


Abstract
Survivin is a novel member of the inhibitor of apoptosis protein (IAP) family. Here we report that the chemotherapeutic drug doxorubicin, a DNA-damaging agent, activates a p53-survivin signaling pathway inducing cell cycle arrest and apoptosis in childhood acute lymphoblastic leukemia (ALL). Treatment of wild-type (wt) p53 ALL cells (EU-3 cell line) with doxorubicin caused accumulation of p53, resulting in dramatic down-regulation of survivin, depletion of cells in G2/M, and apoptosis (increased sub-G1compartment). In contrast, doxorubicin treatment of mutant (mut) p53 cells (EU-6/ALL line) up-regulated survivin and induced G2/M arrest without inducing apoptosis. However, treating EU-6 with anti-survivin antisense resensitized these cells to doxorubicin, resulting in apoptosis. With a p53-null cell line (EU-4), although doxorubicin treatment arrested cells in G2/M, survivin expression was unchanged, and cells underwent only limited apoptosis. However, re-expression of wt-p53 in EU-4 cells could restore the doxorubicin-p53-survivin pathway, resulting in significantly decreased survivin expression and increased apoptosis in these cells after doxorubicin treatment. Following cotransfection of p53-null EU-4 cells with survivin promoter-luciferase constructs and either wt-p53 or different mut-p53 expression vectors, wt-p53 inhibited survivin promoter activity; p53-mediated inhibition could be abrogated by overexpression of murine double minute2 (MDM2) protein. Together, these studies define a novel p53-survivin signaling pathway activated by DNA damage that results in down-regulation of survivin, cell cycle arrest, and apoptosis. Furthermore, our data indicate that loss of wt-p53 function in tumor cells may contribute to up-regulation of survivin and resistance to DNA-damaging agents.
Apoptosis or programmed cell death is activated through signal transduction in response to a variety of stimuli including DNA-damaging agents such as ionizing radiation and doxorubicin (Raff, 1992; Thompson, 1995). Regulation of apoptosis is delicately balanced by signaling pathways between apoptosis-promoting factors such as p53 and caspases, and antiapoptotic factors such as Bcl-2 and MDM2. Several lines of evidence have shown that the functional interactions between apoptosis-promoting factors and antiapoptotic factors play important roles in the control of cell growth and apoptosis. For example, p53 regulates the expression of MDM2 and Bcl-2 (Barak et al., 1993; Miyashita et al., 1994b), whereas both MDM2 and Bcl-2 inhibit p53-mediated apoptosis (Chiou et al., 1994; Chen et al., 1996). Recently, a group of highly conserved genes termed the inhibitor of apoptosis protein (IAP) family has been recognized. There are currently six human IAP family members, c-IAP1, c-IAP2, XIAP, NAIP, survivin and livin. All of the human IAP family members, with the exception of NAIP, have been shown to inhibit apoptosis by blocking the activation or activity of specific caspases (Rothe et al., 1995; Duckett et al., 1996; Liston et al., 1996;Ambrosini et al., 1997; Kasof and Gomes, 2001).
Survivin is abundantly expressed during normal development and in neoplastic cells but is rarely present in adult tissues, suggesting that deregulation of survivin expression may be involved in tumorigenesis (Ambrosini et al., 1997; Tamm et al., 1998; Carter et al., 2001). Regulation of survivin expression is cell cycle-dependent. In proliferating cells, survivin is expressed at high levels in G2/M phase and is rapidly down-regulated after cell cycle arrest in G1 phase (Li et al., 1998). The G2-specific transcription is commonly due to periodic occupation by specific repressor of regions in the gene promoter called cell cycle-dependent elements (CDE) and cell cycle genes homology regions (CHR). Proteins binding to the CDE/CHR regions in G1 result in a phase-specific repression of activated transcription (Zwicker et al., 1995). It has been demonstrated that cell cycle-dependent expression of survivin is modulated by the CDE/CHR regions in the survivin promoter (Li et al., 1998).
Doxorubicin, a widely used chemotherapeutic drug for treatment of childhood acute lymphoblastic leukemia (ALL) and other neoplasms, is reported to inhibit cell growth in MCF-7 breast cancer cells (Fornari et al., 1994, 1996), and induce cell cycle arrest and cell death in neuroblastoma and neuroepithelioma cells (Di Bartolomeo et al., 2000). Although studies have demonstrated that cell cycle arrest and apoptosis induced in neurotumors by doxorubicin is associated with the expression of p53 (Di Bartolomeo et al., 2000), the molecular mechanism of p53-mediated signaling in these events is not fully understood. Here, we have used ALL cell lines to evaluate the interaction between p53 and survivin in regulating cell cycle and apoptosis following exposure to doxorubicin. We have identified a marked down-regulation of survivin, depletion of G2/M cells, and an increase in sub-G1 cells (apoptosis) in wt-p53+ cells after doxorubicin treatment. In contrast, similar treatment of mut-p53+ cells resulted in up-regulation of survivin and G2/M arrest without apoptosis, although apoptosis was restored in the presence of anti-survivin antisense. Similarly, apoptosis was minimal in p53-null cells following doxorubicin but could be greatly enhanced by transfection-mediated expression of wt-p53 in these cells. These studies characterize a novel p53-survivin signaling pathway activated by DNA damage that results in down-regulation of survivin, cell cycle arrest, and apoptosis. Our results further suggest that loss of wt-p53 function in tumor cells augments resistance to DNA-damaging agents by up-regulating survivin.
Materials and Methods
Cell Lines and Cell Treatment.
The ALL cell lines EU-3, EU-4, and EU-6 were established in our laboratory from three pediatric ALL patients during relapse, respectively. Cultured cells resembled the primary leukemic cells in immunophenotypes (B-cell precursor-ALL) and molecular changes such as p53 mutations (Zhou et al., 1995; Findley et al., 1997). The three lines were chosen for the present study due to their distinct p53 phenotypes: EU-3 expresses wt-p53, whereas EU-6 has a p53 point mutation in position 273; EU-4 does not express p53. These lines were grown in standard culture medium (RPMI 1640 medium containing 10% FBS, 2 mM l-glutamine, 50 U of penicillin, and 50 μg/ml streptomycin) at 37°C in 5% CO2in air. Exponentially growing cells were treated with doxorubicin at 0.1 μg/ml for the indicated times at 37°C and then harvested for total RNA and protein extraction. For cell cycle assay, cell lines were treated with doxorubicin or one of the following reagents: 400 μM mimosine for 24 h to arrest cells in G1, 2 mM thymidine for 16 h to arrest cells in S, or 0.04 μg/ml nocodazole for 24 h to arrest cells in G2/M phase (Hwang et al., 1995). For antisense treatment, a 1 μM concentration of a 20-mer oligonucleotide 5′-CCCAGCCTTCCAGCTCCTTG-3′ antisense targeting the 232–251 nucleotide site in survivin mRNA as described previously (Olie et al., 2000) was delivered in the form of complex with Lipofectin (Invitrogen, Carlsbad, CA).
RNA Preparation and Northern Blot Analysis.
Total cellular RNA was extracted from cell lines before and after treatment with an RNA isolation kit (Biotecx Laboratories, Inc., Houston, TX). Total RNA (5–10 μg per lane) was electrophoresed using a 1% agarose/6% formaldehyde gel and transferred to a nylon filter. To detect survivin, p53, and p21/WAF-1 expression, we used cDNA probes for the corresponding genes. Probes were prepared by a randomized-labeling approach using [α-32P]dCTP. Hybridization was performed in 50% (v/v) formamide/5× SSC/1% SDS/5× Denhardt's/20% dextran sulfate/100 μg/ml sheared salmon sperm DNA at 42°C for 16 h. A final wash was carried out in 1× SSC/0.1% SDS at 65°C for 30 min. The membrane was checked for equal loading by rehybridization with a human β-actin probe. After washing, the filter was autoradiographed for 24 h. Levels of mRNA expression pre- and post-treatment with anti-survivin antisense were compared by densitometry.
Western Blot Assay.
Cells were lysed in a buffer composed of 150 mM NaCl, 50 mM Tris (pH 8.0), 5 mM EDTA, 1% (v/v) nonidet P-40, 1 mM phenylmethylsulfonyl fluoride, 20 μg/ml aprotinin, and 25 μg/ml leupeptin for 30 min at 4°C. After clarification, equal amounts of protein extracts were resolved by SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose filter. After blocking with buffer containing 20 mM Tris-HCl (pH 7.5) and 500 mM NaCl, 5% nonfat milk for 1 h at room temperature, the filter was incubated with specific antibodies to caspase-3 and poly(ADP-ribose) polymerase (PARP) for 1 h at room temperature followed by horseradish peroxidase-labeled secondary antibody. Blots were developed using a chemiluminescent detection system (ECL, Amersham Biosciences UK Ltd., Little Chalfont, Buckinghamshire, UK).
Flow Cytometry.
Flow cytometry was performed to analyze cell cycle position and annexin-V staining. Cells were treated with doxorubicin or cycle inhibitors as described above, and then were pulsed with 30 μM bromodeoxyuridine (BrdUrd; Sigma B5002) for 1 h. Cells (3 × 106) were collected, washed once with phosphate-buffered saline (PBS), and fixed in 70% ethanol (1 h, 4°C). Cells were then washed twice in PBS containing 0.5% Tween 20 and incubated in 1 ml of 0.1 N HCl/0.7% Triton X-100 for 10 min at 4°C. After washing with 5 ml of PBS, cells were heated to 97°C for 10 min in deionized/distilled water and then chilled on ice for 15 min. Cells were washed twice with HBT buffer (PBS/0.5% Tween 20/5% FBS) and incubated with 100 μl of a 1:20 dilution of anti-BrdUrd monoclonal antibody-fluorescein isothiocyanate (BD Biosciences PharMingen, San Diego, CA) in HBT (12.5 μg/ml) for 1 h at room temperature with occasional mixing. Then 1 ml of HBT was added and cells were spun down. The samples were washed with HBT and finally suspended in 0.5 ml of 25 μg/ml propidium iodide (PI) and 40 μg/ml RNase A. After incubating at 4°C for at least 0.5 h, the samples were analyzed using the FACScan (BD Biosciences) and WinList software (Verity Software House Inc., Topsham, ME). For annexin-V staining, cells with or without treatment were washed once with PBS and stained with fluorescein isothiocyanate-annexin-V and PI according to the manufacturer's instructions. Stained cells were analyzed as above.
Transfection and Luciferase Assay.
p53-null EU-4 cells were transiently cotransfected by electroporation with the survivin promoter-luciferase constructs as described previously (Li and Altieri, 1999) and different forms of p53 (wild type and mutants with point mutations at positions 143, 175, 248, and 273, respectively) or MDM2 expression vectors (provided by Dr. B. Vogelstein, Johns Hopkins University, Baltimore, MD). Briefly, 1 × 107 cells in exponential growth were mixed with the corresponding survivin promoter-luciferase construct plus different doses of p53 and/or MDM2 expression vectors and electroporated at 300 V, 950 μF using a Gene Pulser II System (Bio-Rad, Hercules, CA). Transfected cells were resuspended in 10 ml of RPMI 1640 medium containing 10% FBS. At 48 h post-transfection, cell extracts were prepared with 1× lysis buffer (Promega, Madison, WI), and then 20-μl aliquots of the supernatant were mixed with 100 μl of luciferase assay reagent (Promega) and analyzed on a microplate luminometer (Turner Designs, Sunnyvale, CA). Luciferase activity was normalized to β-galactosidase activity as an internal control. For stable p53 gene transfection, EU-4 cells in exponential growth were transfected with either wt- or mut-p53 expression vectors by electroporation. The cells were seeded 48 h post-transfection into culture dishes for the selection of geneticin (G418)-resistant colonies. Colonies were grown in methylcellulose medium containing G418 (500 μg/ml) for 2 to 3 weeks, and clones were picked and grown in RPMI 1640 medium with or without G418 for the duration of the experiments.
DNA Fragmentation Assay.
In addition to the flow-cytometric method, apoptosis was determined by a DNA fragmentation assay, using p53-transfected EU-4 cells with or without doxorubicin treatment. DNA was prepared from each sample using a Puregene DNA isolation kit (Gentra Systems, Inc., Minneapolis, MN). DNA (5 μg) was resolved by electrophoresis on a 1.5% agarose gel containing ethidium bromide, and DNA fragments were visualized under UV light.
Results
p53-Dependent Cell Cycle Response of ALL to Doxorubicin.
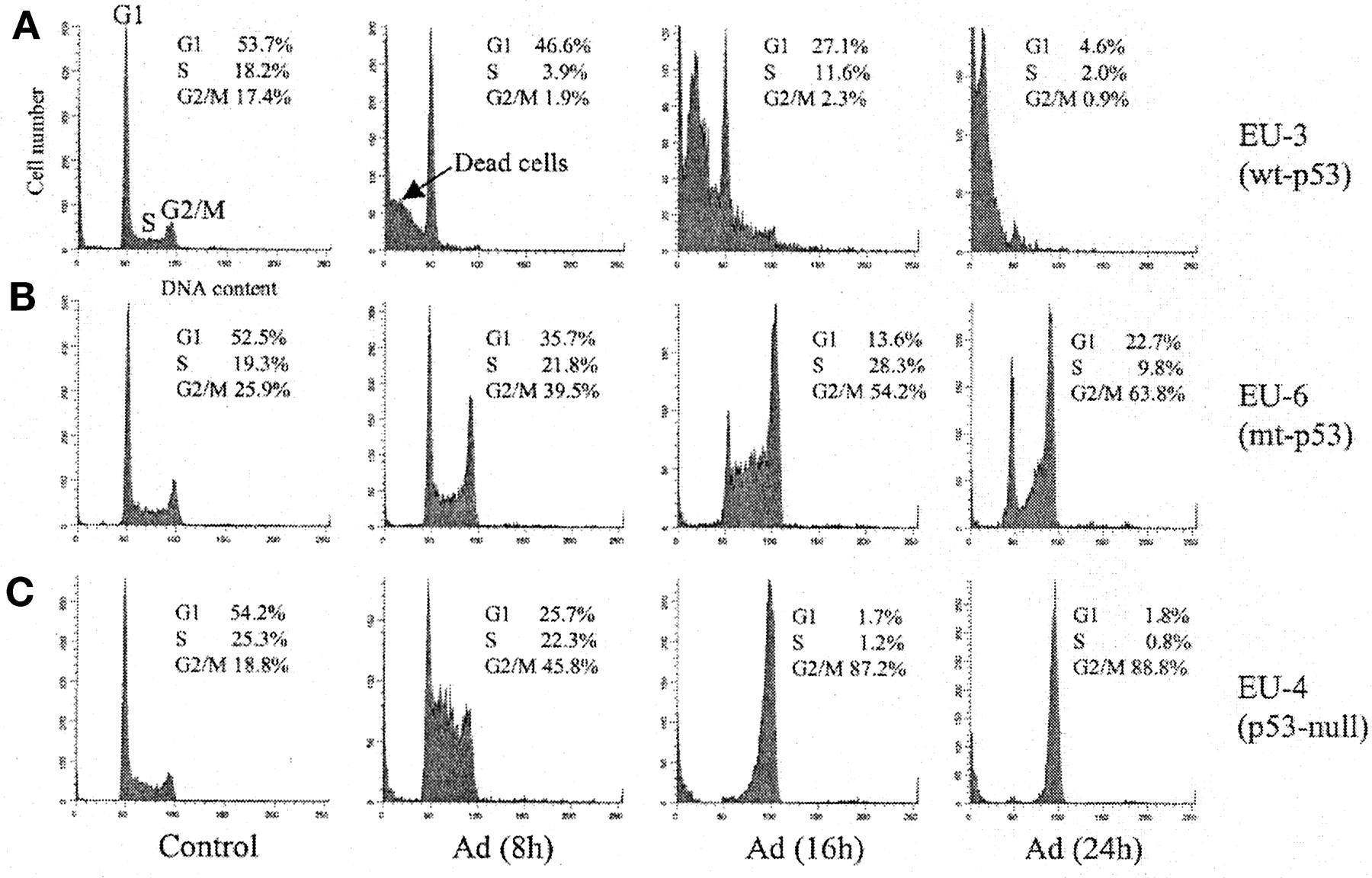
To investigate the cell cycle regulation and sensitivity of ALL cells with different p53 phenotypes in response to doxorubicin treatment, wt-p53+ EU-3 cells, mut-p53+ EU-6 cells, and p53-null EU-4 were treated with 0.1 μg/ml doxorubicin, and their cell cycle distributions were subsequently determined by flow cytometry at various time points following exposure to doxorubicin (Fig.1). BrdUrd incorporation and PI staining detected S-phase cells undergoing DNA synthesis and total DNA content, respectively. As demonstrated in Fig. 1A, EU-3 cells showed a rapid G2/M depletion and extensive cell death (sub-G1 DNA content indicated by an arrow in Fig.1A) at 8 h after doxorubicin treatment. Over 90% of EU-3 cells died after doxorubicin treatment for 24 h as indicated in Fig. 1A or as detected by trypan blue staining (data not shown). In contrast, both mut-p53+ EU-6 and p53-null EU-4 cells appeared to be severely impaired in their ability to arrest in G1. Instead, these cells clearly showed an accumulation in G2/M without cell death (EU-6 cells) or only a slight increase in dead cells (i.e., sub-G1 DNA content) for EU-4 after 24 h of doxorubicin (Fig. 1, B and C).

Cell cycle analysis of ALL cells with different p53 phenotypes after treatment with doxorubicin. wt-p53+ EU-3, mut-p53+ EU-6, and p53-null EU-4 were incubated with doxorubicin (Adriamycin, Ad) at 0.1 μg/ml for the indicated time and analyzed by flow cytometry using BrdUrd (for S phase) and PI (for total DNA content) staining.
Specific Inhibition of Survivin Expression by Doxorubicin in wt-p53+ ALL.
To evaluate a possible interaction between p53 and survivin in doxorubicin-induced cell death, we examined the expression of p53 and survivin in doxorubicin-treated EU-3, EU-6, and EU-4. Additionally, we examined transcription of the p21/WAF-1gene, which is involved in p53-mediated cell cycle arrest (El-Deiry et al., 1993). We found that doxorubicin treatment could induce expression of p53 and p21/WAF-1 in wt-p53+ EU-3 cells accompanied by a dramatic down-regulation of survivin mRNA expression (Fig.2A, left panel). Induction of p53 expression occurred as early as 30 min (not shown) and reached the highest level at 4 to 8 h after doxorubicin treatment. At this time, cells were arrested in G1, and a 5- to 30-fold decrease in survivin expression was observed. At 12 h post-treatment, survivin mRNA was undetectable. In contrast, the expression of p53 mRNA in mut-p53+ EU-6 cells was unchanged, and a significant up-regulation of survivin mRNA was detected at 12 h post-treatment (Fig. 2A, middle panel).

Expression of p53, p21/WAF-1, and survivin mRNA in ALL cells with different p53 phenotypes following exposure to doxorubicin and agents for cell cycle synchronization. A, Northern blot analysis for expression of p53, p21/WAF-1, and survivin in wt-p53+ EU-3, mut-p53+ EU-6, and p53-null EU-4 cells treated with doxorubicin (0.1 μg/ml) for the indicated times. Actin serves as control for equal RNA loading and RNA integrity. Labels below each blot show cell cycle profiles at each time point. B, expression of survivin mRNA and cell cycle profiles in EU-3, EU-6, and EU-4 cells treated either with 400 μM mimosine (Mim) to arrest cells in G1, 2 mM thymidine (Thy) to arrest cells in S, or 0.04 μg/ml nocodazole (Noc) to arrest cells in G2/M phase.
To determine whether this differential regulation of survivin transcription in wt-p53+ versus mut-p53+ cells was due to the specific phase (G1 versus G2/M of cycle) in which the cells arrested, we treated EU-3 and EU-6 cells with mimosine, thymidine, and nocodazole, which arrest cells in G1, S, and G2/M phases, respectively. As shown in Fig. 2B, although increased survivin expression was observed in both wt-p53+ EU-3 and mut-p53+ EU-6 treated with nocodazole (G2/M arrest), significant reduction of survivin mRNA following mimosine (G1arrest) and thymidine (S-phase arrest) was observed in EU-3 cells but not in EU-6 cells. Moreover, although similar patterns of cell cycle arrest were observed in p53-null EU-4 cells treated either with doxorubicin or with synchronization agents, the expression of survivin was unchanged following these treatments (Fig. 2, A and B, right panel). These data suggest that down-regulation of survivin in G1- and S-phase arrested cells is regulated by wt-p53 and is accompanied by cell death.
Down-Regulation of Survivin by Doxorubicin-Induced wt-p53 Accumulation Triggers Apoptosis.
To determine whether cell death following down-regulation of survivin in wt-p53+ ALL cells is due to apoptosis, we evaluated the activation of caspase-3 and cleavage of death substrate PARP in doxorubicin-treated cells, as well as the binding of annexin-V to these cells. We found that activation of caspase-3 and cleavage of PARP in wt-p53+ EU-3 cells was detectable at 8 h and pronounced at 16 h after doxorubicin treatment (Fig.3A, left panel). In contrast, these markers of apoptosis were not observed in mut-p53+ EU-6 cells even 24 h post-treatment (Fig. 3A, middle panel). Consistent with these observations, a significant percentage of EU-3 cells (from 19% to 59%) but not EU-6 cells showed annexin-V binding post-treatment (Fig.3B). With p53-null EU-4 cells, activation of caspase-3 and cleavage of PARP were not detected, although a small subset of cells (from 11% to 20%) underwent apoptosis as detected by flow cytometry (Fig. 3, A and B, right panel).

Time course of apoptosis in wt-p53+ EU-3, mut-p53+ EU-6, and p53-null EU-4 cells treated with doxorubicin. Apoptosis was determined either by Western blot assay for activation of caspase-3 and cleavage of death substrate PARP (A), or by flow cytometry to detect annexin-V staining (B). Cells for Western blot assay were treated with doxorubicin (0.1 μg/ml) for the indicated time, and cells for annexin-V staining were similarly treated for 16 h. Western blotting and flow cytometry were performed as described underMaterials and Methods.
Survivin Targeting by an Antisense Approach Sensitizes mut-p53+ EU-6 Cells to Doxorubicin.
To further evaluate the role of survivin in doxorubicin-induced apoptosis, we treated mut-p53+ EU-6 cells with doxorubicin in the presence of survivin antisense oligonucleotides. As shown in Fig. 4A, up-regulation of survivin gene expression in doxorubicin-treated EU-6 cells was effectively inhibited by anti-survivin antisense, resulting in increased apoptosis: 30% of survivin-antisense-treated cells underwent apoptosis compared with 13% of cells treated with a scrambled oligonucleotide (Fig. 4b).

Anti-survivin antisense sensitizes mut-p53+ EU-6 cells to doxorubicin. Cells were treated with 0.1 μg/ml doxorubicin (Ad) plus Lipofectin and either 1 μM survivin antisense (AS) or scrambled oligonucleotide control (SC), and harvested after a 16-h incubation. A, total RNA was extracted and analyzed for survivin mRNA expression by Northern blot (right). Densitometric scan data for survivin mRNA bands in Northern blots were plotted in the left panel. B, cells treated as described above were analyzed for apoptosis by flow cytometry.
Expression of wt-p53 in p53-Null EU-4 Cells Restores the Doxorubicin-p53-Survivin Signaling Pathway and Sensitizes Cells to Doxorubicin.
To test our hypothesis that expression of wt-p53 in doxorubicin-treated, p53-null EU-4 cells can restore the p53-survivin pathway and induce apoptosis, we transfected wt-p53 and different mut-p53 expression plasmids into EU-4 cells. Cell clones with stably transfected p53 genes were selected with G418, and the expression of p53 mRNA was determined by Northern blotting (Fig.5A). When p53-transfected EU-4 cells were treated with doxorubicin, survivin expression significantly decreased in wt-p53+ EU-4 cells but not in EU-4 cells expressing either theneo gene control or the p53-273 mutant (Fig. 5B). Furthermore, transfected wt-p53+ but not control or mut-p53+ EU-4 cells underwent apoptosis (as detected by DNA fragmentation assay) after doxorubicin treatment (Fig. 5C).

Stable expression of transfected (ectopic) p53 constructs and their effects on the expression of survivin and apoptosis induced by doxorubicin treatment. A, p53-null EU-4 cells were transfected by electroporation with expression vectors containing cDNA for wt-p53 and p53-248 (with 1.8-kb cDNA insert), p53-175 and p53-273 (with 1.3-kb cDNA insert), and with empty neo gene vectors. Clones that stably expressed the transfected cDNA were selected by G418, and mRNA was detected by Northern blotting. EU-3 cells treated with doxorubicin for 5 h served as a positive control for expression of endogenous (native) p53. B, expression of survivin mRNA was detected by Northern blot in EU-4 cells transfected with wt-53, p53-273, and neo after treatment with 0.1 μg/ml doxorubicin for 8 h (T) as compared with untreated control (C). C, DNA fragmentation in p53-transfected EU-4 cells was assayed for apoptosis in the presence (T) and absence (C) of 0.1 μg/ml doxorubicin for 24 h.
wt-p53 Protein Represses Survivin Promoter Activity.
To investigate whether wt-p53 could directly repress survivin expression, we performed survivin promoter-luciferase reporter gene assays (Fig.6). Cotransfection of p53-null EU-4 cells with survivin promoter-luciferase construct pLuc-1430 together with wt-p53 or mut-p53 expression vectors resulted in significant down-regulation of survivin promoter activity by wt-p53 but not by mut-p53 constructs compared with the neo gene control (Fig.6A). Moreover, the inhibitory effect of wt-p53 on the survivin promoter could be reversed by overexpression of MDM2 protein in a dose-dependent manner (Fig. 6B). To map the survivin promoter region responsible for the inhibitory effect of wt-p53 protein, we cotransfected a series of truncated survivin promoter-luciferase constructs together with wt-p53 and p53-273 mutant expression vectors into p53-null EU-4 cells and measured luciferase activity. As shown in Fig. 6C, wt-p53 (but not mut-p53 and neo control) transfectants showed inhibition of survivin promoter activity. Moreover, the survivin promoter region responsible for inhibition of survivin expression by wt-p53 protein is apparently located in the 230-bp basic core promoter region, since the pLuc-230 construct was as efficiently repressed by wt-p53 as the longer survivin promoter-luciferase constructs tested (Fig. 6C).

Effect of p53 on survivin promoter activity in transient transfection assays. A, 10 μg of survivin promoter-luciferase construct (pLuc-1430) was cotransfected into p53-null EU-4 cells with 10 μg of various p53 expression vectors (wild type or mutants with mutation at codon 143, 175, 248, or 273) or empty neo vector, respectively, by electroporation. Electroporation was performed at 300 V and 950 μF. At 48 h post-transfection, cells extracts were analyzed for luciferase activity assay as described under Materials and Methods. B, 10 μg of pLuc-1430 was cotransfected into EU-4 cells with expression vectors of wt-p53 (1, 5, 10, 10, 10, and 10 μg from lane 2 to 7), or of MDM2 (1, 5, 10, and 10 μg from lane 5 to 8) by electroporation, and luciferase activity was measured as in A. C, a series of truncated survivin promoter-luciferase constructs described previously (Li and Altieri, 1999) were cotransfected into EU-4 cells with expression vectors of either wt-53 or p53-273 (mutant) or with an emptyneo gene vector, respectively, by electroporation, and luciferase activity was measured as described in A. Results in Fig. 6. were normalized to β-galactosidase activity and plotted as the means of duplicates of a representative experiment out of at least three independent determinations.
Discussion
In the present study, we have characterized a novel p53-survivin signaling pathway activated by doxorubicin in pediatric ALL cells and resulting in cell cycle arrest and apoptosis. Previous studies have examined the effect of doxorubicin on p53 but not survivin expression.Di Bartolomeo et al. (2000) showed that doxorubicin induces rapid up-regulation of p53 and cell cycle arrest in p53+ neuroblastoma cells, although they did not report whether the phenotype was wild type or mutant. In our studies of ALL cells, doxorubicin up-regulated wt-p53 expression in EU-3 cells, and increased p53 expression was accompanied by down-regulation of survivin and apoptosis. In contrast, untreated mut-p53+ EU-6 cells expressed constitutively high levels of p53 protein and did not show any down-regulation of survivin or apoptosis following treatment. Likewise, doxorubicin-treated p53-null EU-4 cells failed to down-regulate survivin and were resistant to this drug. These results strongly suggest that down-regulation of survivin and doxorubicin sensitivity are wt-p53-dependent.
Interestingly, survivin was markedly up-regulated in mut-p53+ cells but not p53-null cells following doxorubicin. The mechanism for this up-regulation is not clear. Since doxorubicin produced a G2/M arrest in mut-p53+ and p53-null cells, we tested the cell cycle inhibitor nocodazole to examine whether survivin up-regulation was associated with G2/M arrest in the presence of mutant-p53 protein. A slight increase in survivin expression was noted in nocodazole-treated mut-p53+ cells, although less than that seen with doxorubicin. In contrast, treatment with the G1-specific inhibitor mimosine down-regulated survivin, but only in wt-p53+ cells. Down-regulation of survivin was also observed in wt-p53+ cells treated with thymidine, which arrests cells in early S phase, leaving many of the growing DNA chain unfinished, which in many cases mimics DNA damage and induces p53 activation. These results support the idea that down-regulation of survivin was wt-p53-dependent.
It was reported that survivin expression was associated with mutant p53 expression in gastric cancer patients, although the mechanism is unknown (Lu et al., 1998). Our results showed that survivin expression is up-regulated in mut-p53+ EU-6 cells and down-regulated in wt-p53+ EU-3 cells after doxorubicin treatment. Thus, in the present study, regulation of survivin is dependent on p53 phenotype and occurs in a cell cycle-dependent manner following doxorubicin treatment through a doxorubicin-p53-survivin pathway. Loss of normal p53 function due to mutation or gene deletion could therefore result in deregulation of survivin and increased resistance to genotoxic, p53-dependent agents such as doxorubicin.
Based on our finding that wt-p53 is required for survivin down-regulation, we examined the effect of p53 on the survivin promoter using cotransfection and promoter-luciferase reporter-gene assays. In this assay, only wt-p53 could inhibit survivin promoter activity, and blocking p53 activity by overexpression of a cotransfected MDM2 gene abrogated p53-mediated inhibition of the survivin reporter. Additionally, mut-p53 constructs did not activate the survivin promoter, suggesting that mut-p53 protein up-regulates survivin via mechanisms other than transcriptional regulation. For example, certain p53 mutations could exert gain-of-function effects by deregulating survivin protein turnover. By using truncated survivin promoter-luciferase constructs, we were able to map the p53-binding region to the 230-bp basic core promoter region. These results agree with a very recent study showing that wt-p53 can transcriptionally inhibit the survivin promoter (Hoffman et al., 2002).
Survivin is a novel member of the IAP family. Members of this family are believed to protect cells from apoptosis by binding to activated caspases via the IAP (baculovirus IAP repeat) domain, thereby inhibiting their activity (Hinds et al., 1999). Survivin has been shown to bind specifically to and inhibit the activated forms of caspase-3 and -7 (Shin et al., 2001). Activation of caspase-3 or -7 is the distal step in mitochondrial or p53-dependent pathway of apoptosis, which begins with activation of caspase-9 and release of cytochromec from the mitochondria (Soengas et al., 1999). This pathway of apoptosis is referred to as p53-dependent since p53 can repress transcription of certain antiapoptotic genes (including Bcl-2 and nuclear factor-κβ) that prevent activation of this pathway (Miyashita et al., 1994a; Yang et al., 1997; Ryan et al., 2000). Our finding that doxorubicin can suppress survivin transcription via wt-p53 is consistent with this apoptotic function of p53. The role of wt-p53 as a transcriptional repressor of survivin is further supported by our finding that high levels of MDM2 expression can block survivin repression. Although our results indicate that doxorubicin induces apoptosis in ALL cells primarily through the p53-survivin pathway, the presence of a subset (up to 20%) of apoptotic cells in p53-null EU-4 cells post-treatment suggests that doxorubicin may exert a p53-survivin-independent apoptotic effect as well.
The survivin protein is rarely present in normal adult tissues/cells but is abundantly expressed in most human tumors (Ambrosini et al., 1997; Tamm et al., 1998; Carter et al., 2001). The expression of survivin in these tumors has been associated with resistance to chemotherapy, increased aggressiveness, and decreased patient survival rate (Adida et al., 1998, 2000a,b; Tamm et al., 1998; Islam et al., 2000). However, in the ALL cell lines studied here, survivin was expressed at uniformly high levels pretreatment regardless of their sensitivity to doxorubicin. Similarly, we have found that primary leukemic cells derived from pediatric ALL patients that have the wt-p53 phenotype also express high levels of survivin and yet are generally sensitive to doxorubicin (data not shown). Our data suggest that it is the post-treatment fluctuation (i.e., up-regulation or down-regulation) in survivin expression rather than the pretreatment level that determines sensitivity, and that this fluctuation is determined by the p53 phenotype of the ALL cells.
It was reported that down-regulating survivin expression by anti-survivin antisense significantly inhibits cell growth and induces apoptosis in melanoma (Grossman et al., 2001), lung cancer (Olie et al., 2000), and neuroblastoma (Shankar et al., 2001). The importance of survivin up-regulation following doxorubicin treatment suggests that approaches to down-regulate survivin expression should sensitize tumor cells to this agent. Accordingly, we have found that anti-survivin antisense inhibited survivin expression in mut-p53+ ALL cells postdoxorubicin and sensitized these cells to treatment. In contrast, granulocyte-macrophage colony-stimulating factor (GM-CSF) protects leukemic cells against doxorubicin-induced apoptosis (Kaplinsky et al., 1996). Furthermore, GM-CSF up-regulates survivin in leukemic cells (Carter et al., 2001). These results suggest that treatments which increase survivin expression in tumor cells may protect these cells from doxorubicin, and that molecular targeting of survivin should lead to new therapies for ALL and other tumors.
Footnotes
-
Supported by grants from the National Cancer Institute-National Institutes of Health (R01 CA82323), CURE Childhood Cancer, Inc., Children's Healthcare of Atlanta, and a National Cancer Institute Cancer Center Support Grant (CA 16056).
-
DOI: 10.1124/jpet.102.037192
- Abbreviations:
- MDM2
- murine double minute2
- IAP
- inhibitor of apoptosis protein
- CDE
- cell cycle-dependent element
- CHR
- cell cycle genes homology region
- ALL
- acute lymphoblastic leukemia
- wt
- wild type
- mut
- mutant
- FBS
- fetal bovine serum
- SSC
- standard saline citrate
- PARP
- poly(ADP-ribose) polymerase
- BrdUrd
- bromodeoxyuridine
- PBS
- phosphate-buffered saline
- PI
- propidium iodide
- G418
- geneticin
- bp
- base pair
- GM-CSF
- granulocyte-macrophage colony-stimulating factor
- Received April 8, 2002.
- Accepted June 3, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}