


Abstract
Aripiprazole is the first next-generation atypical antipsychotic with a mechanism of action that differs from currently marketed typical and atypical antipsychotics. Aripiprazole displays properties of an agonist and antagonist in animal models of dopaminergic hypoactivity and hyperactivity, respectively. This study examined the interactions of aripiprazole with a single population of human D2 receptors to clarify further its pharmacologic properties. In membranes prepared from Chinese hamster ovary cells that express recombinant D2L receptors, aripiprazole bound with high affinity to both the G protein-coupled and uncoupled states of receptors. Aripiprazole potently activated D2 receptor-mediated inhibition of cAMP accumulation. Partial receptor inactivation using the alkylating agentN-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ) significantly reduced the maximum effect of aripiprazole on inhibition of cAMP accumulation. This effect was seen with concentrations of EEDQ that did not alter the maximal inhibitory effect of dopamine. Consistent with the expected effects of a partial agonist, increasing concentrations of aripiprazole blocked the action of dopamine with maximal blockade equal to the agonist effect of aripiprazole alone. The efficacy of aripiprazole relative to that of dopamine varied from 25% in cells that lacked spare receptors for dopamine to 90% in cells with receptor reserve. These results, together with previous studies demonstrating partial agonist activity at serotonin 5-hydroxytryptamine (5-HT)1A receptors and antagonist activity at 5-HT2A receptors, support the identification of aripiprazole as a dopamine-serotonin system stabilizer. The receptor activity profile may underlie the unique activity of aripiprazole in animals and its antipsychotic activity in humans.
Aripiprazole, 7-{4-[4-(2,3-dichlorophenyl)-1-piperazinyl]butyloxy}-3,4-dihydro-2(1H)-quinolinone, is the first next-generation atypical antipsychotic that is active against positive and negative symptoms of schizophrenia (Petrie et al., 1997; Saha et al., 2001), has a low propensity for extrapyramidal side effects (Petrie et al., 1997; Saha et al., 2001), causes minimal weight gain or sedation (Petrie at al., 1997; Carson et al., 2002), and produces no elevation in serum prolactin levels (Petrie et al., 1997;Saha et al., 2001) or prolongation of QTc interval on ECG (Kane et al., 2000; Carson et al., 2002). The mechanism of action of aripiprazole differs from that of currently marketed typical and atypical antipsychotics. Previous preclinical studies have provided evidence that aripiprazole is a dopamine-serotonin system stabilizer with potent partial agonist activity at dopamine D2 and 5-HT1A receptors and antagonist activity at 5-HT2A receptors (Inoue et al., 1996; Jordan et al., 2001; T. Kikuchi, unpublished observations).
Like many antipsychotics, aripiprazole binds with high affinity to members of the D2 family of dopamine receptors (Kikuchi et al., 1995;Lawler et al., 1999). Whereas currently marketed antipsychotics are believed to exert their effects through antagonism of D2 (and possibly 5-HT2) receptors (see Miyamoto et al., 2000 for a recent review), aripiprazole may exert its effects through partial agonism at D2 receptors. In multiple studies in vivo, aripiprazole has been shown to have potent agonist activity at dopamine autoreceptors. For example, aripiprazole decreases γ-butyrolactone- and reserpine-induced DOPA accumulation (Kikuchi et al., 1995), consistent with a decrease in presynaptic tyrosine hydroxylase activity. The inhibitory effect of aripiprazole on γ-butyrolactone-induced DOPA accumulation is blocked by the D2 receptor antagonist haloperidol. Administration of aripiprazole to laboratory rats results in decreased extracellular levels of dopamine in the striatum and frontal cortex suggestive of decreased release of dopamine (Semba et al., 1995). Finally, the ability of aripiprazole to decrease spontaneous firing of dopaminergic neurons in the ventral tegmentum by activation of D2 autoreceptors was shown by extracellular recording in vivo (Momiyama et al., 1996).
Whereas results of the above studies are consistent with agonist activity of aripiprazole at D2 receptors, in other in vivo studies, aripiprazole displays properties of a D2 receptor antagonist. For example, aripiprazole blocks apomorphine-induced stereotypy and locomotor activity and does not produce stereotypy or increased locomotion when administered alone (Kikuchi et al., 1995). Consistent with blockade of dopamine receptors coupled to the inhibition of prolactin release, administration of aripiprazole to male rats results in a 2-fold increase in levels of serum prolactin (Inoue et al., 1996).
In vivo, partial agonists may act predominantly as agonists or antagonists depending upon the level of endogenous receptor activation. Partial agonist activity of aripiprazole at D2 receptors may explain its antagonist properties in animal models of dopaminergic hyperactivity (e.g., blockade of apomorphine-induced stereotypy) and agonist activity in an animal model of dopaminergic hypoactivity (blockade of increased dopamine synthesis in reserpine-treated rats) (Kikuchi et al., 1995). A variety of efficacy values for aripiprazole at D2-like receptors have been reported using different in vitro preparations where endogenous dopaminergic tone is eliminated. In slices of rat pituitary, aripiprazole decreased spontaneous release of prolactin with an effect approximately 50% that of talipexole, a D2 receptor agonist. Consistent with a partial agonist effect, aripiprazole moderately blocked the effect of talipexole (Inoue et al., 1996). Likewise, in C6 cells that express recombinant rat D2L receptors linked to the inhibition of cAMP accumulation, aripiprazole displayed modest agonist activity with a maximum effect 30% that of dopamine (Lawler et al., 1999). In contrast, in CHO cells that express transfected rat D2L receptors, aripiprazole completely blocked the ability of dopamine to inhibit forskolin-stimulated accumulation of cAMP while having no efficacy alone, consistent with antagonist activity at D2 receptors (Lawler et al., 1999). Similarly, in rat striatal membranes, increased GTPase activity stimulated by the D2 receptor agonist quinpirole is completely blocked by aripiprazole. However, aripiprazole alone does not stimulate GTPase activity (Inoue et al., 1997).
The purpose of this study was to clarify the functional activity of aripiprazole at D2 receptors and to demonstrate how partial agonism in conjunction with modulation of components of the signal transduction pathway may explain the range of activities of aripiprazole at D2 receptors.
Experimental Procedures
Materials.
[125I]7-OH-PIPAT (2200 Ci/mmol) was purchased from PerkinElmer Life Sciences (Boston, MA). [3H]Spiperone was purchased from Amersham Biosciences (Piscataway, NJ). Haloperidol, (+)-butaclamol hydrochloride, S(−)-PPP, terguride, quinpirole hydrochloride, Tris, EDTA, BSA, and polyethylenimine were purchased from Sigma-Aldrich (St. Louis, MO). 5′-guanylylimidodiphosphate was purchased from Calbiochem (San Diego, CA). Tissue culture plates (100 × 20-mm) were purchased from Corning Glassworks (Corning, NY). F-12 Nutrient Mixture (Ham), fetal bovine serum, and G418 sulfate were purchased from Invitrogen (Carlsbad, CA).
Tissue Culture.
CHO cells that express human recombinant D2L receptors (CHO-D2L) have been previously described (Filtz et al., 1993). Cells were grown at 37°C in 5% CO2 as a monolayer in medium consisting of F-12 supplemented with 10% fetal bovine serum and G418 sulfate (500 μg/ml).
Radioligand Binding Assays.
Cells were rinsed twice with phosphate-buffered saline (155 mM NaCl, 3.3 mM Na2HPO4, and 1.1 mM KH2PO4, pH 7.4), and incubated for 5 to 10 min at 4°C in hypotonic lysis buffer consisting of 10 mM Tris (pH 7.4) and 5 mM EDTA. Cells were transferred from plates to polypropylene tubes (16 × 100 mm), homogenized, and centrifuged at 32,000g for 20 min. Pellets were resuspended in buffer consisting of 50 mM Tris (pH 7.7 at 26°C) and 1 mM EDTA, then stored at −80°C until needed. On the day of an experiment, homogenates were thawed, resuspended by homogenization, and centrifuged at 32,000g for 20 min. Following centrifugation, supernatants were discarded, and remaining pellets were resuspended in buffers as detailed below. Binding of [3H]spiperone was carried out in buffer containing 50 mM Tris (pH 7.4 at 25°C), 100 μM GMP-PNP, and 1% DMSO. Homogenates (2–3 μg of protein/tube) were incubated with [3H]spiperone (10–1000 pM) for 90 min at 25°C. Binding of [125I]7-OH-PIPAT was carried out in buffer consisting of 50 mM Tris (pH 7.7 at 25°C), 2 mM MgCl2, 0.1% BSA, 0.025 mN HCl, and 1% DMSO. Homogenates (10 μg of protein/tube) were incubated with [125I]7-OH-PIPAT (200 pM) for 60 min at 37°C. Assays were stopped by addition of cold wash buffer (50 mM Tris, [3H]spiperone or 20 mM Tris, [125I]7-OH-PIPAT). Filtration over glass fiber filters (Whatman GF/B; Whatman, Clifton, NJ) previously soaked in 0.05% polyethylenimine (for [3H]spiperone binding) or 20 mM Tris (for [125I]7-OH-PIPAT binding) was carried out using a Brandel cell harvester (Brandel Inc., Gaithersburg, MD). Nonspecific binding was defined with 2 μM (+)-butaclamol.
Maximum binding (Bmax) andKD values were determined by unweighted linear regression analysis of transformed saturation binding data (Scatchard, 1949). Protein concentrations were determined by the method of Bradford (1976) with BSA as a standard.
Accumulation of cAMP.
Cells were harvested by successive washing and centrifugation in Cell Dissociation Buffer (Invitrogen). The final pellet was resuspended in phosphate-buffered saline containing 0.9 mM CaCl2, 0.5 mM MgCl2, and 0.5% BSA. Approximately 6 × 104 cells were added to each well of a 96-well plate. Cells were exposed to test compounds for 10 min at 37°C in the presence of 10 μM forskolin and 100 μM 3-isobutyl-1-methylxanthine. The reaction was terminated by the addition of 0.15 N HCl. Accumulation of cAMP was measured using the cAMP SPA Direct Screening Assay kit (Amersham Biosciences).
Receptor Inactivation Studies.
Cells were collected in Cell Dissociation Buffer and centrifuged at 100g for 5 min. Cells were resuspended in F-12 media and divided equally into three separate tubes containing either vehicle (0.1% DMSO) or 1 μM or 10 μM EEDQ and incubated for 60 min at 37°C in 5% CO2. Following treatment with EEDQ, cells were washed once by centrifugation and resuspension in F-12 media. Cells were centrifuged, and the pellet was resuspended in phosphate-buffered saline containing 0.9 mM CaCl2, 0.5 mM MgCl2, and 0.5% BSA. Approximately 6 × 104 cells were added to each well of a 96-well plate, and the effects of agonists on forskolin-stimulated accumulation of cAMP were determined as above.
Results
Binding of Agonists and Antagonists to D2 Receptors.
The affinity values of human D2L receptors for agonists and antagonists were determined for receptors labeled with the agonist [125I]7-OH-PIPAT and with the antagonist [3H]spiperone under conditions that promote, respectively, coupling or uncoupling of receptors to G proteins (Burris et al., 1995). Butaclamol and haloperidol, D2 receptor antagonists, bound with slightly higher affinity to the antagonist-labeled noncoupled state of D2L receptors than to the G protein-coupled state labeled with [125I]7-OH-PIPAT (Table1). In contrast, the agonists dopamine and quinpirole bound with 34- to 67-fold higher affinity to the G protein-coupled state of D2 receptors. The partial agonistsS(−)-PPP, terguride, and aripiprazole displayed higher affinity for the G protein-coupled state of D2 receptors than for the noncoupled state (Table 1). The ratio of affinities for the partial agonists was intermediate between those for full agonists and antagonists.
Affinity values of the G protein-coupled and noncoupled states of D2L receptors for agonists, partial agonists, and antagonists
Stimulation of D2 Receptors by Aripiprazole and Other Agonists: Effects of Modulation of Receptor Density.
Slightly higher affinity for the G protein-coupled state compared with the noncoupled state of D2 receptors suggest that aripiprazole is a partial agonist at D2 receptors. The ability of aripiprazole to stimulate D2 receptors was examined directly in CHO cells that express human recombinant D2L receptors coupled to the inhibition of cAMP accumulation. The increase in cAMP accumulation induced by exposure to 10 μM forskolin was potently inhibited by dopamine (Fig. 1). Haloperidol (1 μM), a D2 receptor antagonist, completely blocked the inhibition of cAMP accumulation by dopamine (data not shown). Similar to the effects of dopamine, increasing concentrations of aripiprazole potently inhibited the increase in cAMP accumulation stimulated by forskolin (Fig. 1). Consistent with the activity of a partial agonist, the maximum effect of aripiprazole was approximately 85% that of dopamine. The effect was not unique to CHO cells, as similar results were obtained in HEK-293 cells that express human recombinant D2L cells (Fig. 1, inset).

Cells were incubated for 10 min with 10 μM forskolin and 100 μM 3-isobutyl-1-methylxanthine in the absence or presence of increasing concentrations of dopamine or aripiprazole. cAMP was measured using the Biotrak cAMP Direct Screening Assay System (Amersham Biosciences). Accumulation of cAMP stimulated by forskolin ranged from 3 to 11 pmol/well. Data were fit to a four-parameter logistic equation using GraphPad Prism version 3.0 (GraphPad Software, San Diego, CA). The data shown are the mean ± S.E.M.,n = 8 (CHO-D2L) and 5 (HEK-293-D2L; inset) experiments.
The relationship of the density of D2 receptors to the efficacy of aripiprazole and other partial agonists was examined. Exposure of CHO-D2L cells to increasing concentrations of EEDQ resulted in a progressive decrease in the density of receptors as determined by binding of [3H]spiperone (Fig.2). Exposure of cells to 1 μM EEDQ for 1 h resulted in a greater than 50% decrease in the density of D2L receptors, whereas exposure to 10 μM EEDQ resulted in a nearly 80% decrease in the density of receptors (Fig. 2). The affinity of D2 receptors for [3H]spiperone was not affected by exposure of cells to EEDQ.

The density of D2L receptors was determined by saturation binding of [3H]spiperone to washed membranes prepared from CHO-D2L cells previously incubated in the absence or presence of EEDQ at 37°C for 60 min. Nonspecific binding was determined with 2 μM (+)-butaclamol. KDand Bmax values were determined by fitting data to a one-site binding isotherm.
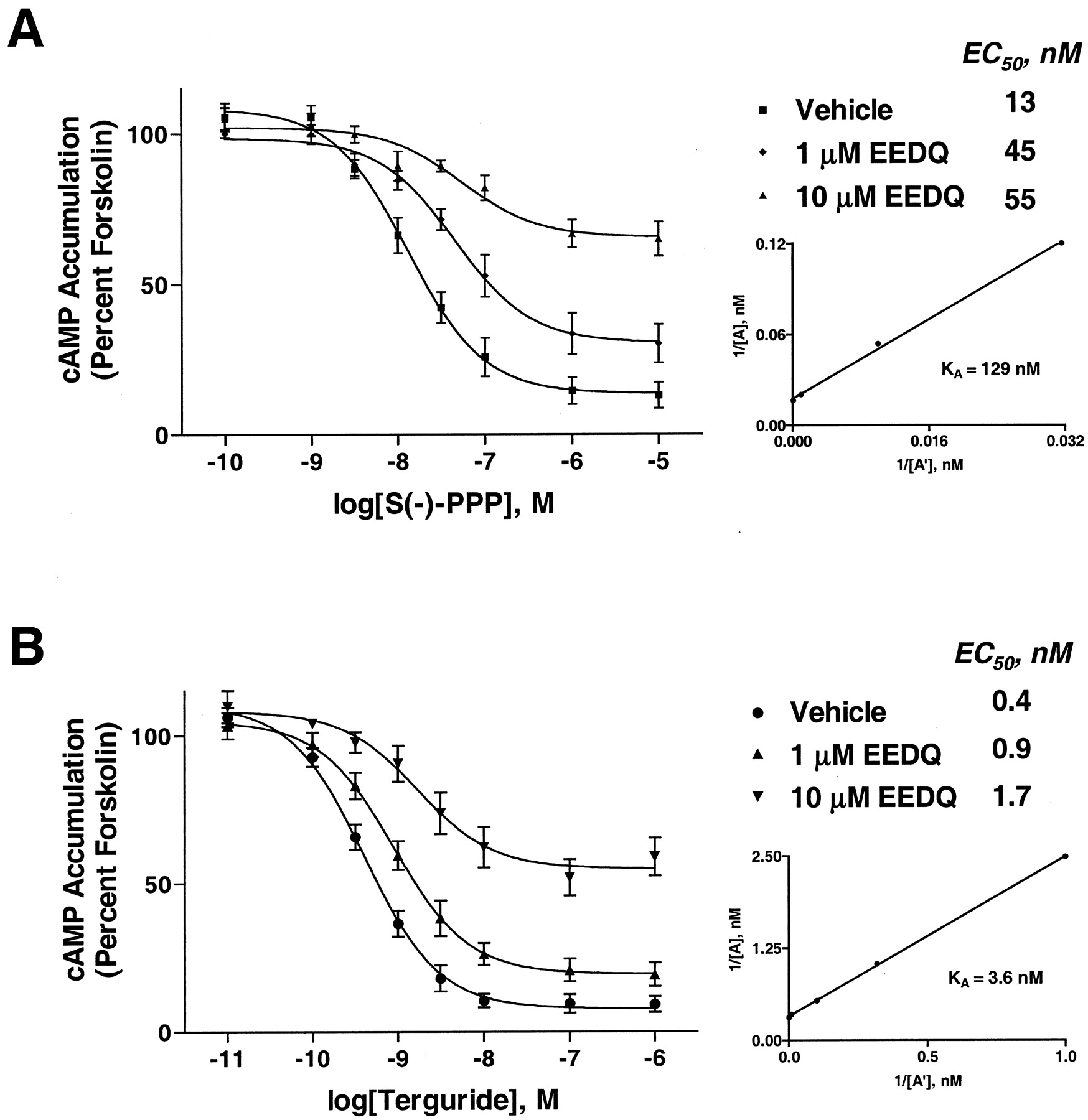
In cells exposed to 1 μM EEDQ, dopamine inhibited forskolin-stimulated cAMP accumulation with a 2.5-fold lower potency than in cells exposed to vehicle (Fig.3A). There was no significant change in the maximum effect of dopamine, consistent with the presence of receptor reserve. The potency of dopamine was further reduced in cells exposed to 10 μM EEDQ (Fig. 3A). Exposure of cells to 10 μM EEDQ resulted in a small decrease in efficacy. The apparent dissociation constant (KA) of dopamine was calculated from a double-reciprocal plot of equieffective concentrations of dopamine determined in cells incubated in the presence and absence of 10 μM EEDQ, according to the method ofFurchgott and Bursztyn (1967). The KAvalue for dopamine was 178 nM (Fig. 3A, inset). In contrast to effects seen with dopamine, the maximum effect of aripiprazole was significantly reduced in cells exposed to 1 μM EEDQ (Fig. 3B). In cells exposed to 10 μM EEDQ, the maximum effect of aripiprazole was further reduced. The efficacy of aripiprazole relative to dopamine was reduced from nearly 90 to 25% upon partial inactivation of D2 receptors with 10 μM EEDQ (Fig. 3, A and B). A double-reciprocal plot of equieffective concentrations of aripiprazole determined in cells incubated in the presence and absence of 1 μM EEDQ yielded aKA of 28 nM (Fig. 3B, inset). As seen with aripiprazole, the maximum effects of S(−)-PPP and terguride were progressively reduced in cells exposed to increasing concentrations of EEDQ (Fig. 4A,B). TheKA values of S(−)-PPP and terguride were 129 and 3.6 nM, respectively (Fig. 4, A and B, inset).

CHO-D2L cells were incubated in the absence or presence of increasing concentrations of EEDQ at 37°C for 60 min. Inhibition of forskolin-stimulated cAMP accumulation by dopamine (A) and aripiprazole (B) was determined in washed cells as in Fig. 1. Accumulation of cAMP stimulated by forskolin ranged from 3 to 15 pmol/well. In cells exposed to 1 and 10 μM EEDQ, the mean ± S.D. level of cAMP accumulation stimulated by forskolin was 105 ± 17 and 115 ± 13%, respectively, of that seen with cells exposed to vehicle. The data shown are the mean ± S.E.M.,n = 7 experiments. Inset, the apparentKA values for dopamine and aripiprazole were determined from a plot of the reciprocals of equieffective concentrations of drugs from cells incubated in the absence (1/[A]) and presence (1/[A′]) of 1 μM EEDQ using the equation:KA = (slope − 1)/y − intercept (Furchgott and Bursztyn, 1967).

CHO-D2L cells were incubated in the absence or presence of increasing concentrations of EEDQ at 37°C for 60 min. Inhibition of forskolin-stimulated cAMP accumulation byS(−)-PPP (A) and terguride (B) was determined in washed cells as in Fig. 1. The data shown are the mean ± S.E.M.,n = 4 experiments. For experiments withS(−)-PPP, accumulation of cAMP stimulated by forskolin ranged from 3 to 11 pmol/well. In cells exposed to 1 and 10 μM EEDQ, the mean ± S.D. level of cAMP accumulation stimulated by forskolin was 98 ± 19 and 111 ± 12%, respectively, of that seen with cells exposed to vehicle. For experiments with terguride, accumulation of cAMP stimulated by forskolin ranged from 3 to 15 pmol/well. In cells exposed to 1 and 10 μM EEDQ, the mean ± S.D. level of cAMP accumulation stimulated by forskolin was 113 ± 10 and 121 ± 11%, respectively, of that seen with cells exposed to vehicle. Inset, the apparent KA values for S(−)-PPP and terguride were determined from a plot of the reciprocals of equieffective concentrations of drugs from cells incubated in the absence (1/[A]) and presence (1/[A′]) of 1 μM EEDQ using the equation: KA = (slope − 1)/y − intercept (Furchgott and Bursztyn, 1967).
KA values for dopamine, aripiprazole,S(−)-PPP, and terguride were used to compare the relative efficacy of agonists as a function of occupancy of receptors. A steep hyperbolic occupancy-effect relationship was seen for dopamine (Fig.5). The response to dopamine was nearly maximal at 20% occupancy of D2 receptors. Inhibition of cAMP accumulation by 50% was achieved with occupancy by dopamine of only 2% of the receptors. Greater levels of occupancy of receptors by terguride, S(−)-PPP, and aripiprazole were required for the same response (Fig. 5).
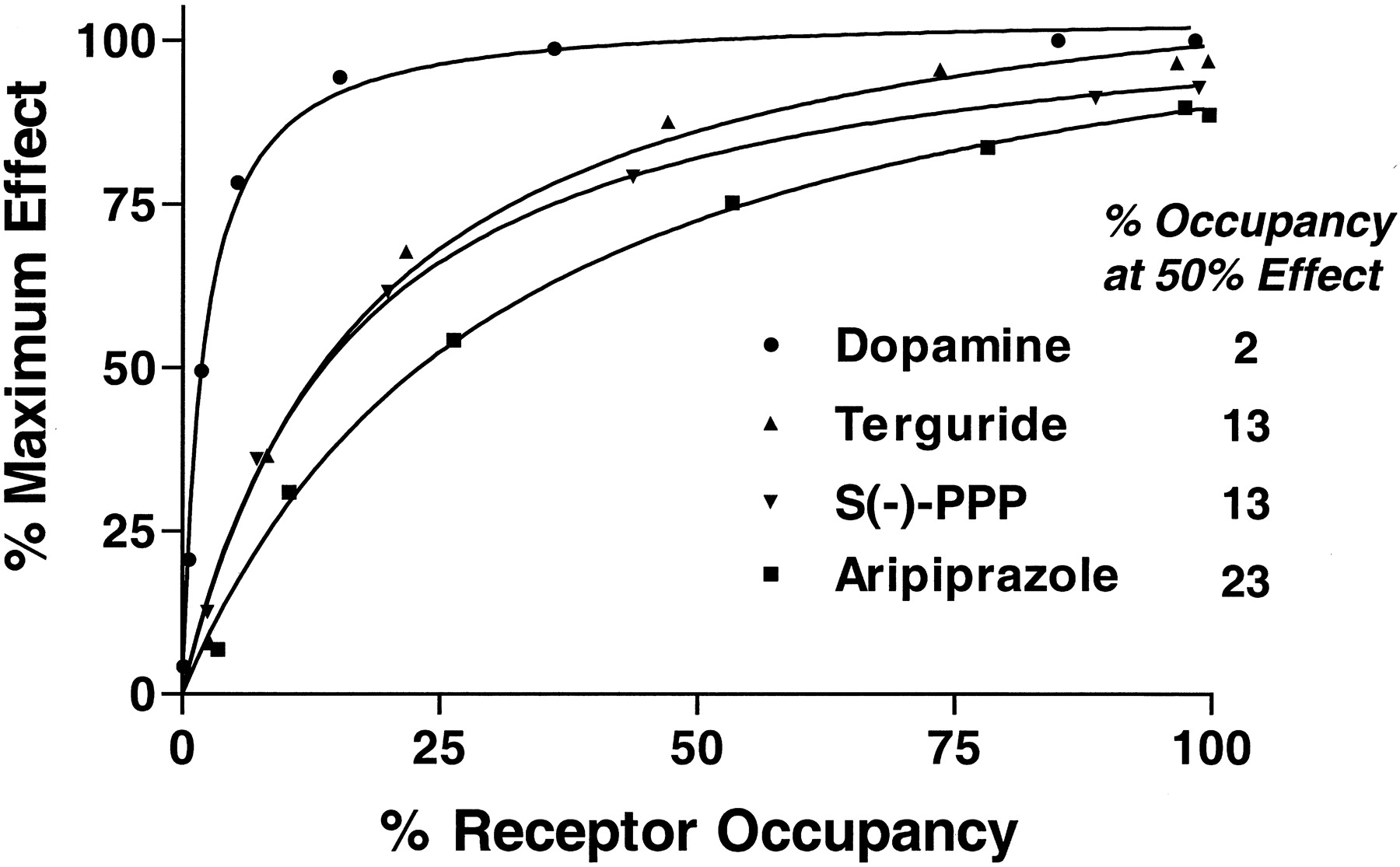
Given the partial agonist effect in cells exposed to 10 μM EEDQ, the ability of aripiprazole to antagonize the effect of dopamine was examined. Dopamine (100 nM) inhibited cAMP accumulation by approximately 60% (Fig. 6). Consistent with the action of a partial agonist, increasing concentrations of aripiprazole blocked the effect of dopamine up to the maximum agonist effect seen with aripiprazole alone (Fig. 6).

CHO-D2L cells were incubated in the presence of 10 μM EEDQ to eliminate receptor reserve. Inhibition of forskolin-stimulated cAMP accumulation by aripiprazole in the absence and presence of 100 nM dopamine was determined in washed cells as in Fig. 1. Accumulation of cAMP stimulated by forskolin ranged from 9 to 12 pmol/well. The data shown are the mean ± range of two experiments.
Discussion
A range of efficacy values for aripiprazole at D2 receptors has been reported in different cell lines and tissues. The present study examined the efficacy of aripiprazole at a single population of human D2 receptors. Multiple factors, including intrinsic activity, receptor density, and coupling efficiency in the signal transduction cascade, contribute to the activity of an agonist in a given system (Kenakin 1997). Agonists with low intrinsic activity may show agonist or antagonist activity depending upon the sensitivity of the method used for detection, the level of basal or endogenous receptor activation, and the molecular properties of the signaling event under investigation (Hoyer and Boddeke, 1993). Given the level of complexity, current techniques cannot unambiguously determine the intrinsic efficacy of an agonist at a receptor (Clarke and Bond, 1998; Kenakin, 1999). One approach that may avoid the variables associated with the signal transduction cascade involves calculation of the ratio of affinity values of agonists for coupled and noncoupled states of D2 receptors (Lahti et al., 1992). D2 receptors exist in multiple states having high and low affinity for agonists (Zahniser and Molinoff, 1978; DeLean et al., 1982). The agonist-preferring high-affinity state of D2 receptors is thought to reflect the active state of receptors and involves the formation of a ternary complex of agonist, receptor, and G protein (Wregget and DeLean, 1984). Whereas antagonists bind with equally high affinity to noncoupled and G protein-coupled states of D2 receptors, agonists typically display higher affinity for the G protein-coupled state.
Comparisons of affinity values were used to predict the efficacy of aripiprazole and some known agonists, partial agonists, and antagonists at D2 receptors. In CHO cells expressing human D2L receptors, the full agonists dopamine and quinpirole bound with greater than 30-fold higher affinity to the G protein-coupled state of D2 receptors than to the noncoupled state. The partial agonists, terguride andS(−)-PPP, although displaying less of a difference between affinity values for the different states, had higher affinity for the G protein-coupled state of D2 receptors. In contrast, the antagonists butaclamol and haloperidol bound with higher affinity to the noncoupled state. Consistent with the properties of a partial agonist, aripiprazole bound with 2-fold higher affinity to the G protein-coupled state of D2 receptors.
Aripiprazole was a partial agonist at human D2L receptors coupled to the inhibition of forskolin-stimulated cAMP accumulation. Results with terguride and S(−)-PPP were consistent with previous reports that these compounds are partial agonists at D2 receptors (Clark et al., 1984; Kehr, 1984; Lahti et al., 1992). For a given receptor-effector system, the density of receptors plays an important role in determining the potency and maximum efficacy of agonists (Kenakin, 1999). The alkylating agent EEDQ, an irreversible antagonist of dopamine receptors (Hamblin and Creese, 1983), has been used previously as a tool to modulate the density of D2L receptors in transfected cells (Filtz et al., 1994). Exposure of CHO-D2L cells to increasing concentrations of EEDQ resulted in a decrease in the density of receptors measured by [3H]spiperone. The efficacy of aripiprazole in CHO-D2L cells ranged from 25 to 90% that of dopamine, depending on the density of receptors. In preliminary studies, the efficacy of dopamine, aripiprazole, (−)-3-PPP, terguride, and OPC-4392 were compared in clonal cell lines that express high (11–18 pmol/mg) and low (0.3–0.9 pmol/mg) densities of D2L and D2S receptors. Aripiprazole was a partial agonist at both D2S and D2L receptors, and density-dependent efficacy comparable to the studies with EEDQ was seen (Y. Tadori, T. Miwa, K. Tottori, K. D. Burris, P. B. Molinoff, F. D. Yocca, and T. Kikuchi, unpublished observations).
Inactivation of a fraction of D2 receptors with EEDQ resulted in a rightward shift in the dose-response curve for dopamine but no change in maximum effect. This suggests a nonlinear relationship between occupancy of receptors and the response to dopamine. Classically, this has been ascribed to the presence of receptor reserve in the tissue (Nickerson, 1956). In contrast, the maximum effects of terguride,S(−)-PPP, and aripiprazole were significantly reduced, with progressive alkylation indicating a lack of receptor reserve for these partial agonists. Occupancy-response curves for the partial agonists were less steep and were right-shifted compared with that seen with dopamine. Interestingly, a nonlinear relationship between occupancy and response appeared to exist for all three partial agonists. Although partial agonists would be expected to yield linear occupancy-response relationships, nonlinear relationships have been reported (Kenakin and Beek, 1984; Kenakin, 1997).
The relative efficacy of aripiprazole was higher in the present study than in some previous reports. In C6 cells expressing rat D2L receptors, the efficacy of aripiprazole for inhibition of isoproterenol-stimulated cAMP accumulation was approximately 30% that of dopamine (Lawler et al., 1999). In the same study, aripiprazole did not significantly inhibit cAMP accumulation in CHO cells expressing rat D2L receptors. The lack of efficacy may be due to a lack of receptor reserve or differences in the coupling of receptors in the cell lines. In contrast to the nearly complete inhibition of forskolin-stimulated cAMP accumulation by dopamine in the present study, dopamine inhibited the response to forskolin by only 50% in CHO cells that expressed transfected rat D2L receptors (Lawler et al., 1999). Receptor reserve has been reported for the effects of agonists at D2 receptors in rat pituitary (Meller et al., 1991). Similar to the present results, aripiprazole is a partial agonist at D2 receptors linked to inhibition of prolactin release in slices of rat pituitary (Inoue et al., 1996).
Differences in receptor reserve may play a role in the varying efficacy of aripiprazole at pre- and postsynaptic D2 receptors. Receptor reserve has been reported for the effects of agonists at presynaptic D2 receptors (Meller et al., 1987) but not for responses mediated by postsynaptic D2 receptors (Meller et al., 1988). Meller et al. (1987)hypothesized that enhanced sensitivity of presynaptic D2 receptor signal transduction (compared with postsynaptic) may underlie the reported autoreceptor selectivity of D2 receptor partial agonists. The present study demonstrated the effects of modulating the density of receptors on the efficiency of signal transduction. In the presence of receptor reserve for dopamine, aripiprazole was an efficacious agonist. In its absence, the relative efficacy of aripiprazole was low, and a predominant antagonist effect was seen. Likewise, although aripiprazole is a potent partial agonist at all D2 receptors, it functions with greater efficacy at presynaptic receptors with substantial receptor reserve and with lower efficacy at postsynaptic D2 receptors with less receptor reserve.
The molecular mechanisms underlying differences in sensitivity at pre- and postsynaptic D2 receptors are not known. The present studies demonstrate the dependence of efficacy on the density of receptors for a single effector system and receptor population. At high levels of receptor expression, a significant amount of receptor reserve exists for full agonists, and partial agonists display high relative efficacy values. In the absence of receptor reserve, partial agonists have the properties of antagonists. In addition to their stoichiometry, the molecular identity of the signaling molecules can modulate the relative efficacies of agonists. The molecular identity of the subtypes of D2-like receptors that mediate pre- and postsynaptic actions of agonists have not been determined. The cataleptic effects of haloperidol reportedly are absent or attenuated in D2L receptor-deficient transgenic mice (Usiello et al., 2000; Wang et al., 2000). In the same studies, D2 receptor-mediated inhibition of dopamine release, locomotor activity, and the firing rate of dopaminergic neurons in the substantia nigra were similar to that seen in wild-type animals, suggesting that D2S receptors can function as autoreceptors.
Many subtypes of G protein-coupled receptors activate a diversity of G protein subtypes, with agonists displaying different efficacies depending upon the signaling system activated (Berg et al., 1998;Clarke and Bond, 1998; Yang and Lanier, 1999). Likewise, D2 receptors couple to multiple effector systems and display subtype selectivity in coupling to G proteins, suggesting the possibility of subtype- and effector-dependent efficacy of agonists (Senogles, 1994; Guiramand et al., 1995; Boundy et al., 1996). Whether differences in receptor density, distinct signaling pathways, or receptor-effector coupling underlie presynaptic dopaminergic receptor reserve remains to be determined.
Previous studies evaluating the clinical utility of D2 receptor partial agonists in the treatment of schizophrenia have not identified an agent with substantial promise (Wetzel and Benkert, 1993;Lahti et al., 1998). The best-studied of these agents,S(−)-3-(3-hydroxyphenyl)-N-n-propylpiperidine [S(−)3-PPP; preclamol], demonstrated efficacy against positive and negative symptoms, but activity was not sustained for longer than 1 week (Lahti et al., 1998), which investigators attributed to desensitization of D2 receptors. In contrast, aripiprazole has demonstrated lasting overall efficacy in the treatment of schizophrenia. In several 4-week, placebo-controlled studies, aripiprazole improved positive and negative symptoms, with efficacy sustained throughout (Petrie et al., 1997; Saha et al., 2001). One possible explanation for the mixed results with previous dopamine partial agonists is the range of values for intrinsic activity and affinity at D2 receptors. Multiple factors govern the efficacy and affinity of agonists, making it difficult to determine the optimal pharmacological properties of a partial agonist in vitro needed to attain antipsychotic efficacy.
The partial agonist properties of aripiprazole at D2 receptors likely contribute to stabilization, rather than blockade, of dopaminergic tone, resulting in a unique clinical profile. The results of this study, in combination with data demonstrating that aripiprazole is a partial agonist at 5-HT1A receptors and an antagonist at 5-HT2A receptors, support the identification of aripiprazole as a dopamine-serotonin system stabilizer. The activity of aripiprazole at D2 receptors, as well as 5-HT1A and 5-HT2A receptors, may contribute to robust and sustained overall efficacy in the treatment of schizophrenia, including activity against both positive and negative symptoms, with minimal risk of extrapyramidal symptoms, sedation, or elevated prolactin levels.
Footnotes
-
↵1 Present address: Palatin Technologies, 175 May Street, Suite 500, Edison, NJ 08837.
-
This work was funded by Bristol-Myers Squibb Company and Otsuka Pharmaceutical Co. Ltd. Results of this study were presented at the XXIInd Collegium Internationale Neuropsychopharmacologicum Congress, July 9–11, 2000, Brussels, Belgium and the VIIIth International Congress on Schizophrenia Research, April 28–May 2, 2001, Whistler, BC, Canada.
-
DOI: 10.1124/jpet.102.033175
- Abbreviations:
- 5-HT
- 5-hydroxytryptamine (serotonin)
- BSA
- bovine serum albumin
- CHO
- Chinese hamster ovary
- DMSO
- dimethyl sulfoxide
- EEDQ
- N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline
- HEK
- human embryonic kidney
- [125I]7-OH-PIPAT
- R-(+)-trans-7-hydroxy-2-(N-n-propyl-N-3′-iodo-2′-propenyl)aminotetralin
- S(−)-3-PPP
- S(−)-3-(3-hydroxyphenyl)-N-n-propylpiperidine, preclamol
- GMP-PNP
- 5′-guanylylimidodiphosphate
- OPC-4392
- 7-(3-[4-(2,3-dimethylphenyl)piperazinyl]propoxy)-2-(1H)-quinolinone
- Received January 15, 2002.
- Accepted March 27, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}