


Abstract
L-Type Ca2+ channel blockers inhibit glucose and KCl-stimulated insulin secretion by pancreatic β cells. However, the role of the two distinct L-type channels expressed by β cells, Cav1.2 and Cav1.3, in this process is not clear. Therefore, we stably transfected INS-1 cells with two mutant channel constructs, Cav1.2DHPi or Cav1.3 DHPi. Whole-cell patch-clamp recordings demonstrated that both mutant channels are insensitive to dihydropyridines (DHPs), but are blocked by diltiazem. INS-1 cells expressing Cav1.3/DHPi maintained glucose- and KCl-stimulated insulin secretion in the presence of DHPs, whereas cells expressing Cav1.2/DHPi demonstrated DHP resistance to only KCl-induced secretion. INS-1 cells were also stably transfected with cDNAs encoding the intracellular loop between domains II and III of either Cav1.2 or Cav1.3 (Cav1.2/II-III or Cav1.3/II-III). Glucose- and KCl-stimulated insulin secretion in Cav1.2/II-III cells were not different from untransfected INS-1 cells. However, glucose-stimulated insulin secretion was completely inhibited and KCl-stimulated secretion was substantially resistant to inhibition by DHPs, but sensitive to ω-agatoxin IVA in Cav1.3/II-III cells. Moreover, the L-type channel agonist FPL 64176 markedly enhanced KCl-stimulated secretion by Cav1.3/II-III cells. Together, our results suggest that Ca2+ influx via Cav1.3 is preferentially coupled to glucose-stimulated insulin secretion in INS-1 cells.
Calcium influx through voltage-dependent Ca2+ channels (VDCCs) plays a crucial role in insulin secretion from pancreatic β cells (Wollheim and Sharp, 1981). Glucose metabolism in β cells causes an increase in ATP/ADP ratio, which closes ATP-dependent potassium channels (Rajan et al., 1990). The resulting membrane depolarization opens VDCCs, and calcium influx triggers insulin secretion. Several distinct VDCCs have been detected in β cells and insulin-secreting cell lines, including high-voltage-activated subtypes (Seino et al., 1992; Ligon et al., 1998) and the low-voltage-activated channels (Zhuang et al., 2000). L-Type VDCCs play a major role in the function of pancreatic β cells because L-type-specific blockers significantly inhibit glucose or depolarization-induced insulin secretion (Devis et al., 1975). Two distinct L-type channels, Cav1.2 and Cav1.3, are expressed in human pancreatic islets (Seino et al., 1992) and in insulin-secreting cells lines (Horvath et al., 1998). However, Cav1.2 and Cav1.3 are both blocked by the same small-molecule drugs (Hockerman et al., 1997; Bell et al., 2001). Thus, the relative contribution of calcium flux through Cav1.2 and/or Cav1.3 to Ca2+-dependent insulin secretion is not clear.
Previously, we reported a double mutation that renders Cav1.2 insensitive to dihydropyridines (DHPs), such as nifedipine and PN200-110, but normally sensitive to block by diltiazem (Hockerman et al., 2000). We used this mutant channel (Cav1.2/DHPi), and the corresponding Cav1.3 mutant (Cav1.3/DHPi), in a novel strategy to study the roles of Cav1.2 (Snutch et al., 1991) and Cav1.3 (Williams et al., 1992) in insulin secretion. When these mutant channels were introduced into the rat pancreatic β-cell line INS-1 (Asfari et al., 1992), endogenous L-type channels were “turned off” with a DHP such as PN200-110, functionally isolating the drug-insensitive Cav1.2 or Cav1.3 mutant. We found that expression of Cav1.3/DHPi but not Cav1.2/DHPi allowed glucose-stimulated insulin secretion that was insensitive to DHPs, but blocked by diltiazem, in INS-1 cells.
Insulin-containing secretory granules and L-type VDCCs are colocalized in β-cells (Bokvist et al., 1995; Qian and Kennedy, 2001). The functional coupling between L-type channels and exocytotic granules may resemble that of non-L-type VDCCs to neurotransmitter release in neurons. An interaction between the intracellular loop linking domains II and III of Cav2.2 or Cav2.1 and SNARE proteins mediates efficient coupling of Ca2+ influx to neurotransmitter vesicle fusion (Sheng et al., 1998). Pancreatic β-cells express isoforms of the corresponding SNARE proteins, which regulate both insulin secretion and calcium channel activity (Jacobsson et al., 1994;Sadoul et al., 1995; Nagamatsu et al., 1996). Functional interactions between syntaxin and both Cav1.3 (Yang et al., 1999) and Cav1.2 (specifically the II-III loop) (Wiser et al., 1999) are reported to play a role in depolarization-induced insulin secretion. To examine the role of the II-III loop of both Cav1.2 and Cav1.3 in coupling Ca2+influx to insulin secretion, we stably transfected INS-1 cells with plasmids encoding the II-III loop of either Cav1.2 or Cav1.3, fused to GFP. Experiments with the resulting cell lines (Cav1.2/II-III cells and Cav1.3/II-III cells) showed that glucose-stimulated insulin secretion was completely abolished in Cav1.3/II-III cells, but was not different from untransfected cells in the Cav1.2/II-III cell line.
Materials and Methods
Cell Culture.
INS-1 cells were cultured as reported previously (Asfari et al., 1992).
Stable Transfection.
INS-1 cells were transfected using GenePorterII (Gene Therapy Systems, San Diego, CA). After 3 days, 100 μg/ml G418 (Promega, Madison, WI) was added to the medium. Colonies were isolated, and subsequently screened by RT-PCR and Western blot.
Plasmids.
Construction of the Cav1.2/DHPi mutant was described previously (Hockerman et al., 2000). The two amino acid mutations in domain IIIS5 of Cav1.3 (Williams et al., 1992) (Thr 1029 to Tyr; Gln 1033 to Met) were mutated using the QuikChange method (Stratagene, La Jolla, CA) to generate Cav1.3/DHPi. Cav1.2/DHPi and Cav1.3/DHPi were subcloned into EGFP vector (BD Biosciences Clontech, Palo Alto, CA). The intracellular II-III loops of Cav1.2 and Cav1.3 were amplified by PCR using Pfu DNA polymerase, followed by ligation into the EGFP vector. All constructs were confirmed by cDNA sequencing.
RT-PCR.
Total RNA was extracted from INS-1 cells using TRIzol (Invitrogen, Carlsbad, CA), and 2 μg were incubated with random primers at 70°C for 5 min, and then put on ice. RNase inhibitor (1 μl), 100 μM dNTPs, 0.01 M dithiothreitol, and 200 U of M-MLV reverse transcriptase (Promega, Madison, WI) were added to the mixture (20 μl final volume) and incubated at 42°C for 30 min, followed by incubation at 85°C for 5 min. Two primer pairs were used for PCR with Taq polymerase (Promega) as follows. Mutant primer set (overlaps mutations in IIIS5): Cav1.2 forward (5′-cta cac tct gct gat gtt c-3′) and reverse (5′-ggg gat cca cgt acc aca ctt tgt act-3′) and Cav1.3 forward (5′-cat gac cct cct gat gtt c-3′) and reverse (5′-cgg gat ccc gcg aag agt tca cca cgt ac-3′). PCR products are 538 bp for CaV1.2/DHPi and 540 bp for Cav1.3/DHPi. GFP primers set: Cav1.2 (5′-agc tgt gta tat gcc ctg g-3′), GFPr (5′-gaa gaa gtc gtg ctg ctt c-3′), Cav1.3 (5′-gtc ctg gct aca gcg acg-3′), and GFPr were used to amplify the channel/EGFP junction. PCR products are 344 bp for Cav1.2/DHPi and 351 bp for Cav1.3/DHPi. PCR products were visualized by ethidium bromide staining after 1% agarose gel electrophoresis in TAE buffer (40 mM Tris-acetate, 2 mM EDTA, pH 8.5).
Western Blot.
Crude membranes from indicated cells were isolated as described previously (Peterson et al., 1997). For whole-cell lysates, indicated cells were incubated in SDS lysis buffer (0.5% SDS, 0.05 M Tris-Cl, 1 mM dithiothreitol, pH 8.0) for 10 min. Lysates were boiled for 5 min and clarified by centrifugation at 26,000g, 4°C for 90 min, and supernatants were collected for Western blot. The proteins were separated by SDS-polyacrylamide gel electrophoresis (5% gels for crude membranes and 12% gels for cell lysates) followed by transfer to nitrocellulose membrane. The membranes were blocked with 5% nonfat milk in Tris-buffered saline at 4°C overnight, washed with 0.1% Tween 20 in Tris-buffered saline, and incubated with polyclonal rabbit anti-GFP antibodies (BD Biosciences Clontech) for 2 to 3 h. The blots were detected by incubation with horseradish peroxidase-conjugated anti-rabbit antibodies and visualized by enhanced chemiluminescence with Hyperfilm (Amersham Pharmacia AB, Uppsala, Sweden). Protein concentrations were determined using the Bradford assay (Bio-Rad, Hercules, CA).
Insulin Secretion and Cellular Insulin Content.
Cells in 12-well dishes were rinsed twice with modified Krebs-Ringer-bicarbonate HEPES buffer (KRBH buffer: 115 mM NaCl, 24 mM NaHCO3, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, 25 mM HEPES, 0.5% BSA, pH 7.4), and incubated in 1 ml KRBH buffer for 30 min at 37°C, 5% CO2. After aspiration of the buffer, the cells were stimulated in KRBH buffer supplemented with 50 mM KCl or 11.2 mM glucose (±indicated drugs) for 30 min at 37°C. The buffer containing secreted insulin was collected and centrifuged at 700g for 3 min to remove any detached cells. The cellular insulin was extracted by incubation in 1 ml of acid-ethanol (ethanol/H2O/HCl, 14:57:3) overnight at 4°C and centrifuged at 700g for 3 min. Supernatants were diluted 10-fold with glycine-NaOH buffer (0.2 M glycine, 0.5% bovine serum albumin, pH 8.8) before insulin assay. Assays were performed using the High Range Rat Insulin ELISA kit (ALPCO, Windham, NH) according to the manufacturer's instructions.
Electrophysiology.
Cells were cultured on plastic coverslips for 2 days (Nalge Nunc, Naperville, IL). Whole-cell barium currents were recorded at room temperature using an Axopatch 200B amplifier (Axon Instruments, Inc., Foster City, CA) and filtered at 1 kHz (six-pole Bessel filter, −3 dB). Electrodes were pulled from borosilicate glass (VWR, West Chester, PA) and fire polished to resistances of 2 to 6 MΩ. Voltage pulses were applied and data were acquired using pClamp8 software (Axon Instruments, Inc.). Nifedipine and diltiazem (Sigma-Aldrich, St. Louis, MO) were applied to the recording chamber in bath saline at 0.5 ml/min. The bath saline contained 150 mM Tris, 10 mM BaCl2, and 4 mM MgCl2. The intracellular solution contained 130 mM N-methyl-d-glucamine, 10 mM EGTA, 60 mM HEPES, 2 mM MgATP, and 1 mM MgCl2. The pH of both solutions was adjusted to 7.3 with methanesulfonic acid.
Data Analysis.
Data were analyzed using ClampFit 8.1 (Axon Instruments, Inc.) and SigmaPlot 2001 (SPSS Science, Chicago, IL). Data are shown as mean values ± standard error. Statistical significance was determined using Student's t test.
Results
Molecular Characterization of Cav1.2/DHPi and Cav1.3/DHPi Stable Cell Lines.
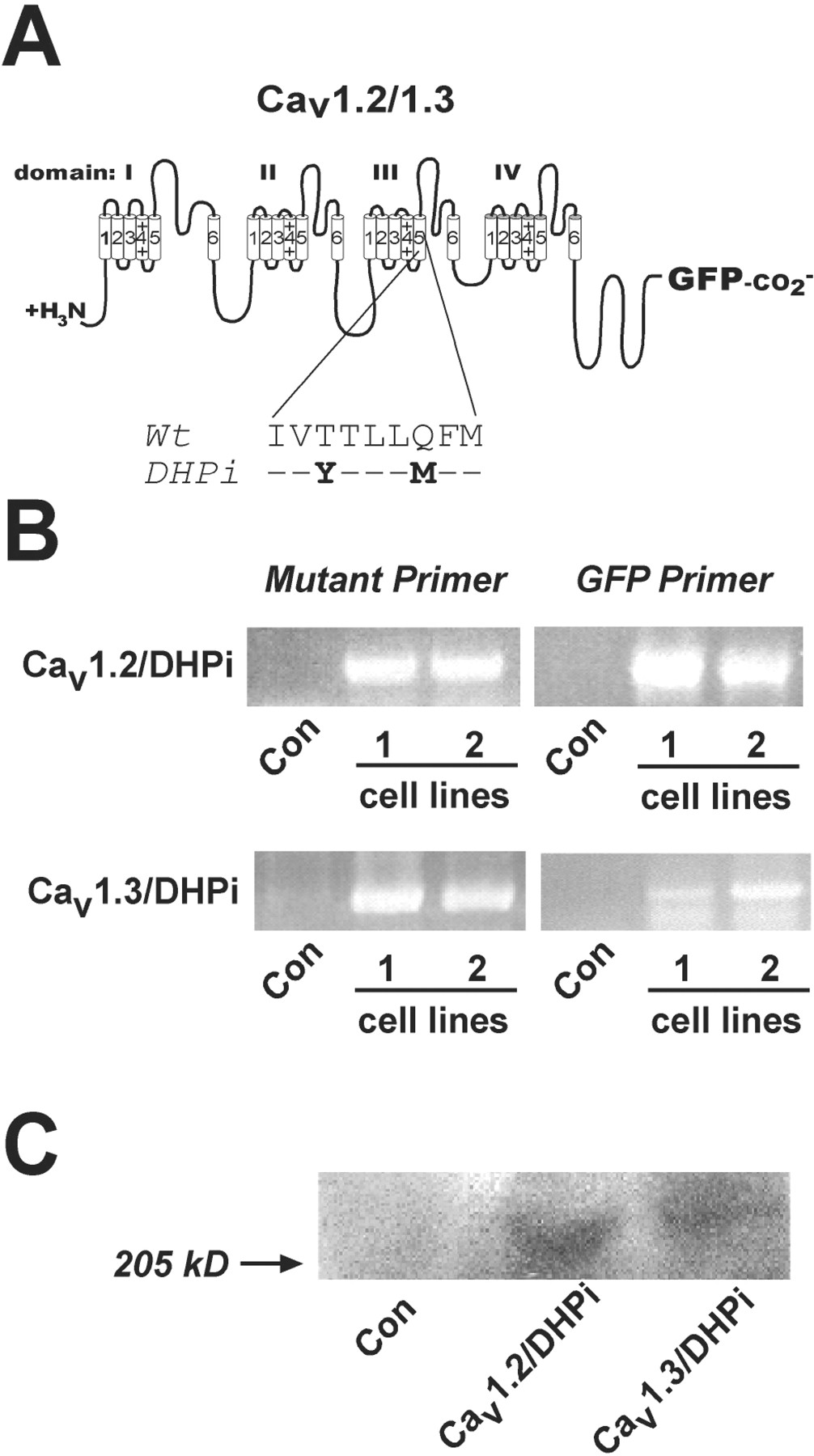
The Cav1.2/DHPi and Cav1.3/DHPi mutant constructs incorporate Tyr for Thr and Met for Gln substitutions in IIIS5 that render them insensitive to DHPs, but do not affect sensitivity to diltiazem block (Hockerman et al., 2000) (Fig.1A). INS-1 cells were transfected with either Cav1.2/DHPi or Cav1.3/DHPi fused to GFP. After selection in G418, colonies were screened by RT-PCR. Because both Cav1.2 and Cav1.3 are endogenously expressed in INS-1 cells, we used oligonucleotide primers complementary to the mutations in transmembrane domain IIIS5, or primers complementary to GFP cDNA in the PCR reactions. As shown in Fig. 1B, RT-PCR reactions using RNA from two different clones from each transfection, and both primer pairs, amplified DNA fragments of the expected size. Control reactions using the same primers with RNA from untransfected INS-1 cells did not amplify the corresponding DNA fragments. We further characterized one clone from each transfection by Western blot. Because antibodies to Cav1.2 or Cav1.3 would detect endogenous channels, we used an anti-GFP antibody to detect the mutant channels. Figure 1C shows that anti-GFP antibodies detect a protein migrating with a molecular mass of approximately 240 kDa in crude membrane fractions from Cav1.2/DHPi cells, and a slightly larger protein in crude membranes prepared from Cav1.3/DHPi cells. Neither protein was detected by anti-GFP antibodies in crude membrane fractions from untransfected INS-1 cells.

Stable expression of Cav1.2/DHPi or Cav1.3/DHPi in INS-1 cells. A, amino acids mutated in Cav1.2/DHPi and Cav1.3/DHPi. Both constructs included GFP fused to the C-terminal tail. B, RT-PCR of stable INS-1 cells expressing Cav1.2/DHPi or Cav1.3/DHPi. Primers complementary to the IIIS5 mutations or GFP were used as described under Materials and Methods. RNA from untransfected INS-1 cells served as control. C, Western blot analysis. Crude membrane fractions from untransfected INS-1 cells and selected cell lines subjected to SDS-polyacrylamide gel electrophoresis and blotted with anti-GFP antibodies. A protein of ∼240 kDa was detected in both stable cell lines, but not in untransfected INS-1 cells.
Electrophysiological Characterization of Cav1.2/DHPi and Cav1.3/DHPi Cell Lines.
Both Cav1.2/DHPi and Cav1.3/DHPi cell lines were characterized using whole-cell patch-clamp recordings of Ba2+ currents (IBa). Stable expression of the Cav1.2/DHPi or Cav1.3/DHPi channels did not significantly increase theIBa density compared with untransfected INS-1 cells (Fig. 2B). Both 1 μM PN200-110 and 10 μM nifedipine block approximately 13% ofIBa in untransfected INS-1 cells, and coapplication of 50 μM diltiazem with 10 μM nifedipine does not significantly reduce current amplitude (Fig. 2, A and C). The fraction of current blocked by application of 10 μM nifedipine to Cav1.2/DHPi cells or Cav1.3/DHPi cells under voltage clamp (Fig. 2, A and C) was not different from untransfected INS-1 cells. However, coapplication of 50 μM diltiazem along with 10 μM nifedipine to Cav1.2/DHPi cells or Cav1.3/DHPi cells further reducedIBa. Thus, the pharmacological profile of IBa in Cav1.2/DHPi and Cav1.3/DHPi cells confirms the functional expression of the mutant channels. Furthermore, the IBa density (pA/pF) is not significantly different in the Cav1.2/DHPi and Cav1.3/DHPi cells lines compared with untransfected INS-1 cells (Fig. 2B). Thus, stable tranfection of INS-1 cells with the mutant α1 subunits alone, does not lead to a significant increase in whole-cell barium current.

Electrophysiological characterization of Cav1.2/DHPi and Cav1.3/DHPi cells.IBa was measured using whole-cell voltage-clamp. Currents were elicited by 100-ms depolarizations from −60 to +10 mV. A, representative traces from untransfected INS-1 cells, Cav1.2/DHPi cells, and Cav1.3/DHPi cells. Nifedipine (Nif; 10 μM) blocked DHP-sensitive current. Nifedipine (10 μM) plus diltiazem (dilt; 50 μM) blocked Cav1.2/DHPi or Cav1.3/DHPi channels. B, barium current density in INS-1,Cav1.2/DHPi, and Cav1.3/DHPi cells. Total whole-cell current (pA/pF) measured (mean ± S.E.;n = 4–6) is shown. There is no statistically significant difference among the three cell lines tested. C, relative fractional blockade of IBa by drugs. In untransfected INS-1 cells, 13.5 ± 3% (n = 4) of total barium current was blocked by 10 μM nifedipine, whereas 12 ± 2.5% (n = 6) was blocked by 1 μM PN200-110. Coapplication of 50 μM diltiazem did not significantly reduce DHP-resistant current. In Cav1.2/DHPi and Cav1.3/DHPi cells, coapplication of 50 μM diltiazem blocked significantly more current than 10 μM nifedipine alone [46 ± 4% (n = 5) and 40 ± 4.5% (n = 5), respectively (∗, p< 0.05)].
Insulin Secretion in Cav1.2/DHPi and Cav1.3/DHPi Cells.
We tested the ability of the Cav1.2/DHPi and Cav1.3/DHPi cells to secretion insulin in response to either KCl or glucose stimulation, in the presence or absence of a DHP channel blocker. Glucose- and KCl-induced insulin secretion in untransfected INS-1 cells is completely blocked by the DHP PN200-110 (0.1 μM) (Fig.3A). Glucose-stimulated insulin secretion in Cav1.2/DHPi cells is completely blocked by 1 μM PN200-110 or 1 μM PN200-110 + 500 μM diltiazem (Fig. 3B). However, glucose-stimulated insulin secretion in Cav1.3/DHPi cells is substantially resistant to 1 μM PN200-110, but inhibited by the addition of 500 μM diltiazem. In contrast, KCl-induced insulin secretion is resistant to PN200-110 in both Cav1.3/DHPi and Cav1.2/DHPi cells (Fig. 3C). Figure 3D summarizes the results of these experiments by comparing the percentage of glucose- and KCl-stimulated insulin secretion resistant to 1 μM PN200-110 in both Cav1.2/DHPi and Cav1.3/DHPi cells. In Cav1.3/DHPi cells, ∼42% of glucose-stimulated insulin secretion was resistant to PN200-100, whereas glucose did not stimulate insulin secretion from Cav1.2/DHPi cells in the presence of PN200-110. Upon KCl stimulation, ∼44% of insulin secretion was resistant to PN200-110 in Cav1.3/DHPi cells, whereas a significantly lower percentage of insulin secretion (∼29%) was resistant to PN200-110 in Cav1.2/DHPi cells. Thus, although the percentage of DHP-resistant insulin secretion was the same regardless of the stimulus in Cav1.3/DHPi cells, DHP-resistant insulin secretion in Cav1.2/DHPi cells was dependent upon KCl stimulation. The portion of DHP-sensitive secretion in both cell lines suggests that some endogenous L-type channels remain functionally coupled to secretion.
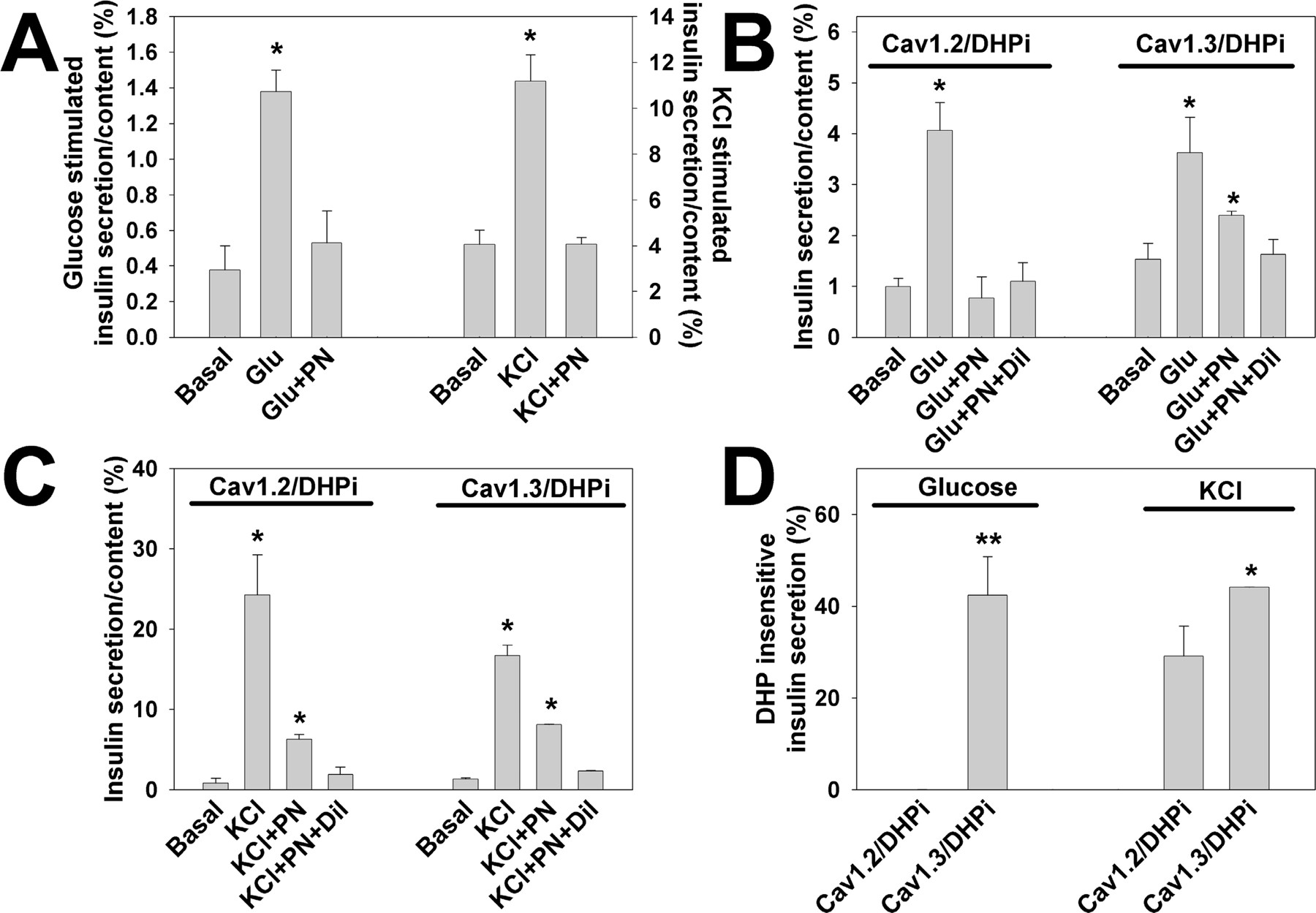
Insulin secretion in Cav1.2/DHPi and Cav1.3/DHPi cells. A, glucose- (11 mM) and KCl (50 mM)-stimulated secretion in INS-1 cells. In untransfected INS-1 cells, 0.1 μM PN200-110 completely blocked glucose- or KCl-induced insulin secretion [glucose: basal = 0.38 ± 0.14% (n = 3); 11 mM glucose = 1.38 ± 0.12% (n = 3) KCl: basal = 4.06 ± 0.63% (n = 3); 50 mM KCl = 11.18 ± 1.15% (n = 3)] (∗, p < 0.05 compared with basal). B, glucose-stimulated secretion in Cav1.2/DHPi or Cav1.3/DHPi cells (11 mM; Glu). Cav1.2/DHPi: basal = 0.99 ± 0.16% (n = 3); Glu = 4.06 ± 0.55% (n = 3). PN200-110 (1 μM) completely inhibited glucose stimulated insulin secretion in Cav1.2/DHPi cells (Glu + PN). With Cav1.3/DHPi cells, a significant fraction of glucose-stimulated insulin secretion was resistant to 1 μM PN200-110, but blocked by 500 μM diltiazem (Glu + PN + Dil). Cav1.3/DHPi: basal = 1.53 ± 0.32% (n = 3); Glu = 3.63 ± 0.7% (n = 3); Glu + PN = 2.4 ± 0.08% (n = 3); Glu + PN + Dil = 1.63 ± 0.3% (n = 3) (∗, p < 0.05 compared with basal). C, KCl-stimulated secretion. Both Cav1.2/DHPi cells and Cav1.3/DHPi cells demonstrated PN200-110-resistant insulin secretion in response to KCl stimulation. Cav1.2/DHPi: basal = 0.83 ± 0. 6% (n = 3); KCl = 24.3 ± 5% (n = 3); KCl + PN = 6.28 ± 0.6% (n = 3); KCl + PN + Dil = 1.91 ± 0.9% (n = 3). Cav1.3/DHPi: basal = 1.34 ± 0.13% (n = 3); KCl = 16.7 ± 1.3% (n = 3); KCl + PN = 8.1 ± 0.04% (n = 3); KCl + PN + Dil = 2.3 ± 0.1% (n = 3) (∗, p < 0.05 compared with basal). D, DHP-insensitive insulin secretion in Cav1.2/DHPi and Cav1.3/DHPi cells. In Cav1.3/DHPi cells, 42.4 ± 8.4% (n = 3) of the glucose-induced insulin secretion was resistant to 1 μM PN200–110, whereas no DHP-insensitive insulin secretion was detected in Cav1.2/DHPi cells (−0.06 ± 0.12%, n = 3, ∗∗, p < 0.01). With KCl stimulation, Cav1.3/DHPi supported a significantly higher level of DHP-insensitive insulin secretion (44.1 ± 0.05%, n = 3) than Cav1.2/DHPi (29.1 ± 6.5%, n = 3). DHP-insensitive secretion: percentage of secretion in 1 μM PN200-110 less basal divided by percentage of secretion without drug, less basal.
Characterization of Cav1.2/II-III and Cav1.3/II-III Cells.
The intracellular II-III loops of several Cav channels are known to bind to other signaling proteins, thus efficiently coupling Ca2+ influx to a cellular response (Sheng et al., 1994; Rettig et al., 1996; Grabner et al., 1999). Therefore, we hypothesized that overexpression of the Cav1.3 II-III loop should inhibit glucose-stimulated insulin secretion if an interaction between this domain and another protein is critical for this specific signaling pathway. To test this hypothesis, we created stable INS-1 cell lines that express the intracellular II-III loop of either Cav1.2 or Cav1.3 fused via the C terminus to GFP (Cav1.2/II-III and Cav1.3/II-III) (Fig.4A). Western blot analysis of Cav1.2/II-III and Cav1.3/II-III cells using anti-GFP antibodies detected proteins of ∼43 and ∼45 kDa, respectively, consistent with the expected molecular mass of each fusion protein (Fig. 4B). Neither protein was detected in untransfected INS-1 cells.
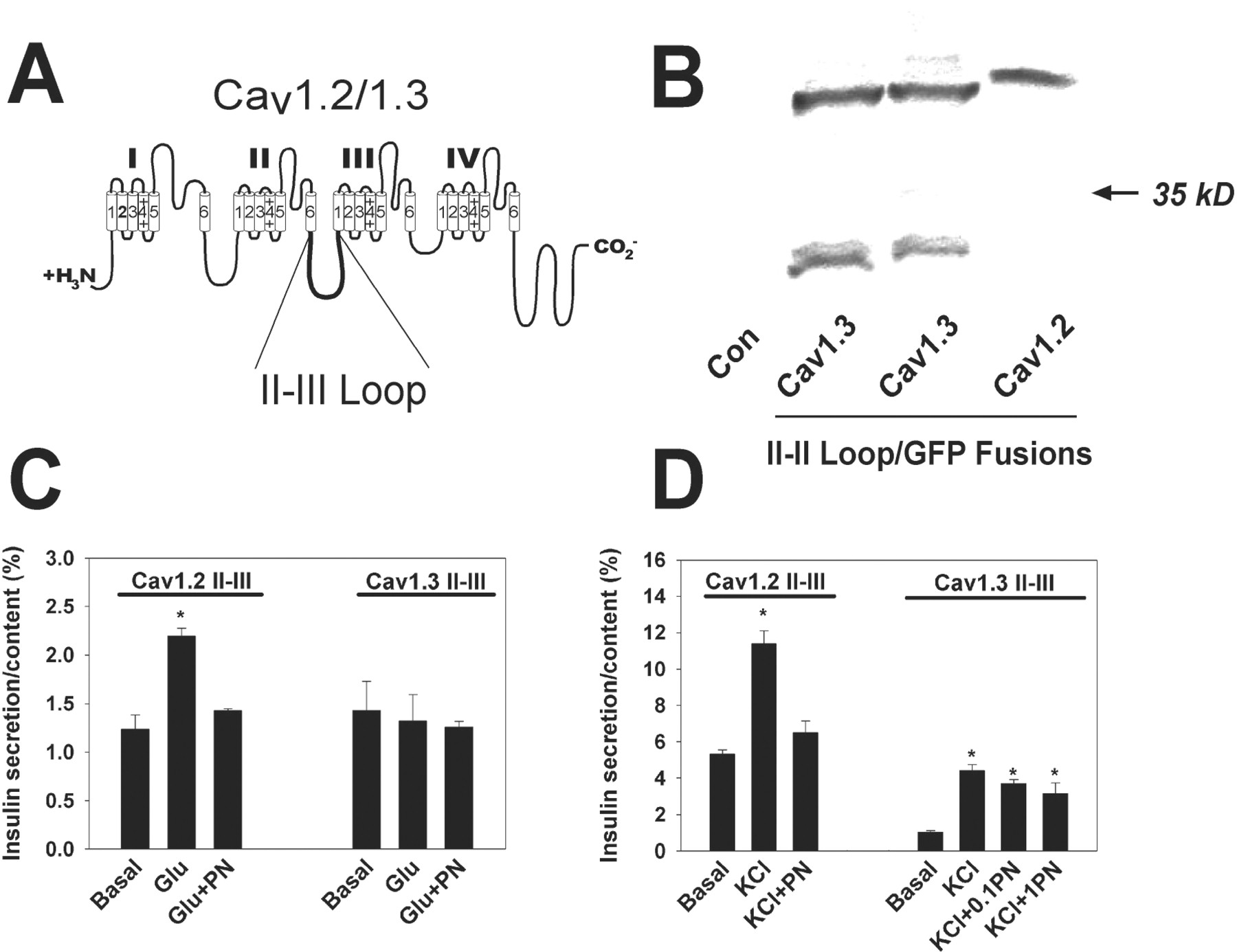
Insulin secretion in Cav1.2/II-III and Cav1.3/II-III cells. A, intracellular II-III loop of Cav1.2 and Cav1.3. Both Cav1.2/II-III and Cav1.3/II-III were subcloned into the EGFP vector such that GFP was fused to the C terminus of each and stably transfected into INS-1 cells. B, stable expression of Cav1.2 II-III or Cav1.3 II-III in INS-1 cells. Western blot of whole-cell lysates with anti-GFP antibodies detected proteins of 43 and 45 kDa corresponding to Cav1.3 II-III/GFP and Cav1.2 II-III/GFP, respectively. The lower molecular mass band from Cav1.3/II-III cells most likely represents a small amount of proteolytic cleavage of the fusion to generate a fragment containing GFP and little, if any, of the Cav1.3 II-III loop. C, glucose (11 mM; Glu) did not stimulate insulin secretion in Cav1.3/II-III cells (n = 3). In Cav1.2/II-III cells, glucose-stimulated insulin secretion was similar to untransfected cells (basal = 1.23 ± 0.15%; Glu = 2.2 ± 0.08%) and completely blocked by 0.1 μM PN200-110 (PN) (n = 3). D, both Cav1.2/II-III and Cav1.3/II-III cells secreted insulin in response to 50 mM KCl stimulation (KCl). KCl-stimulated secretion in Cav1.2/II-III cells was blocked by 0.1 μM PN200-110 (PN) (basal = 5.33 ± 0.24%; KCl = 11.4 ± 0.7%; KCl + PN = 6.51 ± 0.63% (n = 3)). KCl-stimulated secretion in Cav1.3/II-III cells was resistant to 0.1 μM (0.1 PN) and 1.0 μM PN200-110 (1PN) [basal = 1.03 ± 0.09%; KCl = 4.42 ± 0.32%; KCl + 0.1PN = 3.7 ± 0.21%; KCl + 1PN = 3.16 ± 0.57% (n = 3)] (∗, p < 0.05 compared with basal).
Insulin Secretion in Cav1.3/II-III and Cav1.2II-III Cells.
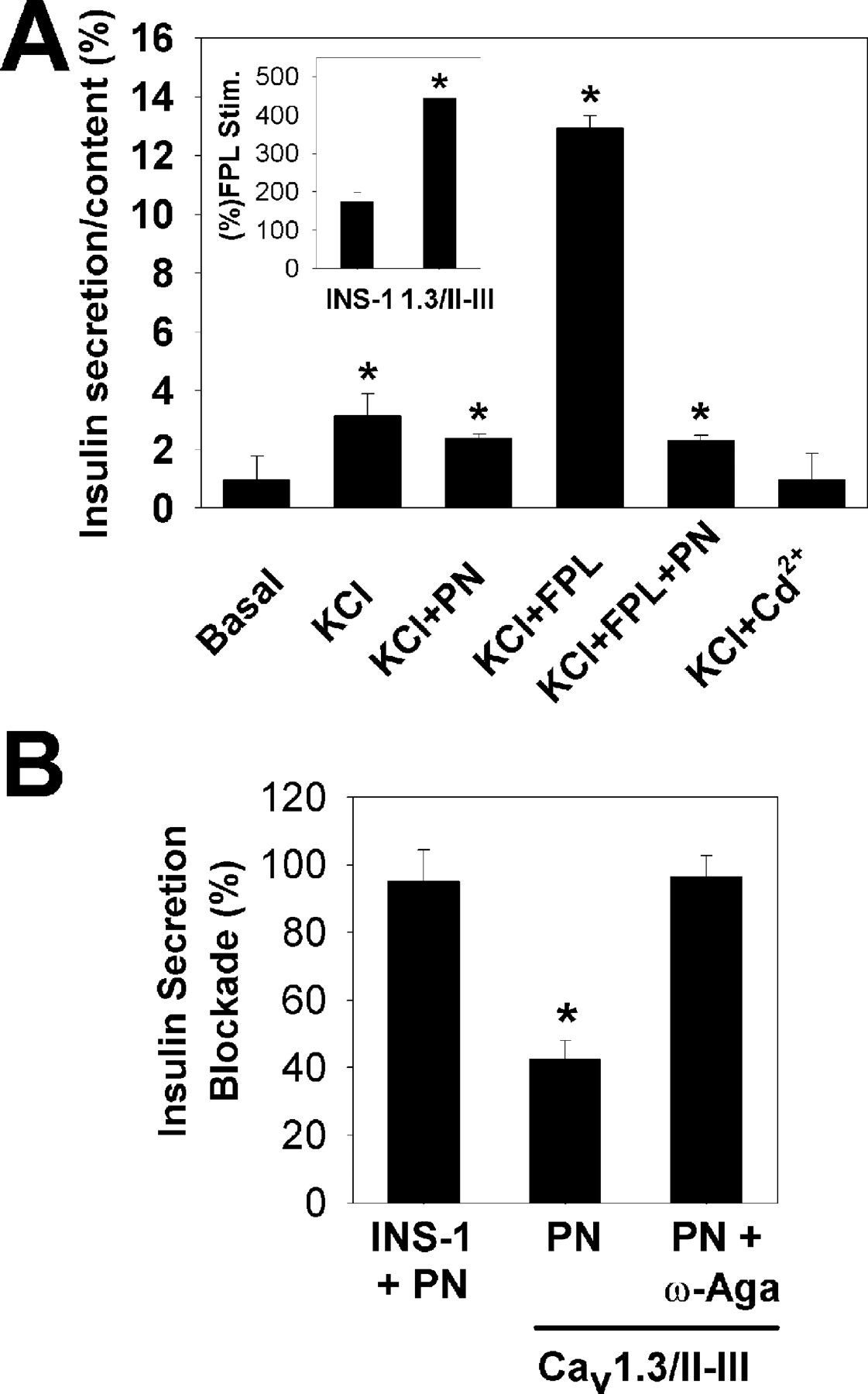
Fig. 4C shows that glucose-stimulated insulin secretion was maintained and completely inhibited by PN200-110 in Cav1.2/II-III cells. However, in Cav1.3/II-III cells, glucose did not stimulate insulin secretion. Upon KCl stimulation (Fig. 4D), Cav1.2/II-III cells again demonstrated normal insulin secretion that was completely blocked by PN200-110. However, KCl-stimulated insulin secretion by Cav1.3/II-III cells was sharply decreased compared with untransfected INS-1 or Cav1.2/II-III cells, and it was substantially resistant to block by PN200-110. Analysis ofIBa in Cav1.3/II-III cells detected a normal level of L-type channel activity (data not shown). Thus, we asked whether enhancing the Ca2+ influx via the endogenous L-type channels could increase KCl-stimulated secretion. Figure5A shows the effect of the L-type channel agonist FPL 64176 on KCl-stimulated insulin secretion in Cav1.3/II-III cells. As before, KCl evoked a modest level of insulin secretion, which was not sensitive to PN200-110. When FPL 64176 (10 μM) was applied to Cav1.3/II-III cells, KCl-stimulated secretion was sharply increased (>440%; Fig. 5A, inset). This increase was completely blocked by 1 μM PN200-110. In contrast, when FPL 64167 is applied to untransfected INS-1 cells, insulin secretion is increased by only 174% (Fig. 5A, inset). These data suggest that overexpression of the Cav1.3 II-III loop may spatially separate the endogenous Cav1.3 channels from the cell's secretory machinery. Furthermore, the DHP-resistant secretion evoked by KCl was completely blocked by 10 μM Cd2+ (Fig.5A), a nonselective Cav blocker (Hille, 1995), and by 1 μM ω-agatoxin IVA (Mintz et al., 1992), a specific Cav2.1 blocker (Fig. 5B).

Endogenous L-type Ca2+ channels are functional in Cav1.3/II-III cells. A, stimulation with 50 mM KCl (KCl) induces insulin secretion from Cav1.3/II-III cells, which is largely resistant to 1 μM PN200-110 (PN). FPL 64176 (10 μM; KCl + FPL) markedly potentiated KCl-induced insulin secretion in Cav1.3/II-III cells. The potentiation was blocked by 1 μM PN200-110 (KCl + FPL + PN). The DHP-resistant insulin secretion was completely blocked by 10 μM Cd2+ (KCl + Cd2+) (∗, p < 0.05 compared with basal). Inset, The increase in KCl-stimulated insulin secretion with 10 μM FPL 64176 in Cav1.3/II-III cells was greater (443 ± 1.5%) than in untransfected INS-1 cells (174 ± 25%) (∗,p < 0.05 compared with INS-1 cells). B, PN200-110 (1 μM) blocked virtually all KCl-stimulated insulin secretion in INS-1 cells (INS-1 + PN; 95 ± 9.3%), but only 42.5 ± 5.5% of KCl-stimulated secretion in Cav1.3/II-III cells (PN; ∗, p < 0.05). However, addition of both 1 μM PN200-110 and 1 μM ω-agatoxin IVA to Cav1.3/II-III cells completely blocked KCl-stimulated secretion (PN + ω-Aga; 96.4 ± 6.3).
Discussion
Cav1.3 Is Preferentially Linked to Glucose-Stimulated Insulin Secretion in INS-1 Cells.
By using INS-1 cells that stably express DHP-insensitive Cav1.2 or Cav1.3 channels, we were able to pharmacologically isolate these two distinct channels subtypes and to study the contribution of each to insulin secretion. The Cav1.2 (rat brain; Snutch et al., 1991) and Cav1.3 (human brain; Williams et al., 1992) clones used in this study are nearly identical to the respective channel subtypes isolated from human pancreatic islets (Seino et al., 1992). Electrophysiological characterization of both Cav1.2/DHPi and Cav1.3/DHPi cells demonstrated that the mutant channels were functionally expressed, and that total Ca2+ channel activity was not grossly different from untransfected INS-1 cells (Fig.2). Because only the pore-forming α1 subunits were used in the construction of the Cav1.2/DHPi and Cav1.3/DHPi cell lines, it is likely that the endogenous auxiliary channel subunits were limiting, and thus controlled the number of Cav α1 subunits expressed at the cell surface. Using this model system, we found that both channel subtypes were able to mediate DHP-resistant insulin secretion in response to KCl depolarization, whereas only Cav1.3 was able to mediate DHP-resistant secretion in response to glucose. Expression of the drug-resistant mutants did not seem to grossly disrupt the endogenous secretory pathway. In both Cav1.2/DHPi and Cav1.3/DHPi cells, the majority of secretion was blocked by low concentrations of PN200-110, indicating the contribution of endogenous L-type channels to excitation-secretion coupling. A previous study attempted to assess the roles of both Cav1.2 and 1.3 channels in mediating insulin secretion using Cav1.3 knockout mice (Platzer et al., 2000), but that study did not indicate a role for Cav1.3 in insulin secretion. However, a subsequent article suggested that the pancreatic β-cells of the mouse strain used to generate the Cav1.3 knockouts did not express Cav1.3 (Barg et al., 2001). Understanding how Cav1.3 is coupled to insulin secretion is of therapeutic interest because the predominant Ca2+channel in human pancreatic β cells is Cav1.3 (Seino et al., 1992). This study also suggests that drug-insensitive channels may facilitate study of channel regulation or other channel-mediated events in the native cell type or tissue.
Intracellular II-III Loop of Cav1.3 Plays a Critical Role in Glucose-Stimulated Insulin Secretion.
We further demonstrated the critical role of Cav1.3 in glucose-stimulated insulin secretion using INS-1 cells that stably express the intracellular II-III loop of either Cav1.2 or Cav1.3. Cav1.2/II-III cells were not different from untransfected INS-1 cells in both KCl- and glucose-stimulated insulin secretion. However, the endogenous L-type channels in Cav1.3/II-III cells seemed to be largely uncoupled from insulin secretion. This uncoupling completely abolished insulin secretion in response to glucose, and markedly reduced KCl-stimulated secretion. The observation that 1 μM ω-agatoxin IVA could inhibit the DHP-resistant fraction of KCl-stimulated insulin secretion in these cells suggests that the Cav2.1 channels endogenous to INS-1 cells can effectively couple to KCl- but not glucose-stimulated insulin secretion. However, the endogenous L-type channels in Cav1.3/II-III cell are functional because in the presence of FPL 64176, KCl-stimulated secretion is restored to levels similar to untransfected INS-1 cells. We speculate that the increased Ca2+ influx induced by FPL 64176 compensates for a greater distance between the channel and the Ca2+ sensor of the secretory machinery. A similar mechanism was proposed to explain inhibition of evoked neurotransmitter release upon injection of a peptide corresponding to the II-III loop of Cav2.2 into the presynaptic cell of a cultured synapse preparation (Rettig et al., 1997). This inhibition was reversed by increasing the extracellular Ca2+ concentration. This result suggests that the intracellular II-III loop of Cav1.3 plays a prominent role in linking the Cav1.3 channel specifically to glucose-stimulated secretion, and that overexpressed Cav1.3 II-III loop competitively inhibits a key protein-protein interaction. A binding partner for the Cav1.3 II-III loop has not yet been clearly identified. Syntaxin has been shown to interact with the II-III loop of Cav1.2 (Wiser et al., 1999) and to colocalize with Cav1.3 channels in pancreatic β-cells (Yang et al., 1999). However, neither of these studies specifically examined the role of syntaxin/L-type channel interactions in glucose-stimulated insulin secretion. Because we found that Cav1.3, but not Cav1.2, can contribute to glucose-induced secretion in INS-1 cells, our results suggest that L-type channel/syntaxin interactions may not be sufficient to efficiently couple glucose-induced membrane depolarization to insulin secretion. The II-III loops of the channels used in this study are only 43% identical, and the II-III loop of Cav1.3 contains two consensus protein kinase A phosphorylation sites not present in Cav1.2. In addition, the Cav1.3 II-III loop contains two potential SH-3 domain binding sites (PXXP motifs; Mayer, 2001) that are not conserved in Cav1.2. Identification of the protein(s) that interacts with the II-III loop of Cav1.3 to mediate this specific coupling will give further insight into the mechanism of glucose-stimulated insulin secretion.
Potential Mechanism for Specificity of Cav1.3 Coupling to Insulin Secretion.
Although interactions between SNARE proteins and voltage-gated Ca2+ channels are clearly important for vesicular exocytosis of neurotransmitters, it seems unlikely that such interactions alone could account for the preferential linkage of Cav1.3 to glucose-stimulated insulin secretion that we observed. Because glucose induces smaller depolarization of membrane potential in INS-1 cells than 30 mM KCl (Kennedy et al., 1998; Antunes et al., 2000), one potential explanation for our results is that Cav1.3 channels are activated at more negative potentials than Cav1.2 channels. Several studies have concluded that various splice variants of Cav1.3 activate at more negative potentials than Cav1.2 (Koschak et al., 2001; Scholze et al., 2001; Xu and Lipscombe, 2001). However, the voltage dependence of activation for the Cav1.3 and Cav1.2 clones used in this study are virtually identical when measured in human embryonic kidney 293 cells (Bell et al., 2001; Gage et al., 2002). Furthermore, we have not observed any significant difference in the current-voltage relationship of the DHPi fraction of current between the Cav1.2/DHPi and Cav1.3/DHPi cell lines using conventional whole recordings (G. Liu and G. H. Hockerman, unpublished observations). However, we cannot rule out the possibility that glucose metabolism by intact INS-1 cells can shift the activation threshold of Cav1.3 to more negative potentials (Smith et al., 1989). Therefore, it is unlikely that differences in Cav1.2 and Cav1.3 channel activation threshold can explain our results unless this property of these channels is differentially modulated in intact INS-1 cells.
An alternative explanation for our results is that Ca2+ influx via Cav1.3 channels is specifically coupled to an intracellular signaling mechanism that is not activated by Ca2+ influx via Cav1.2. Although INS-1 cells retain glucose-induced insulin secretion, RINm5F cells, another rat β-cell line, do not, although both cell lines express Cav1.3 (Safayhi et al., 1997; Horvath et al., 1998). Interestingly, RINm5F cells do not express the endoplasmic reticulum Ca2+ release channel RYR2, whereas INS-1 cells do (Gamberucci et al., 1999). The role of Ca2+ release from internal stores in insulin secretion is not clear. However, a recent report concluded that L-type Ca2+ channel activity initiates Ca2+ induced Ca2+ release via RYR2 in mouse β-cells (Lemmens et al., 2001). The L-type Ca2+ channel from skeletal muscle, Cav1.1, is coupled to RYR1 in the sarcoplasmic reticulum, via the intracellular II-III loop (Grabner et al., 1999). Thus, it is attractive to speculate that a similar coupling between Cav1.3 and intracellular Ca2+ release may mediate a specific role for Cav1.3 in glucose-stimulated insulin secretion.
In summary, we have shown that Cav1.3 is preferentially linked to glucose-stimulated insulin secretion in INS-1 cells and that the intracellular loop between homologous domains II and III is likely involved in mediating this specificity. It will be of interest to investigate the mechanism for this specificity, and specifically, to determine whether the II-III loop of Cav1.3 interacts with other proteins in this process.
Acknowledgments
PN200-110 was the gift of Novartis (Basel, Switzerland). The human brain Cav1.3 clone was provided by SIBIA Neurosciences (San Diego, CA), now Merck Research Laboratories. We thank Dr. Ming Li for the gift of INS-1 cells and helpful discussions regarding their culture.
Footnotes
-
This work was supported by Grant 1119990378 from the American Diabetes Association (to G.H.H.).
-
DOI: 10.1124/jpet.102.046334
- Abbreviations:
- VDCC
- voltage-dependent calcium channel
- DHP
- dihydropyridine
- DHPi
- dihyrdropyridine-insensitive
- SNARE
- solubleN-ethylmaleimide-sensitive factor attachment protein receptor
- GFP
- green fluorescent protein
- RT-PCR
- reverse transcription-polymerase chain reaction
- EGFP
- enhanced green fluorescent protein
- PCR
- polymerase chain reaction
- bp
- base pair(s)
- KRBH
- Krebs-Ringer-bicarbonate HEPES buffer
- IBa
- barium current
- Received October 30, 2002.
- Accepted December 30, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}