


Abstract
Buprenorphine (BUP) is an oripavine analgesic that is beneficial in the maintenance treatment of opiate-dependent individuals. Although BUP has been studied extensively, relatively little is known about norbuprenorphine (norBUP), a major dealkylated metabolite of BUP. We now describe the binding of norBUP to opioid and nociceptin/orphanin FQ (ORL1) receptors, and its effects on [35S]guanosine-5′-O-(γ-thio)triphosphate ([35S]GTPγS) binding mediated by opioid or ORL1 receptors and in the mouse acetic acid writhing test. Chinese hamster ovary cells stably transfected with each receptor were used for receptor binding and [35S]GTPγS binding. NorBUP exhibited high affinities for μ-, δ-, and κ-opioid receptors withK i values in the nanomolar or subnanomolar range, comparable to those of BUP. NorBUP and BUP had low affinities for the ORL1 receptor with K i values in the micromolar range. In the [35S]GTPγS binding assay, norBUP displayed characteristics distinct from BUP. At the δ-receptor, norBUP was a potent full agonist, yet BUP had no agonist activity and antagonized actions of norBUP and DPDPE. At μ- and κ-receptors, both norBUP and BUP were potent partial agonists, with norBUP having moderate efficacy and BUP having low efficacy. At the ORL1 receptor, norBUP was a full agonist with low potency, while BUP was a potent partial agonist. In the writhing test, BUP and norBUP both suppressed writhing in an efficacious and dose-dependent manner, giving A50 values of 0.067 and 0.21 mg/kg, s.c., respectively. These results highlight the similarities and differences between BUP and norBUP, each of which may influence the unique pharmacological profile of BUP.
Buprenorphine (BUP) (Fig. 1), an oripavine derived from thebaine, has antinociceptive activities in animal pain models and is used clinically for treatment of moderate-to-severe pain (Cowan and Lewis, 1995). BUP is 25 to 40 times more potent than morphine as an analgesic after parenteral injection (Cowan et al., 1977; Lewis, 1995). Since it is associated with less physical dependence and reinforcing effects than many other opioids (Negus and Woods, 1995), BUP is being developed as a potential pharmacotherapy for opioid abuse and dependence.
Chemical structures of BUP and norBUP.
[3H]BUP administered in vivo preferentially labels μ-opioid receptor sites (Sadee et al., 1982). Although BUP was reported to have pharmacological effects on δ- and κ-opioid receptors (Belcheva et al., 1993, 1996; Pick et al., 1997), its analgesic effects have been ascribed mostly to actions on the μ-receptor (Dum and Herz, 1981; Kamei et al., 1997). At the biochemical level, BUP is a partial agonist at the μ-opioid receptor (Traynor and Nahorski, 1995; Selley et al., 1998; Toll et al., 1998;Lee et al., 1999), or an antagonist (Romero et al., 1999). At the κ-opioid receptor, BUP acts as a low-efficacy partial agonist (Zhu et al., 1997) or as an antagonist (Romero et al., 1999) or shows no agonist activity (Toll et al., 1998). At the δ-opioid receptor, BUP shows no agonistic effects (Toll et al., 1998; Lee et al., 1999; Romero et al., 1999). BUP exhibited pure antagonism at rat brain ORL1 receptors, but acted as a partial agonist or a full agonist at the recombinant human ORL1 receptor (Wnendt et al., 1999; Hashimoto et al., 2000; Hawkinson et al., 2000).
Norbuprenorphine (norBUP) (Fig. 1), the N-dealkylated product of BUP, is a major metabolite of BUP in humans and rats (Cone et al., 1985; Garrett and Chandran, 1990). Ohtani et al. (1989) showed that norBUP was measurable in plasma between 2 to 3 h after a sublingual dose of BUP in a human volunteer. Kuhlman et al. (1998)demonstrated that the mean steady-state plasma concentration of norBUP exceeded that of buprenorphine after daily administration of sublingual buprenorphine to humans, whereas Tzeng et al. (2000) reported that plasma levels of BUP and norBUP declined in a multiple exponential manner in rats.
Although the pharmacology of BUP has been studied extensively, relatively little is known about norBUP. Ohtani et al. (1995) reported that norBUP was considerably less hydrophobic than BUP, and the intrinsic analgesic activity of i.c.v. norBUP was about one-fourth that of BUP in the rat tail-flick test. Additionally, these researchers have reported that norBUP does not readily cross the blood-brain barrier into the rat brain. Intravenous administration of norBUP at 1 to 3 mg/kg decreased respiratory rate, whereas BUP had no effect up to 3 mg/kg (Ohtani et al., 1997). The respiratory depression induced by norBUP appeared to be mediated by μ-opioid receptors in the lung rather than in the brain (Ohtani et al., 1997).
In the present study, we compared the pharmacological profiles of BUP and norBUP on opioid and nociceptin/orphanin FQ (ORL1) receptors. We determined their binding properties to these receptors, and their potencies and efficacies in stimulating these receptors to enhance [35S]GTPγS binding in Chinese hamster ovary (CHO) cells stably transfected with the μ-, δ-, or κ-opioid receptor or ORL1 receptor. In addition, we compared the antinociceptive effects of norBUP and BUP in an in vivo assay—the mouse acetic acid writhing test.
Experimental Procedures
Materials.
Norbuprenorphine was purchased from Ultrafine (Manchester, England). (−)-Buprenorphine HCl and U50,488H were supplied by the National Institute on Drug Abuse. (+)-Buprenorphine HCl was synthesized by Drs. N. A. Grayson and K. C. Rice of the National Institute of Digestive and Kidney Disease, National Institutes of Health. [15,16-3H]Diprenorphine (56 Ci/mmol), [leucyl-3,4,5-3H]nociceptin/orphanin FQ (N/OFQ) (87.7 Ci/mmol), [35S]guanosine-5′-O-(3-thio)triphosphate ([35S]GTPγS) (1250 Ci/mmol), and [3H]cAMP (30–40 Ci/mmol) were obtained from NEN Life Science Products, Inc. (Boston, MA). DAMGO and nociceptin/OFQ were purchased from Phoenix Pharmaceuticals, Inc. (Belmont, CA) The following compounds were obtained as indicated: naloxone HCl (Sigma, St. Louis, MO), GTPγS (Boehringer-Mannheim, Indianapolis, IN), geneticin (G418) (Cellgro, Mediatech, Inc., Herndon, VA), and DPDPE (ICI, Downingtown, PA). Bovine serum albumin (BSA; 1 mg/ml) was added to all BUP solutions, and 0.05 mg/ml bacitracin and 1 mg/ml BSA were included in all buffers for nociceptin/OFQ.
CHO Cell Lines.
CHO cells stably transfected with the rat μ-opioid receptor (Chen et al., 1993) was established as we described previously (Chen et al., 1995). CHO cells stably expressing the mouse δ-opioid receptor (Evans et al., 1992) were kindly supplied by Dr. Ping-Yi Law, Department of Pharmacology, University of Minnesota School of Medicine, Minneapolis, MN. CHO cells with stable expression of the human κ-opioid receptor (Zhu et al., 1995) was established previously (Zhu et al., 1997). CHO cells stably transfected with the human ORL1 receptor were a gift from Dr. Lawrence Toll, SRI International, Menlo Park, CA (Adapa and Toll, 1997).
Cell Membrane Preparation.
Membranes were prepared according to a modified procedure of (Zhu et al., 1997). Cells were washed twice and harvested in Versene solution (EDTA 0.54 mM, NaCl 140 mM, KCl 2.7 mM, Na2HPO4 8.1 mM, KH2PO4 1.46 mM, and glucose 1 mM) and centrifuged at 500g for 3 min. The cell pellet was suspended in buffer A [5 mM Tris (pH 7.4), 5 mM EDTA, 5 mM EGTA, and 0.1 mM phenylmethylsulfonyl fluoride], passed through a 26-gauge
Receptor Binding.
Ligand binding experiments were carried out with [3H]diprenorphine for opioid receptors and [3H]nociceptin/OFQ for the ORL1 receptor.
Saturation binding of [33H]diprenorphine to μ-, δ-, and κ-opioid receptors was performed with at least six concentrations of [33H]diprenorphine (ranging from 25 pM to 1–2 nM), and K d andB max values were determined. Competition inhibition by BUP, norBUP, or (+)-BUP of [3H]diprenorphine (0.4 nM) binding to opioid receptors was performed in the absence or presence of various concentrations of each drug. Binding was carried out in 50 mM Tris-HCl buffer containing 1 mM EGTA (pH 7.4) at room temperature for 1 h in duplicate in a final volume of 1 ml with ∼10 to 20 μg of membrane protein. Naloxone (10 μM) was used to define nonspecific binding. Bound and free [3H]diprenorphine were separated by filtration under reduced pressure with GF/B filters presoaked with 0.1 mg/ml BSA and 0.2% polyethyleneimine. Radioactivity on filters was determined by liquid scintillation counting. Each experiment was performed in duplicate and repeated at least three times. Binding data were analyzed with the EBDA program (McPherson, 1983). K i values of each drug were determined (Cheng and Prusoff, 1973).
Saturation binding of [3H]nociceptin/OFQ to the ORL1 receptor was performed with at least six concentrations of [3H]nociceptin/OFQ (ranging from 25 pM to 2 nM), and K d andB max values were determined. Competition inhibition by BUP, norBUP, or (+)-BUP of [3H]nociceptin/OFQ (0.3 nM) binding to the ORL1 receptor was performed, and K i values of each drug were determined. Nociceptin/OFQ (150 nM) was used to define nonspecific binding.
[35S]GTPγS Binding.
Determination of [35S]GTPγS binding to G proteins was carried out using a modified procedure of Zhu et al. (1997). Immediately before the [35S]GTPγS binding assay, membranes were thawed at 37°C, chilled on ice, and diluted with buffer C [50 mM HEPES (pH 7.4), 100 mM NaCl, 5 mM MgCl2, and 1 mM EDTA]. Membranes (10 μg) were incubated in buffer C containing [35S]GTPγS (200 pM, 300,000–500,000 dpm) and 15 μM GDP with or without a ligand (10−12 to 10−4 M) in a total volume of 0.5 ml for 60 min at 30°C. Nonspecific binding was defined by incubation in the presence of 10 μM GTPγS. Bound and free [35S]GTPγS were separated by filtration with GF/B filters under reduced pressure. Radioactivity on filters was determined by liquid scintillation counting. EC50values and maximal responses (E max) of drugs were determined by curve fitting to the equation for a sigmoidal curve E = [E max/]1 + ([D]/EC50)n + basal level, where E is effect produced by a certain concentration of the drug, [D],E max is the maximal response elicited by the drug, and n is a fitting parameter.
Determination of Adenylate Cyclase Activity.
The cAMP assay is based on the method described by Cote et al. (1982). CHO cells expressing the ORL1 receptor were added to assay tubes containing isobutylmethylxanthine, forskolin, and N/OFQ in serum-free Dulbecco's modified Eagle's medium (final volume 250 μl, final 2.5 mM isobutylmethylxanthine, and 25 μM forskolin). For the basal level, forskolin and agonist were omitted. Assay mixtures were incubated at 37°C for 10 min, and the reaction was terminated by placing the tubes in boiling water for 5 min. The amounts of cAMP in the sample tubes were determined with the cAMP binding protein method. Briefly, [3H]cAMP (∼30,000 dpm in 0.02 M citrate phosphate buffer, pH 5.0) was added to all sample tubes and standard tubes (from 1.25–40 pmol of cAMP) on ice. cAMP binding protein partially purified from bovine adrenal glands was added to each tube at an amount that gave 10,000 to 20,000 dpm [3H]cAMP binding in the absence of cold cAMP, except the blanks. The mixture (final 170 μl) was incubated on ice or at 4°C for at least 2 h. Bound and free [3H]cAMP were separated by absorption of free [3H]cAMP by charcoal (100 μl of 10% Norit A, 4% BSA, and 1% Antifoam A) and centrifugation. Radioactivity of bound [3H]cAMP in an aliquot of the supernatant was determined by liquid scintillation counting. The amounts of cAMP were calculated based on the standard curve.
Acetic Acid Writhing Test.
Male Swiss albino mice (24–27 g;n = 7–12) were injected s.c. with either saline or test agent. After 20 min, acetic acid (0.6%) was injected i.p. (0.25 ml/25 g). A further 5 min later, the number of writhes was counted for 10 min. The number of writhes in each test period was then normalized to the mean number shown by the control group, and antinociceptive-50 (A50) values were obtained by nonlinear regression analysis.
Results
Determination of Receptor Expression Levels of CHO-μ, CHO-δ, and CHO-κ, and CHO-ORL1 Cells.
The rat μ-opioid receptor, mouse δ-opioid receptor, human κ-opioid receptor, and human ORL1 receptor were stably transfected into CHO cells. Saturation binding of [3H]diprenorphine to μ-, δ-, and κ-opioid receptors and [3H]nociceptin/OFQ to the ORL1 receptor was performed on membranes, andK d andB max values were determined. [3H]Diprenorphine exhibited high affinities for μ-, δ-, and κ-opioid receptors, withK i values of 0.14 ± 0.03 nM, 0.33 ± 0.04 nM, and 0.15 ± 0.03 nM, respectively (mean ± S.E.M., n = 3–5) and theB max values of 2.1 ± 0.5, 1.1 ± 0.3, and 1.3 ± 0.2 pmol/mg of protein, respectively (mean ± S.E.M., n = 3–5). [3H]nociceptin/OFQ bound to the ORL1 receptor with high affinity with a K i value of 0.14 ± 0.02 nM and a B maxvalue of 5.0 ± 0.7 pmol/mg of protein (mean ± S.E.M., n = 3).
Binding Affinities of norBUP and BUP to Opioid Receptors and ORL1 Receptor.
Competitive inhibition of [3H]diprenorphine to opioid receptors or [3H]nociceptin/OFQ to ORL1 receptor by BUP, norBUP, and (+)-BUP was conducted to determine binding affinities of these ligands to μ-, δ-, and κ-opioid receptors or the ORL1 receptor (Fig. 2, Table1). NorBUP exhibited high affinities for μ-, δ-, and κ-opioid receptors, withK i values in inhibiting [3H]diprenorphine binding in the subnanomolar and nanomolar range with a ratio of 1:45:13 for μ:δ:κ. In contrast, norBUP had a low affinity for the ORL1 receptor, with aK i value in inhibiting [3H]N/OFQ binding in the micromolar range. Similarly, BUP had high affinities for μ-, δ-, and κ-opioid receptors, with K i values in the subnanomolar range and had a low affinity for the ORL1 receptor, with aK i value in the micromolar range. (+)-BUP did not bind to μ-, δ-, or κ-opioid receptors or the ORL1 receptor.
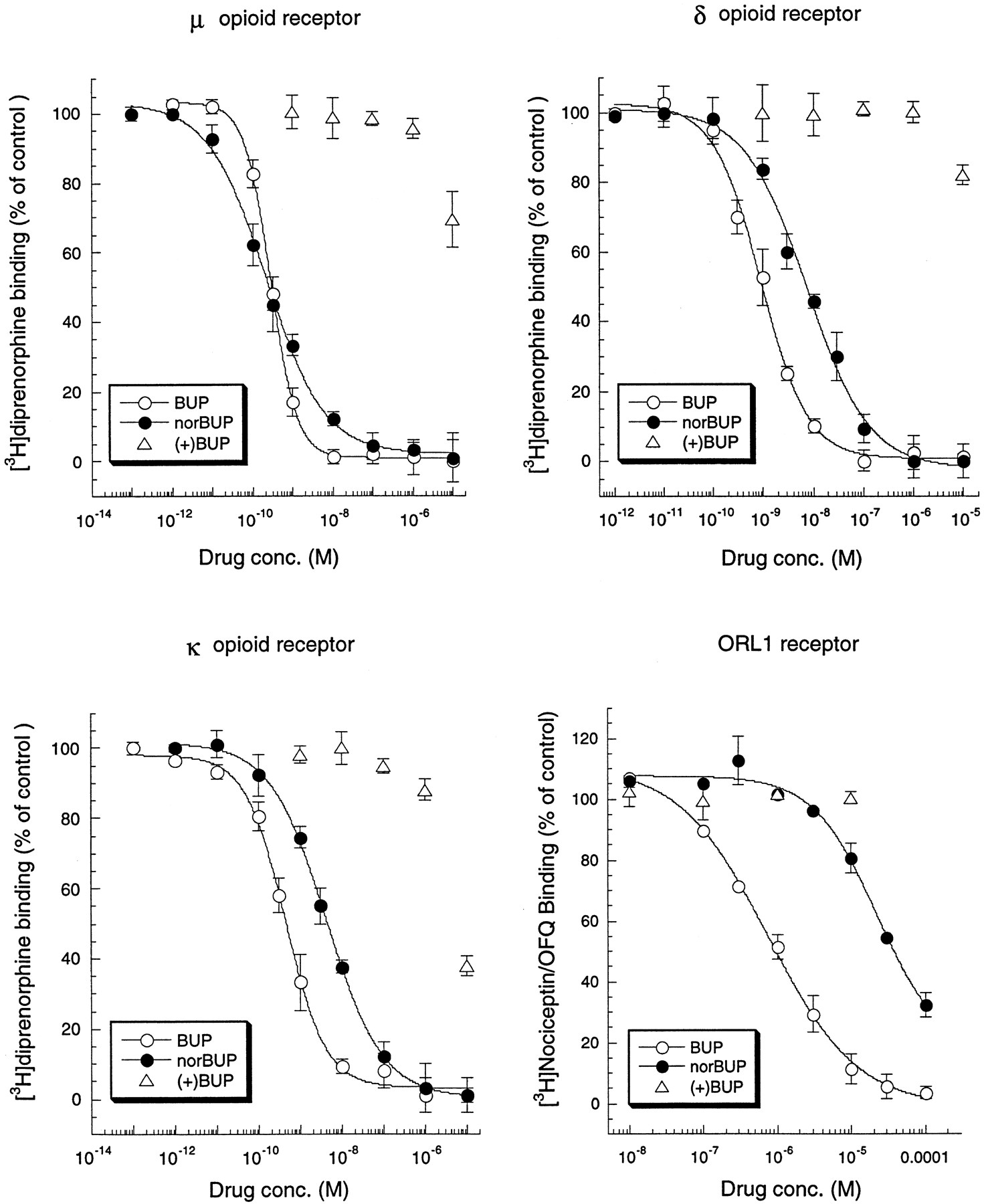
Competitive inhibition by BUP, norBUP, and (+)-BUP of [3H]diprenorphine binding to μ-, δ-, and κ-opioid receptors and [3H]nociceptin/OFQ binding to the ORL1 receptor. Membranes were prepared from CHO cells stably transfected with the μ-, δ-, or κ-opioid receptor or the ORL1 receptor. Binding of opioid receptors and the ORL1 receptor was carried out with ∼0.4 nM [3H]diprenorphine and ∼0.3 nM [3H]nociceptin/OFQ, respectively, in the presence and absence of various concentrations of BUP, norBUP, and (+)-BUP as described under Experimental Procedures. Specific [3H]diprenorphine and [3H]nociceptin/OFQ binding was ∼7,600 dpm/tube and ∼12,000 dpm/tube, respectively, and the corresponding nonspecific binding was 400 to 600 dpm/tube and 1,000 dpm/tube, respectively. Data were normalized to percentage of specific binding. Each value represents the mean ± S.E.M of at least three independent experiments performed in duplicate. ApparentK i values are shown in Table 1.
Apparent K i values (nM) of BUP and norBUP for the opioid receptors and the ORL1 receptor
Potencies and Efficacies of norBUP and BUP in the [35S]GTPγS Binding Assay.
[35S]GTPγS binding to membranes of CHO cells stably transfected with the μ-, δ-, and κ-opioid receptor or the ORL1 receptor in response to norBUP, BUP, and (+)-BUP was examined, with a prototypical full agonist as the control for each receptor (Fig.3). EC50 values and maximal responses are shown in Table 2. NorBUP was a partial agonist at μ- and κ-receptors and a full agonist at the δ-receptor, with EC50 values in the nanomolar range, whereas it was a full agonist at ORL1 receptor with low potency, with EC50 values in the micromolar range. BUP was a partial agonist at the μ- and ORL1 receptors, with EC50 values in the nanomolar range. In addition, BUP demonstrated weak agonism at the κ-receptor giving a maximal response of 10% and was inactive at the δ-receptor. (+)-BUP did not activate μ-, δ-, or κ-opioid receptors or the ORL1 receptor.

Stimulation of [35S]GTPγS binding to membranes of CHO cells stably transfected with the μ-, δ-, κ-opioid or the ORL1 receptor by BUP, norBUP, and a full agonist for each receptor. [35S]GTPγS binding was performed as described under Experimental Procedures. Nonspecific binding, estimated using 10 μM cold GTPγS, was ∼500 dpm and was subtracted from each datum. Basal [35S]GTPγS binding in the absence of added drugs was 3000 to 5000 dpm. Data were normalized to percentage of the basal [35S]GTPγS binding. Each value represents the mean ± S.E.M. of at least three independent experiments performed in duplicate. EC50 values and maximal responses are shown in Table 2.
EC50 values and maximal effects of BUP and norBUP in stimulating [35S]GTPγS binding to membranes of CHO cells stably transfected with the μ-, δ-, or κ-opioid or the ORL1 receptor
Effects of BUP and norBUP on ORL1 Receptor-Mediated Inhibition of Forskolin-Stimulated Adenylate Cyclase.
Our finding that BUP is a partial agonist at the ORL1 receptor in stimulating [35S]GTPγS binding differs from those ofWnendt et al. (1999) and Hashimoto et al. (2000). These researchers demonstrated that BUP was a potent full agonist at the ORL1 receptor in inhibiting forskolin-stimulated adenylate cyclase activity. The difference may be due to the different functional assays used. We thus determined potencies and efficacies of BUP and norBUP at the ORL1 receptor in inhibiting forskolin-stimulated adenylate cyclase activity. As shown in Fig. 4, BUP and norBUP inhibited forskolin-stimulated adenylate cyclase to similar extents as N/OFQ, indicating that both compounds are full agonists in this assay. The EC50 values were determined to be 26.2 nM and 1360 nM, respectively, indicating that BUP is approximately 52 times more potent than norBUP.

Inhibition of cAMP accumulation by nociceptin/OFQ, BUP, and norBUP in CHO cells stably transfected with the ORL1 receptor. CHO cells stably expressing ORL1 receptors were incubated in serum-reduced medium with 10 μM forskolin and 1 mM isobutylmethylxanthine with and without different concentrations of drugs. cAMP contents were determined as described underExperimental Procedures and normalized to percentage of the forskolin-stimulated cAMP level. Each value represents the mean ± S.E.M. of at least three independent experiments performed in duplicate.
Antagonistic Effect of Buprenorphine at the δ-Opioid Receptor.
Since BUP had high affinity for the δ-receptor, yet no agonist activity, we examined whether BUP had antagonistic effects on norBUP-stimulated [35S]GTPγS binding to membranes of CHO-δ cells. BUP shifted the dose-response curves of norBUP to the right in a parallel fashion (Fig.5A). The potency of BUP in antagonizing the action of norBUP at the δ-receptor was determined. Schild analysis was performed, and the slope was determined to be 1.09, indicating that BUP is a competitive antagonist at the δ- receptor (Fig. 5B). The pA2 value of BUP was determined to be 9.31 ± 0.14 (0.49 nM) (n = 3).

A, antagonistic effects of BUP on norBUP-stimulated [35S]GTPγS binding to membranes of CHO cells stably transfected with δ-opioid receptor. B, Schild plot of BUP in antagonizing norBUP-induced increase in [35S]GTPγS binding. [35S]GTPγS binding was performed as described under Experimental Procedures, with various concentrations of norBUP in the absence or presence of a fixed concentration (10−9, 3 × 10−9, 10−8, or 3 × 10−8 M) of BUP. Nonspecific binding, estimated using 10 μM cold GTPγS, was ∼500 dpm and was subtracted from each datum. Basal [35S]GTPγS binding in the absence of added drugs was 3000 to 5000 dpm. Data were normalized to percentage of the basal [35S]GTPγS binding. Each value represents the mean ± S.E.M. of at least three independent experiments performed in duplicate.
Antinociceptive Activity of norBUP and BUP.
NorBUP was a relatively potent analgesic, suppressing writhing with an efficacy similar to that of BUP (Fig. 6). The antinociceptive activity of norBUP was dose-dependent, giving an A50 value of 0.21 mg/kg, approximately 3 times greater than that of BUP.

Antinociceptive effects of BUP, norBUP, and (+)-BUP in the mouse acetic acid writhing test. The mice (n= 7–12) were pretreated s.c. with either saline or test compound. After 20 min, the mice received 0.6% acetic acid i.p., and a further 5 min later, the number of writhes was counted for 10 min. Each point represents the mean (±S.E.M.) percentage inhibition of writhing relative to the saline-control group.
Discussion
In the present study, we have shown that norBUP has a distinctly different pharmacological profile from BUP although both have high affinities for μ-, δ-, and κ- opioid receptors and low affinities for the ORL1 receptor. NorBUP is a full agonist at the δ-receptor, whereas BUP is an antagonist. Both are partial agonists at μ- and κ-receptors, with norBUP having higher efficacy than BUP. NorBUP and BUP are less efficacious at the κ-receptor relative to the μ-receptor. To the best of our knowledge, this represents the first characterization of pharmacological activities of norBUP at cloned μ-, δ-, and κ- opioid receptors and the ORL1 receptor.
NorBUP was a more efficacious but slightly less potent partial agonist than BUP at the μ-receptor, with an 81% maximal [35S]GTPγS binding response and an EC50 of 1.5 nM. Our finding that BUP stimulated [35S]GTPγS binding to CHO-μ membranes with an E max of 38% and an EC50 of 0.08 nM is consistent with several previous reports. In these reports, BUP was a potent partial agonist at the μ-opioid receptor and enhanced [35S]GTPγS binding to varying extents:E max of about 50% in C6 glioma cells expressing the rat μ-receptor (Lee et al., 1999), 73% in human neuroblastoma SH-SY5Y cells (Traynor and Nahorski, 1995), 43% in CHO cells stably transfected with the mouse μ-receptor, and 10% in the rat thalamus (Selley et al., 1998). In addition, with inhibition of forskolin-stimulated adenylate cyclase as the endpoint, BUP displayed agonism at the mouse μ-receptor expressed in HEK293 cells (Blake et al., 1997) and the human μ-receptor expressed in CHO cells (Yu et al., 1997). In contrast, BUP failed to stimulate [35S]GTPγS binding in guinea pig caudate membranes and showed antagonism under certain conditions (Romero et al., 1999). The differences in the efficacy of BUP in these preparations may be related to several factors, including different species of μ-opioid receptors used, the number of μ-opioid receptors, the repertoire and level of G proteins to which the receptor can be coupled, and the receptor/G protein ratio.
NorBUP acted as a full agonist at the δ-opioid receptor with an EC50 value of 30.4 nM. In contrast, BUP had no agonistic activity and was a potent competitive antagonist against effects of norBUP on [35S]GTPγS binding at the δ-opioid receptor. The pA2 value (0.49 nM) of BUP at δ-receptors is similar to itsK i value (0.42 nM). Our observation that BUP has no agonist activity at the δ-opioid receptor is in accord with previous reports (Toll et al., 1998; Lee et al., 1999;Romero et al., 1999). Thus, depending on the concentrations of BUP and its metabolite norBUP, BUP administration can have varying activities in vivo at the δ-receptor, ranging from being predominantly antagonistic to largely agonistic. Since chronic buprenorphine increased δ2-binding sites in some brain regions without changing δ1-sites (Belcheva et al., 1993, 1996), buprenorphine may have different actions on the two subtypes of δ-receptor in vivo.
We have shown that norBUP is a potent partial agonist at the κ-opioid receptor with an E max of 60% and an EC50 of 7.2 nM in stimulating [35S]GTPγS binding, having higher efficacy than BUP. That BUP acted as a partial agonist with low efficacy at the κ-opioid receptor is similar to our previous report (Zhu et al., 1997). In contrast, BUP failed to stimulate [35S]GTPγS binding (Toll et al., 1998; Romero et al., 1999) and inhibited κ-agonist-promoted [35S]GTPγS binding with high potency (Romero et al., 1999). In addition, BUP is a potent antagonist at the κ-receptor in several in vivo tests (Leander, 1987, 1988; Negus and Dykstra, 1988). The difference in efficacy of buprenorphine in different preparations may be attributed to variations in the receptor level, the population and level of G proteins, and receptor/G protein ratio as mentioned above.
NorBUP was found to be a full agonist at the ORL1 receptor with low potency, having an EC50 >1 μM in both [35S]GTPγS binding and inhibition of forskolin-stimulated adenylate cyclase. In contrast, BUP was a partial agonist at the ORL1 receptor, with an EC50 of 35 nM and an E max of 60% in stimulating [35S]GTPγS binding (see Table 2) and BUP acted as a full agonist with an EC50 of 26 nM when evaluated by inhibition of forskolin-stimulated adenylate cyclase. The difference in the efficacy of BUP in the two endpoints may be due to the possibility that full inhibition of adenylyl cyclases requires only partial activation of G proteins. Our results with BUP on the ORL1 receptor are consistent with those of Wnendt et al. (1999), Bloms-Funke et al. (2000), Hashimoto et al. (2000), and Hawkinson et al. (2000). Using [35S]GTPγS binding as the functional measure, Bloms-Funke et al. (2000) and Hawkinson et al. (2000) showed that BUP exhibited partial agonism at the human ORL1 receptor. In contrast, with inhibition of forskolin-stimulated adenylate cyclase as the endpoint, BUP was reported to be a full agonist at the ORL1 receptor (Wnendt et al., 1999; Hashimoto et al., 2000).
Comparison between the EC50 values of norBUP and BUP in stimulating [35S]GTPγS binding (see Table 2) and their K i values in inhibiting [3H]diprenorphine binding to μ-, δ-, and κ-opioid receptors (see Table 1) reveals that EC50 values for norBUP or BUP are similar or greater than their corresponding K ivalues. In contrast, for the ORL1 receptor, theK i values of norBUP or BUP are more than 4 times greater than their corresponding EC50 values. These results indicate that there are spare receptors for norBUP and BUP in their stimulation of the ORL1 receptor, but not opioid receptors, to activate G proteins.
(+)-BUP does not bind to or activate μ-, δ-, and κ-opioid receptors or the ORL1 receptor, indicating that binding of BUP to these receptors is stereospecific.
Results from the acetic acid writhing test showed that norBUP, injected s.c. in mice, has comparable antinociceptive efficacy to BUP, with BUP being about 3 times more potent than norBUP. As noted previously (Cowan et al., 1977), the dose-response relationship for BUP is sigmoidal in this procedure. In contrast, BUP produces bell-shaped curves in several other rodent antinociceptive assays (e.g., Rance et al., 1980; Dum and Herz, 1981; Bryant et al., 1983; Wheeler-Aceto and Cowan, 1991; Woods et al., 1992). Thus, BUP is associated with antinociception at low doses, yet higher doses are often less effective. N/OFQ displays pronociceptive or hyperalgesic activity in different animal pain models after intracerebroventricular administration (Meunier et al., 1995;Reinscheid et al., 1995; Hara et al., 1997), which has been attributed to an inhibition of stress-induced analgesia (Mogil et al., 1996a,b). As hypothesized by Wnendt and colleagues (Wnendt et al., 1999;Bloms-Funke et al., 2000), the ORL1 agonistic activity of BUP may contribute to its bell-shaped dose-response curve in, for example, the mouse tail-flick test. BUP is a potent partial agonist at the μ-receptor, and norBUP is a potent partial agonist at the μ- and κ-receptors and a potent full agonist at the δ-receptor, with EC50 values in the subnanomolar or nanomolar range, which contribute to the antinociceptive effect of low doses of BUP administered in vivo. At the ORL1 receptor, BUP is a potent partial agonist in stimulating [35S]GTPγS binding and a full agonist in inhibiting forskolin-stimulated adenylate cyclase, with EC50 values of about 30 nM. The action of BUP at the ORL1 receptor at higher concentrations may counter the antinociception produced by BUP and norBUP on opioid receptors, resulting in less antinociception at higher doses and thus the bell-shaped dose-response curves. This hypothesis is currently being tested. Although norBUP is a full agonist at the ORL1 receptor, its low potency (EC50 of 1.4 μM) makes it less likely to contribute significantly to the action on ORL1 receptors in vivo.
In conclusion, BUP and norBUP displayed distinct pharmacological properties at μ-, δ-, and κ-opioid receptors and the ORL1 receptor. Since norBUP is a major metabolite of BUP in vivo and its mean steady-state plasma concentration is comparable to or even exceeds that of BUP following sublingual BUP (Kuhlman et al., 1998), it is likely to contribute to the overall pharmacological effects of BUP in vivo.
Footnotes
-
Send reprint requests to: Dr. Lee-Yuan Liu-Chen, Department of Pharmacology, Temple University School of Medicine, 3420 N. Broad St., Philadelphia, PA 19140. E-mail: lliuche{at}astro.temple.edu
-
This work was supported in part by grants from the National Institute on Drug Abuse (DA04745, DA11263, DA13429, and T32 DA07237).
- Abbreviations:
- BUP
- buprenorphine
- norBUP
- norbuprenorphine
- ORL1
- nociceptin/orphanin FQ receptor
- [35S]GTPγS
- [35S]guanosine-5′-O-(3-thio)triphosphate
- DAMGO
- Tyr-d-Ala-Gly-N-(Me)Phe-Gly-ol
- DPDPE
- Tyr-d-Pen-Gly-Phe-d-Pen-OH (disulfide bridge between d-Pen2 andd-Pen5)
- CHO cell
- Chinese hamster ovary cell
- (−)-U50,488H
- (trans)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidiny)-cyclohexyl]benzeneacetamide methanesulfonate
- N/OFQ
- nociceptin/orphanin FQ
- BSA
- bovine serum albumin
- Received October 5, 2000.
- Accepted January 17, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}