20–30-μm tip diameter) and monitored through an operating microscope. The muscles were sutured and the wound closed after injection. Animals were returned to their cage for 3 to 10 days, which is sufficient time to permit retrograde tracer being transported to the RVM. This retrograde labeling and spinal dorsal horn injection technique has been validated in our recent studies (Pan et al., 2002, 2004). To verify the injection site of FluoSpheres in the thoracic spinal cord, the spinal cord was taken out after sacrificing the animal and sectioned at the injection level. The spinal cord slices were then viewed under a microscope equipped with fluorescence illumination to locate the injection site of FluoSpheres in the spinal dorsal horn (Fig. 1). The animals were between 2 and 3 weeks of age at the time of the final electrophysiological experiments. It was technically impossible to use rats greater than 3 weeks of age because it becomes nearly impossible to visualize a labeled cell in the RVM after this time. For the same reason, whole-cell recordings in the RVM have always used brain slices from young animals (Pan et al., 1997; Marinelli et al., 2002).

Identification of a retrogradely labeled spinally projecting RVM neuron. A, FluoSphere-labeled RVM neuron viewed with fluorescent illumination. B, photomicrograph of the same neuron shown in A with an attached recording electrode (⁁) in the slice viewed with differential interference contrast optics. Scale bars, 25 μm (A and B). C, photomicrograph showing the FluoSphere injection site (red) at the spinal dorsal horn in one rat. Note that the bright field and fluorescence images were taken from the same tissue section and superimposed to show the location and diffusion of the FluoSphere injection. Scale bar, 500 μm.
Slice Preparation. Three to 12 days after fluorescent dye injection, the rats were rapidly decapitated under halothane anesthesia. The brain was quickly removed and placed in ice-cold, oxygenated (95% O2, 5% CO2) sucrose artificial cerebral spinal fluid (aCSF) for approximately 2 min. A tissue block containing the RVM was cut and glued onto the stage of the vibratome (Technical Product International, St. Louis, MO). Coronal slices containing the RVM (250–300 μm in thickness) were cut from the tissue block in ice-cold, oxygenated sucrose aCSF. The slices were preincubated in oxygenated nonsucrose aCSF at 36°C for at least 1 h before being transferred into the recording chamber. The sucrose aCSF was composed of the following: 234 mM sucrose, 3.6 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 1.2 mM NaH2PO4, 12.0 mM glucose, and 26.0 mM NaHCO3. The nonsucrose aCSF contained 126 mM NaCl, 2.5 mM KCl, 1.3 mM MgSO4, 2.4 mM CaCl2, 1.2 mM NaH2PO4, 11.0 mM glucose, and 25.0 mM NaHCO3 (pH 7.4; osmolarity 295–300 mOsM).
Recordings of Postsynaptic Currents of RVM Neurons. Recordings of miniature and evoked excitatory and inhibitory postsynaptic currents were performed using whole-cell voltage-clamp method as described previously in this laboratory (Pan et al., 2002, 2004). The electrode for the whole-cell recordings was pulled with a puller (P-97; Sutter Instrument, Novato, CA) using borosilicate glass capillaries (1.2 mm o.d.; 0.86 mm i.d.; WPI, Sarasota, FL). The resistance of the pipette for both miniature and evoked current protocols was about 5 MΩ when filled with an internal solution containing 110 mM CsSO4, 0.5 mM CaCl2, 2.4 mM MgCl, 5.0 mM BAPTA, 10.0 mM Hepes, 5.0 mM Na2ATP, 0.33 mM GTP-Tris salt, and 5.0 mM tetraethylammonium-Cl; adjusted to pH 7.2 and osmolarity 280–290 mOsM. The G protein inhibitor guanosine 5′-O-(2-thiodiphosphate) (1 mM) was added to the internal solution in the evoked protocol to prevent the potential postsynaptic effect of opioids (Pan et al., 2002, 2004). For recording of evoked responses, 10 mM QX314 was added into the internal recording solution to suppress the action potential generation from the recorded cell. Also, the internal pipette solution contained 0.2% biocytin (Sigma-Aldrich, St. Louis, MO) to label the recorded neuron for later use in the immunocytochemical labeling (Pan et al., 2002, 2004).
The brain slice was placed in a glass-bottomed chamber (Warner Instruments, Hamden, CT) and fixed with a grid or parallel nylon threads supported by a U-shaped stainless steel weight. The slice was perfused at 3.0 ml/min at 36°C maintained by an inline solution heater and a temperature controller (TC-324; Warner Instruments). RVM neurons were visualized in the triangular midline region dorsal to the pyramidal tract. Fluorescent labeled RVM cells were briefly identified in the slice with epifluorescence (rhodamine filter) on a fixed stage microscope (BX50WI; Olympus, Tokyo, Japan). The neurons were then viewed with Nomarski optics through a water immersion objective. The tissue image was captured and enhanced through a charge-coupled device camera and displayed on a video monitor (Fig. 1). After the labeled neuron was identified, the cell was recorded in the whole-cell configuration, as we described in detail previously (Pan et al., 2002, 2004). Recordings of postsynaptic currents began 5 to 10 min after whole-cell access was established and the current reached steady state. The neurons recorded met the following electrophysiological criteria that were considered as healthy neurons: the action potential amplitude greater than 60 mV [for those cells used for miniature inhibitory postsynaptic currents (mIPSCs)/miniature excitatory postsynaptic currents (mEPSCs) before tetrodotoxin (TTX) application], and the resting membrane potential more negative than–50 mV.
mIPSCs and mEPSCs were recorded at a holding potential of 0 and–70 mV, respectively (Pan et al., 2002). All mIPSCs were recorded in the presence of TTX (1 μM) and 6-cyano-7-nitroquinoxa-line-2,3-dione (CNQX, 20 μM), and mEPSCs were recorded in the presence of TTX and bicuculline (20 μM). The evoked postsynaptic currents in the labeled RVM neurons were induced by electric stimulation (0.1 ms, 0.1–0.5 mA, and 0.2 Hz) through a bipolar tungsten electrode connected to a stimulator (S48; Grass Instruments, W. Warwick, RI). The stimulating electrode was placed 200 to 800 μm away from the recorded neuron. Evoked inhibitory postsynaptic currents (eIPSCs) and evoked excitatory postsynaptic currents (eEP-SCs) were recorded at a holding potential of 0 and–70 mV, respectively (Pan et al., 2002). We did not further examine whether the evoked synaptic responses were monosynaptic. Currents were measured using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Signals were filtered at 1 to 2 kHz, digitized at 10 kHz (DigiData 1320A; Axon Instruments) and recorded into a Pentium computer using the pClamp 8.01 program.
Experimental Protocols. The resting membrane potential and the input resistance were continuously monitored throughout the recording period. Recordings were abandoned if the input resistance changed more than 15% (Pan et al., 2002, 2004). To determine the effect of DAMGO on the spontaneous mIPSCs/mEPSCs in labeled RVM neurons, 1 μM DAMGO was perfused into the slice for 2 min after recording the mIPSCs/mEPSC for 3 min as the baseline control. To ensure the specific effect of DAMGO, the effect of 1 μM DAMGO was examined in the presence of the specific μ-opioid antagonist, H-d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP, 1 μM) (Vaughan et al., 1997). To determine the effect of DAMGO on evoked IPSCs/EPSCs in labeled RVM neurons, eIPSCs/eEPSCs were measured before and during perfusion of 1 μM DAMGO as described above. Additionally, to determine the types of neurotransmitters mediating IPSCs/EPSCs, the effect of 20 μM bicuculline or 20 μM CNQX on miniature and evoked IPSCs or EPSCs was tested. Because the stimulating electrode was placed close to the recorded cell within the RVM, it was impossible to determine the exact input pathway to the recorded cell in this thin slice preparation.
CNQX and bicuculline were obtained from Sigma-Aldrich. DAMGO and CTAP were purchased from Bachem Biosciences (King of Prussia, PA), and TTX and QX314 were obtained from Alomone Labs (Jerusalem, Israel). The concentrations of the above-mentioned agonists and antagonists used in this study have been determined in previous studies (Vaughan et al., 1997; Marinelli et al., 2002; Pan et al., 2002, 2004). Because the “run-down” frequently occurred within 20 to 30 min after the membrane rupture, we were unable to test several concentrations of DAMGO in this brainstem slice preparation.
Immunocytochemical Labeling of GABA and Tryptophan Hydroxylase (TPH) in Recorded RVM Neurons. After recording, the tissue slice was fixed by submersion in 4% paraformaldehyde in PBS (pH 7.4) and kept in 4°C for 7 to 10 days. The tissue was then placed in cryoprotectant solution at -20°C until use. The sections were cut to 50 μm in thickness and collected free-floating in 0.1 M PBS. For GABA and TPH (a marker for serotonergic neurons) double immunofluorescence labeling, the first primary antibody (anti-GABA) was enhanced with tyramide signal amplification, and conventional immunofluorescence labeling was then performed with the second primary antibody (anti-tryptophan hydroxylase) (Marinelli et al., 2002). The sections were rinsed in Tris-buffered saline (0.1 M Tris-HCl and 0.15 M NaCl, pH 7.8), incubated in 3% H2O2 in 10% methanol for 30 min to quench endogenous peroxidase and blocked in 0.1 M Tris-HCl, 0.15 M NaCl, and 0.5% blocking reagent for 30 min. Sections were incubated with rabbit anti-GABA primary anti-body (Sigma-Aldrich; dilution 1:1000) diluted in 0.1 M Tris-HCl, 0.15 M NaCl, and 0.5% blocking reagent buffer for 2 h at room temperature and 48 h at 4°C. Subsequently, sections were rinsed in 0.1 M Tris-HCl, 0.15 M NaCl, and 0.05% Tween 20 buffer and incubated with peroxidase-conjugated goat anti-rabbit IgG secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA; dilution 1:200) for 1.5 h at room temperature and incubated in fluorescein isothiocyanate-conjugated to tyramide for 10 min. Then, the sections were washed and incubated with mouse anti-TPH (Sigma-Aldrich; dilution 1:400) for2hat room temperature and overnight at 4°C. The sections were incubated with Alexa Fluor-594 conjugated to goat anti-mouse IgG (Molecular Probes; dilution 5 μg /ml) for 1.5 h at room temperature. To identify biocytin-labeled cells, the sections were rinsed for 10 min in TNT after the double labeling and incubated with streptavidin conjugated to Alexa Fluor-350 (Molecular Probes; dilution 5 μg/ml) for 1.5 h at room temperature (Pan et al., 2002, 2004). Finally, the sections were mounted on slides, dried, and coverslipped. All sections were examined on a confocal scanning microscope (Leica, Wetzlar, Germany) and areas of interest were photodocumented and digitally merged. In the higher magnification images, the colocalization was indicated by the color change and represents colocalization, because the optical section thickness (<1 μm) of a confocal image is thin enough to minimize the possibility of superimposition of stained neurons. Because the recordings were performed in thin slices using the visualized guidance technique, all the recorded cells were located within the RVM, and we did not further map the detailed distribution of the recorded cells.
Data Analysis. Data are presented as means ± S.E.M. The amplitude and frequency of mIPSCs and mEPSCs of labeled RVM neurons were analyzed off-line with a peak detection program (Mini-analysis; Synaptosoft Inc., Leonia, NJ). The cumulative probability of the amplitude and interevent interval of mIPSCs and mEPSCs was compared using the Komogorov-Smirnov test, which estimates the probability that two distributions are similar, as we described previously (Pan et al., 2002, 2004). Analyses of the effects of drugs on the amplitude of eIPSCs and eEPSCs were performed using Clampfit (Axon Instruments). Neurons were considered to be responsive to DAMGO if the synaptic responses to DAMGO were altered >20%. The effects of drug treatment on postsynaptic currents were determined by either a paired t test or repeated measures analysis of variance with Dunnett's post hoc test. P < 0.05 was considered statistically significant.
Results
Effect of DAMGO on eIPSCs in Labeled RVM Neurons. The effect of 1 μM DAMGO on eIPSCs was studied in a total of 16 labeled cells. In 11 of 16 (69%) cells, DAMGO significantly decreased the amplitude of eIPSCs from 392.32 ± 64.26 to 210.52 ± 33.19 pA (P < 0.05, Fig. 2, A and B). DAMGO had no significant effect on the eIPSC amplitude in the remaining five neurons (550.25 ± 63.62 versus 532.01 ± 38.57 pA; Fig. 2C).

Effect of 1 μM DAMGO on eIPSCs in labeled RVM neurons. A, original tracings of eIPSCs in a labeled RVM neurons during control, application of 1 μM DAMGO, and washout. The stimulus artifact is removed for clarity. B, summary data showing the inhibitory effect of 1 μM DAMGO on eIPSCs in 11 cells. C, summary data showing lack of effect of 1 μM DAMGO on eIPSCs in another five neurons. Data presented as means ± S.E.M. *, P < 0.05 compared with control.
To determine whether GABA mediated the IPSCs in the RVM, the effect of 20 μM bicuculline on eIPSCs was further examined in the above-mentioned 15 labeled cells. The eIPSCs in the majority of neurons (67%; 10 of 15) tested was abolished by bicuculline (Fig. 3A). Bicuculline reduced the amplitude of eIPSCs by 44 to 90% in another five cells. In four of these five neurons, we further examined the effect of the glycine receptor antagonist strychnine. The remaining eIPSCs was completely blocked by further administration of 5 μM strychnine in these four cells (Fig. 3B). We also verified the specificity of the effect of DAMGO on the μ-opioid receptors using a selective μ-receptor antagonist, CTAP, in another seven labeled neurons. CTAP (1 μM) alone had no significant effect on eIPSCs. In the presence of CTAP, DAMGO failed to change significantly the amplitude of eIPSCs in any of these seven cells tested (from 232.4 ± 41.2 to 212.8 ± 28.6 pA; P > 0.05).

Effect of bicuculline and strychnine on evoked IPSCs in labeled RVM neurons. A, representative tracings showing a complete blockade of evoked IPSCs by 20 μM bicuculline in one cell. B, representative tracings showing the effect of 20 μM bicuculline plus 5 μM strychnine (STR) on evoked IPSCs in another cell. The stimulus artifact was removed from the tracing for clarity.
Effect of DAMGO on eEPSCs in Labeled RVM Neurons. The effect of 1 μM DAMGO on eEPSCs was examined in 17 labeled cells. DAMGO decreased the eEPSC amplitude in nine neurons by 56% (from 436.71 ± 68.28 to 193.16 ± 41.87 pA; P < 0.05; Fig. 4, A and B), but had no significant effect on eEPSCs in the remaining eight neurons (880.75 ± 293.21 versus 843.73 ± 284.24 pA; Fig. 4C).

Effect of 1 μM DAMGO on eEPSCs in labeled RVM neurons. A, original tracings of eEPSCs in a labeled RVM neuron during control, application of 1 μM DAMGO, and washout. The stimulus artifact is removed for clarity. B, summary data showing the inhibitory effect of 1 μM DAMGO on eEPSCs in nine cells. C, summary data showing lack of effect of 1 μM DAMGO on eIPSCs in another eight neurons. Data presented as means ± S.E.M. *, P < 0.05 compared with control.
To determine whether glutamate mediated the EPSCs in the labeled RVM neurons, the effect of 20 μM CNQX on eEPSCs was further tested in these nine cells. CNQX abolished eEPSC in two cells (Fig. 5A) and reduced the eEPSC amplitude between 22 and 67% in the remaining seven cells (Fig. 5B). In these seven cells, we further tested the effect of the nicotinic receptor antagonist mecamylamine. Addition of 10 μM mecamylamine completely eliminated the eEPSC in five of these seven cells (Fig. 5B). In the remaining two neurons, neither CNQX nor mecamylamine had any effect on the amplitude of eEPSCs (data not shown). In another seven labeled RVM cells, DAMGO did not produce any significant effect on the eEPSC amplitude in the presence of 1 μM CTAP (from 179.6 ± 41.2 to 177.4 ± 34.2 pA; P > 0.05).

Effect of CNQX and mecamylamine on evoked EPSCs in labeled RVM neurons. A, representative tracings a showing a complete blockade of evoked EPSCs by 20 μM CNQX in one cell. B, representative tracings showing the effect of 20 μM CNQX plus 10 μM mecamylamine (MCM) on evoked EPSCs in a different neuron. The stimulus artifact is removed for clarity.
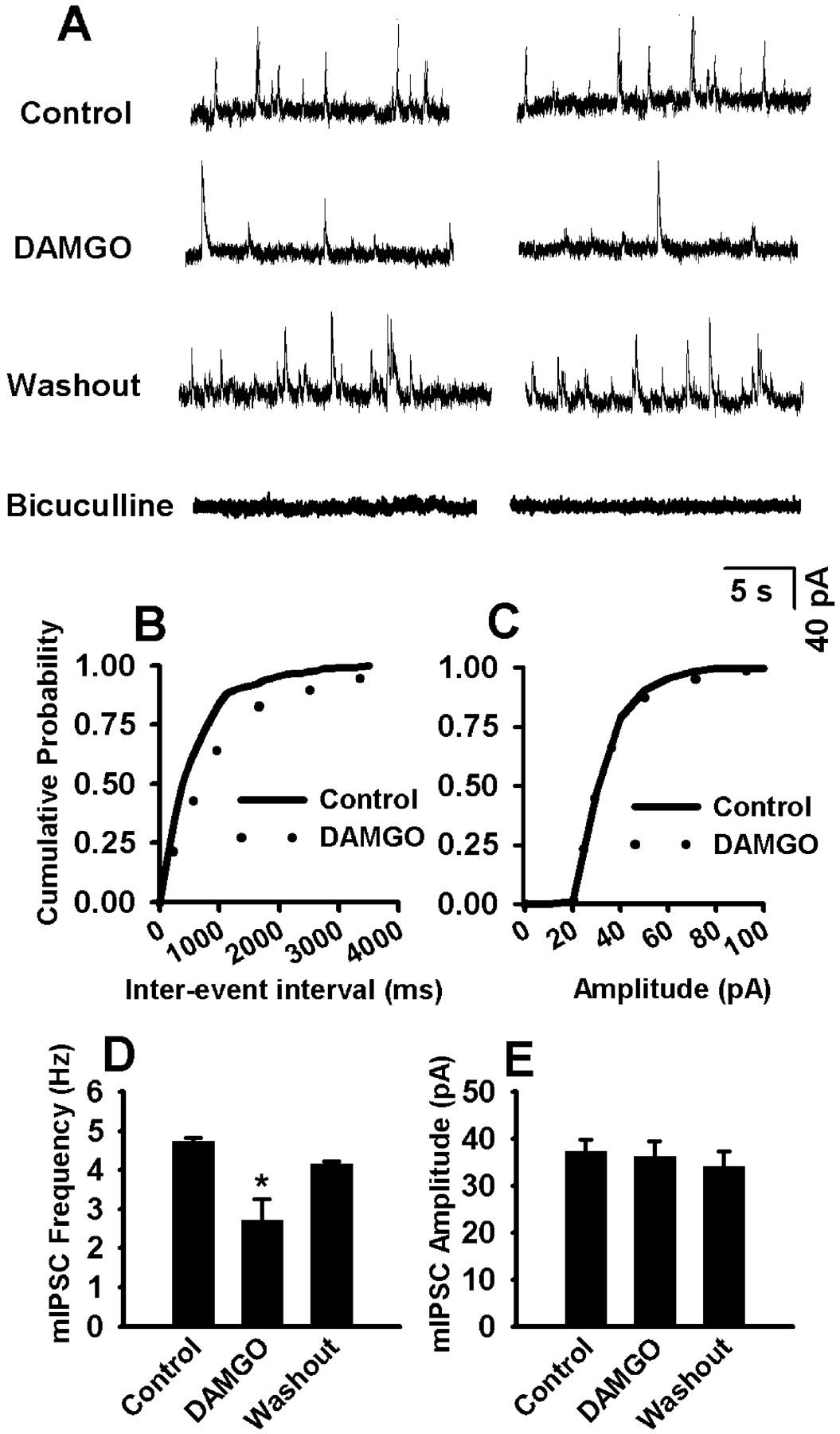
Effect of DAMGO on mIPSCs in Labeled RVM Neurons. In the presence of TTX and CNQX, 1 μM DAMGO significantly decreased the frequency of mIPSCs in all labeled RVM neurons examined (from 4.74 ± 0.74 to 2.72 ± 0.53 Hz; n = 12; P < 0.05; Fig. 6). DAMGO did not, however, significantly alter the amplitude of mIPSCs in these neurons (37.26 ± 2.53 versus 36.21 ± 3.19 pA; Fig. 6). The cumulative probability analysis of mIPSCs revealed that the distribution pattern of the interevent interval shifted toward the right in response to DAMGO, whereas the distribution pattern of the amplitude was not changed by DAMGO (Fig. 6, B and C). The effect of DAMGO on mIPSCs was further analyzed by measuring the decay time constant. The decay phase of the mIPSCs was best fit with a double exponential function. DAMGO did not change significantly the fast (8.10 ± 1.29 versus 8.75 ± 1.48 ms) and slow (26.85 ± 5.29 versus 29.99 ± 4.62 ms) components of the decay time constant.

Effect of DAMGO on mIPSCs in labeled RVM neurons. A, representative tracing showing mIPSCs during control, application of 1 μM DAMGO, or 20 μM bicuculline in a labeled RVM neuron. B and C, cumulative probability plot showing the distribution of interevent interval and the peak amplitude of this neuron during control and DAMGO application. D and E, summary data showing the effect of 1 μM DAMGO on the frequency and amplitude of mIPSCs in 12 labeled cells. Data presented as means ± S.E.M. *, P < 0.05 compared with control.
The frequency of mIPSCs in labeled RVM neurons was completely abolished by 20 μM bicuculline in five of eight (62.5%) cells examined (Fig. 6A). Bicuculline reduced the mIPSC frequency by approximately 20 and 90% in another two cells. In the remaining cell, bicuculline alone resulted in a 77% reduction (from 2.81 to 0.36 Hz) in the mIPSC frequency, and addition of 5 μM strychnine further reduced the mIPSC frequency from 0.36 to 0.12 Hz.
Effect of DAMGO on mEPSCs in Labeled RVM Neurons. The effect of 1 μM DAMGO, in the presence of TTX and bicuculline, on mEPSCs was examined in a total of 27 labeled RVM neurons. DAMGO decreased the mEPSC frequency (from 4.20 ± 0.68 to 2.35 ± 0.41 Hz; P < 0.05; Fig. 7, A–C) in 15 of 27 (55%) neurons. The amplitude of mEPSCs recorded from these 15 cells was not altered significantly (27.37 ± 1.45 versus 26.28 ± 1.75 pA; Fig. 7, A–C) by DAMGO. The decay phase of mEPSCs was best fit with a single exponential function. The decay time constant of mEPSCs did not differ significantly (4.30 ± 0.55 versus 4.69 ± 0.72 ms) between control and DAMGO application. The cumulative probability analysis of mEPSCs in the 15 DAMGO-responsive cells indicated that the distribution pattern of the interevent interval of mEPSCs shifted to the right in response to DAMGO (Fig. 7B). However, the amplitude of mEPSCs was not affected by DAMGO in these 15 cells (Fig. 7B). In 12 of 27 (45%) labeled RVM neurons, DAMGO did not significantly change the frequency (2.42 ± 0.95 versus 3.27 ± 0.91 Hz) and amplitude (25.12 ± 1.04 versus 25.45 ± 1.18 pA; Fig. 7D) of mEPSCs.

Effect of DAMGO on mEPSCs in labeled RVM neurons. A, representative tracing showing mEPSCs during control, application of 1 μM DAMGO, or 20 μM CNQX in a labeled RVM neuron. B, cumulative probability plot showing the distribution of interevent interval and the peak amplitude of this neuron during control and DAMGO application. C, summary data showing the effect of 1 μM DAMGO on the frequency and amplitude of mIPSCs in 15 labeled cells. D, summary data showing lack of effect of 1 μM DAMGO on the frequency and amplitude of mIPSCs in another 12 labeled cells. Data presented as means ± S.E.M. *, P < 0.05 compared with control.
Perfusion of 20 μM CNQX completely eliminated mEPSCs in eight of 10 (80%) cells examined (Fig. 7A). CNQX reduced the mEPSC frequency by 50 and 86% in another two cells. In these two cells, addition of the nicotinic receptor antagonist mecamylamine (10 μM) further reduced mEPSC frequency in the presence of 20 μM CNQX. In one neuron, CNQX reduced the mEPSC frequency from 0.48 to 0.24 Hz, and mecamylamine abolished the remaining mEPSC. In another cell, CNQX reduced the mEPSC frequency from 2.97 to 0.42 Hz, and 10 μM mecamylamine also completely blocked the remaining mEPSC. In these two cells, DAMGO decreased the frequency of mEPSCs by 17% (from 0.57 to 0.48 Hz) and 53% (from 1.22 to 0.57 Hz).
GABA and TPH Immunoreactivity of Recorded RVM Cells. After whole-cell recording, biocytin-labeled cells were then processed for double-immunolabeling of GABA and TPH. In total, 60 labeled RVM neurons was recovered and used for analysis. Of these 60 cells, 50% (n = 30) of the cells had neither GABA nor TPH immunoreactivity. About 35% (n = 21; Fig. 8A) of labeled RVM neurons were TPH-positive, and 15% (n = 9) had GABA immunoreactivity. Eight percent (5 of 60) of the labeled neurons were double-labeled with both GABA and TPH (Fig. 8B).

Confocal images of GABA (green) and TPH (red) immunoreactivity in biocytin (blue)-labeled RVM neurons. A, one biocytin-labeled RVM neuron with TPH-, but not GABA-immunoreactivity. Note that this labeled cell is surrounded with GABA-positive nerve terminals. B, colocalization of GABA and TPH in a separate biocytin-labeled neuron. Images are in all cases single confocal optical sections. Scale bar, 10 μm.
The labeled cells were further examined based on their responses to DAMGO. Fifty-three percent of the cells that responded in some way (i.e., IPSCs and EPSCs) to DAMGO contained TPH immunoreactivity. On the other hand, in DAMGO-unresponsive cells, 22% of them had TPH immunoreactivity. In both DAMGO-responsive and -unresponsive cells, the proportion of GABA-positive neurons was the same (11%).
Discussion
This is the first study investigating the effect of μ-opioid receptor stimulation on excitatory and inhibitory synaptic inputs to spinally projecting RVM neurons. We found that the μ-receptor agonist DAMGO decreased evoked and miniature IPSCs in at least 70% of the cells examined. DAMGO also had a similar inhibitory effect on both evoked and miniature EPSCs in 53 to 75% of labeled RVM neurons. Furthermore, the IPSCs were largely GABAergic in nature mediated by the GABAA receptor because the IPSCs were eliminated by bicuculline in most cells. On the other hand, many of the EPSCs were effectively blocked by CNQX, a glutamate non-NMDA receptor antagonist. It is noteworthy that in some labeled cells, the glycine receptor antagonist strychnine reduced IPSCs, whereas the nicotinic receptor antagonist mecamylamine attenuated EPSCs. Together, these data provide important new information that μ-opioid receptor activation attenuates both excitatory and inhibitory synaptic transmission in most spinally projecting RVM neurons.
The RVM is a major brain site for the descending inhibition of nociception and opioid analgesia. For example, the RVM contains μ-, δ-, and κ-opioid receptors (Bowker and Dilts, 1988; Gutstein et al., 1998; Kalyuzhny and Wessendorf, 1998). The μ-opioid receptor is located on both cell bodies and nerve terminals in the RVM (Kalyuzhny et al., 1996; Kalyuzhny and Wessendorf, 1998; Marinelli et al., 2002). Morphine injected into the RVM results in antinociception in rats (Llewelyn et al., 1986; Jones and Gebhart, 1988). Furthermore, morphine application to the RVM not only results in an increase in the tail-flick latency but also inhibits ON-cells and excites OFF-cells (Gao et al., 1998). Stimulation of OFF-cells by morphine is probably due to inhibition of putative GABAergic interneurons in the RVM (Fields et al., 1983b; Pan et al., 1997). In this regard, it has been shown that morphine inhibits NRM neurons that express glutamate decarboxylase, the enzyme responsible for synthesizing GABA (Li and Wang, 2001). The main purpose of this study was to determine how the μ-receptor activation affects major inhibitory and excitatory synaptic inputs to spinally projecting RVM neurons. A distinct feature of our study is that we specifically studied RVM cells that project to the dorsal horn of the spinal cord. This was achieved by retrograde labeling of RVM neurons through FluoroSpheres injected into the spinal cord. This is an important approach because the RVM neurons are heterogeneous. Although spinally projecting RVM neurons may be ultimately involved in the analgesic action of opioids in the RVM, the majority of previous experiments did not directly examine the functional connection between the RVM and the spinal cord. Marinelli et al. (2002) recently examined the postsynaptic effect of μ-opioids on neurons in the RVM that project to the spinal cord. However, the presynaptic action of opioids was not examined in that study.
In the present study, DAMGO inhibited both GABA and glutamate release onto many spinally projecting RVM neurons. Because the μ-opioid receptor is coupled to inhibitory G proteins, we included the G protein inhibitor guanosine 5′-O-(2-thiodiphosphate) in the internal solution to eliminate the potential postsynaptic effect of DAMGO (Pan et al., 2002, 2004). We found that the inhibitory actions of DAMGO were similar in evoked and spontaneous quantal (miniature) release of neurotransmitters in these cells. There are extensive GABAergic interneurons that probably release GABA onto the output neurons in the RVM. The origin of glutamatergic terminals in the RVM is not clear although a likely site is the PAG (Spinella et al., 1999). Previous studies have shown that DAMGO decreases both GABA and glutamate release in the PAG and dorsal raphe nucleus (Jolas and Aghajanian, 1997; Kishimoto et al., 2001). Although the mechanisms responsible for the inhibitory effect of DAMGO on both GABA and glutamate release in the RVM are not fully known, opioids inhibit GABA release in the PAG through the opening of voltage-dependent presynaptic K+ channels that are controlled by phospholipase A2, arachidonic acid, and 12-lipoxygenase pathways (Vaughan et al., 1997). The effect of DAMGO on glutamate release may be mediated by blocking presynaptic calcium channels (Vlaskovska et al., 1997).
A salient finding of our study is that in addition to glutamate and GABA, endogenous glycine and acetylcholine also play a role in fast synaptic transmission in the RVM. In the present study, although mIPSCs and eIPSCs were mediated by the GABAA receptor in the majority of cells, the glycine receptor antagonist reduced the IPSCs in some labeled RVM neurons. The functional role of glycine in the RVM remains unclear. It has been shown that microinjection of bicuculline, but not strychnine, increases the tail-flick latency and blocks OFF-cell pause (Heinricher et al., 1991; Heinricher and Kaplan, 1991). We also observed that in a small population of cells, the EPSCs were partially mediated by activation of the nicotinic receptor. It has been shown that the postsynaptic nicotinic receptors mediate excitatory postsynaptic potentials or currents in the spinal cord, hippocampus, neocortex, and supraoptic nucleus (Frazier et al., 1998; Chu et al., 2000; Bradaia and Trouslard, 2002; Hatton and Yang, 2002). Although the α4 subunit of the nicotinic receptor is expressed in the NRM (Bitner et al., 1998), to our knowledge, this is the first study that provides functional evidence that nicotinic receptors are involved in the excitatory synaptic transmission in the brainstem. Despite the fact that some EPSCs and IPSCs were mixed synaptic inputs, we found that DAMGO consistently inhibited both excitatory and inhibitory inputs to labeled RVM neurons. It should be noted that in a few labeled cells, the eEPSCs were insensitive to blockade of non-NMDA and nicotinic acetylcholine receptors. Thus, other neurotransmitters also contribute to fast excitatory synaptic transmission in the RVM. A likely candidate is ATP, which activates purinergic P2X receptors and opens a nonspecific ion channel. In this regard, the P2X receptor is found throughout the brain, including the rostral ventrolateral medulla (Kanjhan et al., 1999), and the fast synaptic transmission in the cortex and rostral ventrolateral medulla may involve activation of the P2X receptor (Ralevic et al., 1999; Pankratov et al., 2002).
Our immunocytochemistry data further revealed the heterogeneity of cell types in the RVM. Whereas 35% of recorded cells contained TPH, the GABAergic cells (15%) are far fewer than serotonergic cells in this population of RVM neurons. Furthermore, colocalization of GABA and TPH occurred in 8% of labeled RVM neurons. Our finding that GABA and TPH were colocalized in some spinally projecting RVM cells is consistent with a previous study showing colocalization of glutamate decarboxylase and 5-HT in these cells (Millhorn et al., 1987). We also found that compared with cells unresponsive to DAMGO, there were more TPH-positive neurons in the group of cells that responded to DAMGO. Importantly, both our data and the study by Marinelli et al. (2002) suggest that the majority of spinally projecting RVM neurons is not serotonergic. The involvement of 5-HT in the descending modulation of nociception remains controversial, and it has been shown that 5-HT does not play an important role in opioid analgesia in the RVM (Chiang and Pan, 1985; Gao et al., 1998). Because 50% of the labeled neurons contained neither GABA nor TPH, the neurochemical identity of these spinally projecting RVM neurons and the mechanisms involved in the cellular action of opioids seem to be complex and warrant further investigation.
In summary, using the retrograde labeling technique and whole-cell recordings in a brain slice preparation, we found that DAMGO significantly reduced the excitatory and inhibitory synaptic inputs to many spinally projecting RVM neurons through activation of the μ-opioid receptor. Furthermore, we observed that many spinally projecting RVM neurons contained neither GABA nor TPH. Collectively, this study provides important new information about the synaptic mechanisms involved in the opioid action in the RVM.
Footnotes
-
This study was supported by Grants GM64830 and NS45602 from the National Institutes of Health.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
DOI: 10.1124/jpet.103.064808.
-
ABBREVIATIONS: RVM, rostral ventromedial medulla; PAG, periaqueductal gray; NRM, nucleus raphe magnus; 5-HT, 5-hydroxytryptamine; NMDA, N-methyl-d-aspartate; DAMGO, [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin; aCSF, artificial cerebrospinal fluid; mIPSC, miniature inhibitory postsynaptic current; mEPSC, miniature excitatory postsynaptic current; TTX, tetrodotoxin; CNQX, 6-cyano-7-nitroquinoxaline-2,3-dione; eIPSC, evoked inhibitory postsynaptic current; eEPSC, evoked excitatory postsynaptic currents; CTAP, H-d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2; TPH, tryptophan hydroxylase; PBS, phosphate-buffered saline.
- Received December 22, 2003.
- Accepted December 31, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}