


Abstract
We examined the role of the sarcolemmal and mitochondrial ATP-sensitive potassium (KATP) channel in a rat model of myocardial infarction after stimulation with the selective δ1-opioid receptor agonist TAN-67. Hearts were subjected to 30 min of regional ischemia and 2 h of reperfusion. Infarct size was expressed as a percentage of the area at risk. TAN-67 significantly reduced infarct size/area at risk (29.6 ± 3.3) versus control (63.1 ± 2.3). The sarcolemmal-selective KATP channel antagonist HMR 1098, administered 10 min before TAN-67, did not significantly attenuate cardioprotection (26.0 ± 7.3) at a dose (3 mg/kg) that had no effect in the absence of TAN-67 (56.3 ± 4.3). Pretreatment with the mitochondrial selective antagonist 5-hydroxydecanoic acid (5-HD) 5 min before the 30-min occlusion completely abolished TAN-67-induced cardioprotection (54.3 ± 2.7), but had no effect in the absence of TAN-67 (62.6 ± 4.1), suggesting the involvement of the mitochondrial KATP channel. Additionally, we examined the antiarrhythmic effects of TAN-67 in the presence or absence of 5-HD and HMR 1098 during 30 min of ischemia. Control animals had an average arrhythmia score of 10.40 ± 2.41. TAN-67 significantly reduced the arrhythmia score during 30 min of ischemia (2.38 ± 0.85). 5-HD and HMR 1098 in the absence of TAN-67 produced an insignificant decrease in the arrhythmia score (8.80 ± 2.56 and 4.20 ± 1.07, respectively). 5-HD administration before TAN-67 treatment abolished its antiarrhythmic effect (4.71 ± 1.11). However, HMR 1098 did not abolish TAN-67-induced protection against arrhythmias (1.67 ± 0.80). These data suggest that δ1-opioid receptor stimulation is cardioprotective against myocardial ischemia and sublethal arrhythmias and suggest a role for the mitochondrial KATP channel in mediating these cardioprotective effects.
Recent evidence suggests that agonists of the opioid receptor induce cardioprotection. This observation was first made by Schultz et al. (1995) in an in vivo rat model of myocardial infarction. They subsequently determined that this was a δ1-, but not μ- or κ-opioid receptor-mediated event (Schultz et al., 1998a). Subsequently, we showed that opioids, via a Gi/o protein-linked mechanism, induce cardioprotection that is sensitive to inhibitors of the ATP-sensitive potassium (KATP) channel (Schultz et al., 1998b). Similarly, Miki et al. (1998) have found that cardioprotection induced by morphine in the rabbit myocardium is mediated by protein kinase C (PKC).
Cardioprotection via opioids may be an integral player in ischemic preconditioning (IPC), where brief periods of ischemia and reperfusion before a sustained ischemic stress paradoxically protect the heart.Murry et al. (1986) first demonstrated this effect in the intact dog. Since this investigation, much research has focused on the mechanism of IPC. IPC can be induced pharmacologically via stimulation of numerous Gi protein-coupled receptors, including opioids (Schultz et al., 1998b). These pharmacological agents may induce a multiple kinase-mediated mechanism, similar to that of IPC, including PKC (Speechly-Dick et al., 1994; Ytrehus et al., 1994; Ping et al., 1997; Fryer et al., 1999b), tyrosine kinase (Kukreja and Qian, 1997;Fryer et al., 1998), members of the mitogen-activated protein (MAP) kinase family (Weinbrenner et al., 1997; Ping et al., 1998), and MAP kinase-activated protein kinase 2 (Maulik et al., 1996, 1998).
The end effector of IPC is thought to be a potassium channel negatively modulated by ATP, the KATP channel. This was first proposed by Gross and Auchampach (1992) who demonstrated in dogs that the KATP channel inhibitor glibenclamide abolished IPC-induced cardioprotection. Since the demonstration of functional KATP channels in the mitochondrial inner membrane (Inoue et al., 1991), investigators have questioned whether this ion channel, as opposed to the sarcolemmal KATP channel, may be the mediator of IPC (for review, see Gross and Fryer, 1999). Yao and Gross (1994) first suggested that an intracellular site may be responsible for protection from IPC because cardioprotection was independent of action potential shortening. Selective inhibitors of these channels have recently become available and we have demonstrated that inhibition of the mitochondrial, but not sarcolemmal, KATP channel blocks IPC in the rat heart (Fryer et al., 2000).
Opioid receptor agonists also may possess antiarrhythmic properties. Recent evidence by Yu et al. (1999) suggests that stimulation of the κ-opioid receptor with U50,488H is protective against arrhythmias and intracellular calcium oscillations induced by β-adrenoceptor stimulation of a cAMP-dependent, PKC independent pathway. Similarly,Pepe et al. (1997) have demonstrated that the δ-opioid receptor agonist leucine enkephalin inhibited β1-adrenoceptor stimulation of cAMP, and that β1-adrenoceptor/δ-opioid receptor “cross talk” occurs via a pertussis toxin-sensitive G protein. Because cAMP production is thought to be a major factor contributing to arrythmias (Podzuweit et al., 1981), this may suggest that δ-opioid receptor stimulation is antiarrhythmic. However, Murphy and Murphy (1999)recently suggested that peripheral opioid receptor blockade with methylnaltrexone reduced the incidence of both ventricular fibrillation and arrhythmia score during 40 min of coronary artery occlusion. Similarly, investigators have reported that agonists of the opioid receptor or opioid peptides may increase the number of arrhythmias resulting from myocardial ischemia (Lee et al., 1992; Wu et al., 1993). The effects of stimulation of the δ1-opioid receptor on cardiac arrhythmias were examined in this study.
Our data demonstrate that the mitochondrial KATPchannel is an important component of opioid receptor-induced cardioprotection and that the δ1-opioid receptor agonist TAN-67 induces potent antiarrhythmic and antifibrillatory effects during sustained ischemia that also can be abolished with an antagonist of the mitochondrial KATP channel.
Materials and Methods
General Surgical Preparation.
This study was performed in accordance with the guidelines of the Animal Care Committee of the Medical College of Wisconsin, which is accredited by the American Association of Laboratory Animal Care. Male Wistar rats, 350 to 450 g, were used for all phases of this study. The rats were anesthetized via i.p. administration of Inactin (100 mg/kg), a long-acting barbiturate. A tracheotomy was performed, and the trachea was intubated with a cannula connected to a rodent ventilator (model CIV-101; Columbus Instruments, Columbus, OH, or model 683; Harvard Apparatus, South Natick, MA). The rats were ventilated with room air supplemented with O2 at 60 to 65 breaths/min. Atelectasis was prevented by maintaining a positive end-expiratory pressure of 5 to 10 mm H2O. Arterial pH, pCO2, and pO2 were monitored at control, 15 min of occlusion, and 60 and 120 min of reperfusion by a blood gas system (AVL 995 pH/blood gas analyzer) and maintained within a normal physiological range (pH 7.35 to 7.45; pCO2 25 to 40 mm Hg; and pO2, 80–110 mm Hg) by adjusting the respiratory rate and/or tidal volume. Body temperature was maintained at 38°C by the use of a heating pad and bicarbonate was administered i.v. as needed to maintain arterial blood pH within normal physiological levels.
The right carotid artery was cannulated to measure blood pressure and heart rate via a Gould PE50 or PE23 pressure transducer connected to a Grass (model 7) polygraph. The right jugular vein was cannulated for saline, bicarbonate, and drug infusion. A left thoracotomy was performed at the 5th intercostal space followed by a pericardiotomy and adjustment of the left atrial appendage to reveal the location of the left coronary artery. A ligature (6-0 prolene) was passed below the left descending vein and coronary artery from the area immediately below the left atrial appendage to the right portion of the left ventricle. The ends of the suture were threaded through a propylene tube to form a snare. Pulling the ends of the suture taut and clamping the snare onto the epicardial surface with a hemostat elicited occlusion of the coronary artery and resulted in regional left ventricular ischemia. Epicardial cyanosis and subsequent decrease in blood pressure verified coronary artery occlusion. Reperfusion of the heart was initiated via unclamping the hemostat and loosening the snare and was confirmed by visualizing an epicardial hyperemic response. Heart rate and blood pressure were allowed to stabilize before the following protocols were initiated.
Study Groups and Experimental Protocols.
Rats were randomly assigned to one of six groups (Fig.1). All groups underwent a 30-min coronary artery occlusion and 2 h of reperfusion. The control group (Con) underwent only a 30-min coronary artery occlusion and subsequent 2 h of reperfusion. TAN-67 (10 mg/kg) was infused for 15 min before prolonged ischemia and reperfusion. The mitochondrial selective KATP channel antagonist 5-hydroxydecanoic acid (5-HD) or the sarcolemmal selective KATP channel antagonist HMR 1098 were administered in the presence or absence of TAN-67. HMR 1098 (3 mg/kg), was administered 15 min before the control protocol. The effects of HMR 1098 on TAN-67-induced cardioprotection were examined by administering HMR 1098 10 min before TAN-67 administration. Similarly, the effect of pretreatment with 5-HD in the absence or presence of TAN-67 was examined. 5-HD was administered 5 min before 30 min of coronary artery occlusion and 2 h of reperfusion or during the last 5 min of the TAN-67 infusion. We have previously demonstrated that 3 mg/kg HMR 1098 and 10 mg/kg 5-HD are optimal dosages to determine the effectiveness of inhibiting either the sarcolemmal or mitochondrial KATP channel in ischemic preconditioning (Fryer et al., 2000). Additionally, we have previously demonstrated that 5-HD is maximally effective at inhibiting cardioprotection when administered 5 but not 10 or 30 min before ischemic preconditioning (Fryer et al., 2000).

Protocol bar depicting the experiments used to determine the role of the mitochondrial or sarcolemmal KATPchannel in the cardioprotection from δ1-opioid receptor stimulation. Control animals were subjected to 30 min of coronary artery occlusion and 2 h of reperfusion. TAN-67 was infused 15 min before the control protocol in the presence or absence of 5-HD or HMR 1098.
Determination of Infarct Size (IS).
On completion of the above-mentioned protocols, the coronary artery was reoccluded and the area at risk (AAR) determined by negative staining. Patent blue dye was administered via the jugular vein to effectively stain the nonoccluded area of the left ventricle. The rat was euthanized with a 15% KCl solution. The heart was excised, and the left ventricle was removed from the remaining tissue and subsequently cut into six thin cross-sectional pieces. This allowed for the delineation of the normal area (stained blue) versus the AAR that subsequently remained pink. The AAR was excised from the nonischemic area and the tissues were placed in separate vials and incubated for 15 min with a 1% triphenyltetrazolium chloride (TTC) stain in 100 mM phosphate buffer (pH = 7.4) at 37°C. TTC is an indicator of viable and nonviable tissue (Klein et al., 1981). Tissues were stored in vials of 10% formaldehyde overnight and the infarcted myocardium was dissected from the AAR under the illumination of a dissecting microscope (Cambridge Instruments, Boston, MA). IS and AAR were determined by gravimetric analysis. IS is expressed as a percentage of the AAR (IS/AAR).
Determination of Arrhythmia Score.
We analyzed the antiarrhythmic properties of TAN-67 in the absence or presence of 5-HD or HMR 1098. Arrhythmias were quantitated via a modified scoring system previously validated by Curtis and Walker (1988) in an in vivo model of myocardial ischemia. Our scoring system assigned scores during the ten 3-min intervals of myocardial ischemia. Arrhythmia scores were assigned as follows: 0 = <10 premature ventricular contractions (PVCs)/3-min period; 1 = 10 to 50 PVCs/3-min period; 2 = >50 PVCs/3-min period; 3 = 1 episode of ventricular fibrillation (VF)/3-min period; 4 = 2 to 5 episodes of VF/3-min period; and 5 = >4 episodes of ventricular fibrillation/3-min period.
Exclusion Criteria.
A total of 38 rats successfully completed the above-mentioned protocols for IS investigation and 51 rats were analyzed for arrhythmia determination. Rats were excluded from data analysis if they exhibited severe hypotension (<30 systolic blood pressure) or if we were unable to maintain adequate blood gas values within a normal physiological range due to metabolic acidosis.
Drugs.
Inactin (thiobutabarbital sodium) and 5-HD were purchased from Research Biochemicals (Natick, MA). TTC was purchased from Sigma Chemical Co. (St. Louis, MO). TAN-67 was kindly synthesized and furnished by Dr. Hiroshi Nagase of Toray Industries, Kanagawa, Japan. HMR 1098, the sodium salt of HMR 1883, was a generous gift from Hoechst-Marion-Roussel (Frankfurt, Germany). Inactin and HMR 1098 were dissolved in distilled water. 5-HD was dissolved in saline. All drugs were dissolved in approximately 0.9 ml of vehicle for administration at all doses.
Statistical Analysis.
All values are expressed as mean ± S.E. ANOVA with Newman-Keuls post hoc test were used to determine whether any significant differences existed among groups for hemodynamics, left ventricular weight (LV), IS, AAR, AAR/LV, IS/AAR, and number of arrhythmias. Significant differences were determined atP < .05.
Results
Hemodynamics.
Table 1summarizes heart rate, mean arterial blood pressure (MBP), and rate-pressure product in all groups determined at baseline, 15 min of coronary artery occlusion, and 120 min of reperfusion. Blood pressure was maintained at baseline after either TAN-67, 5-HD, or HMR 1098 treatment. No significant differences were found at baseline or 15-min postcoronary artery occlusion for all parameters. Heart rate, MBP, and rate-pressure product at 2 h of reperfusion was not significantly different between groups versus control and TAN-67; however, MBP for the group administered HMR 1098 in the presence of TAN-67 was significantly elevated from control and TAN-67-treated rats (P < .05).
Hemodynamics
IS and AAR.
There were no significant differences in any group versus control for LV (in grams) or AAR expressed as a percentage of the left ventricle (Fig. 2). Control animals (n = 9) exhibited an IS/AAR of 63.1 ± 2.3 (Fig. 3). A 15-min infusion of TAN-67 (n = 7) significantly reduced IS (29.6 ± 3.3). HMR 1098 (n = 6) and 5-HD (n = 5) in the absence of TAN-67 did not significantly alter IS/AAR (56.3 ± 4.3 and 62.6 ± 4.1, respectively). Similarly, HMR 1098 administered 10 min before a 15-min TAN-67 infusion (n = 6) did not significantly attenuate cardioprotection (26.0 ± 7.3). However, 5-HD treatment during the last 5 min of the 15-min TAN-67 infusion (n = 8) completely abolished cardioprotection (54.3 ± 2.7). These data suggest a mitochondrial KATP channel-sensitive, sarcolemmal KATP channel-insensitive mechanism as a mediator of opioid-induced cardioprotection via the δ1-receptor.

AAR expressed as a percentage of the left ventricle in control (Con) animals and animals treated with the δ1-opioid agonist TAN-67, in the presence or absence of 5-HD or HMR 1098.

IS expressed as a percentage of the AAR in control (Con) animals and animals treated with the δ1-opioid agonist TAN-67 in the presence or absence of 5-HD or HMR 1098. *P < .05 versus Con.
Arrhythmia Score and Incidence of VF (Figs. 4 and 5).
The arrhythmias score in control animals (n = 10) was 10.40 ± 2.41 (Table 2). Nonlethal arrhythmias were significantly reduced via administration of TAN-67 (2.38 ± 0.85;n = 13). 5-HD (n = 10) in the absence of TAN-67 did not reduce or exacerbate arrhythmias induced by myocardial ischemia (8.80 ± 2.56). However, HMR 1098 (n = 5) in the absence of TAN-67 produced a smaller but insignificant number of arrhythmias versus the control group (4.20 ± 1.07). Although insignificant, TAN-67 in the presence of 5-HD retained some antiarrhyhtmic activity (4.71 ± 1.11;n = 7); however, HMR 1098 could not abolish the antiarrhythmic properties of TAN-67 (1.67 ± 0.80;n = 6). Because the method we have chosen to interpret arrhythmia score takes into account both PVCs and VF and because HMR 1098 is thought to be a potent antifibrillatory agent (Billman et al., 1998), we also decided to determine the percentage of animals susceptible to VF. Sixty percent of control animals and 40% of animals administered 5-HD exhibited reversible VF. However, none of the animals treated with HMR 1098 alone exhibited any bouts of VF, confirming that we were using an effective dose of this antifibrillatory compound. Similarly, stimulation of the δ1-opioid receptor with TAN-67 resulted in no bouts of VF when administered either alone or in combination with 5-HD or HMR 1098.
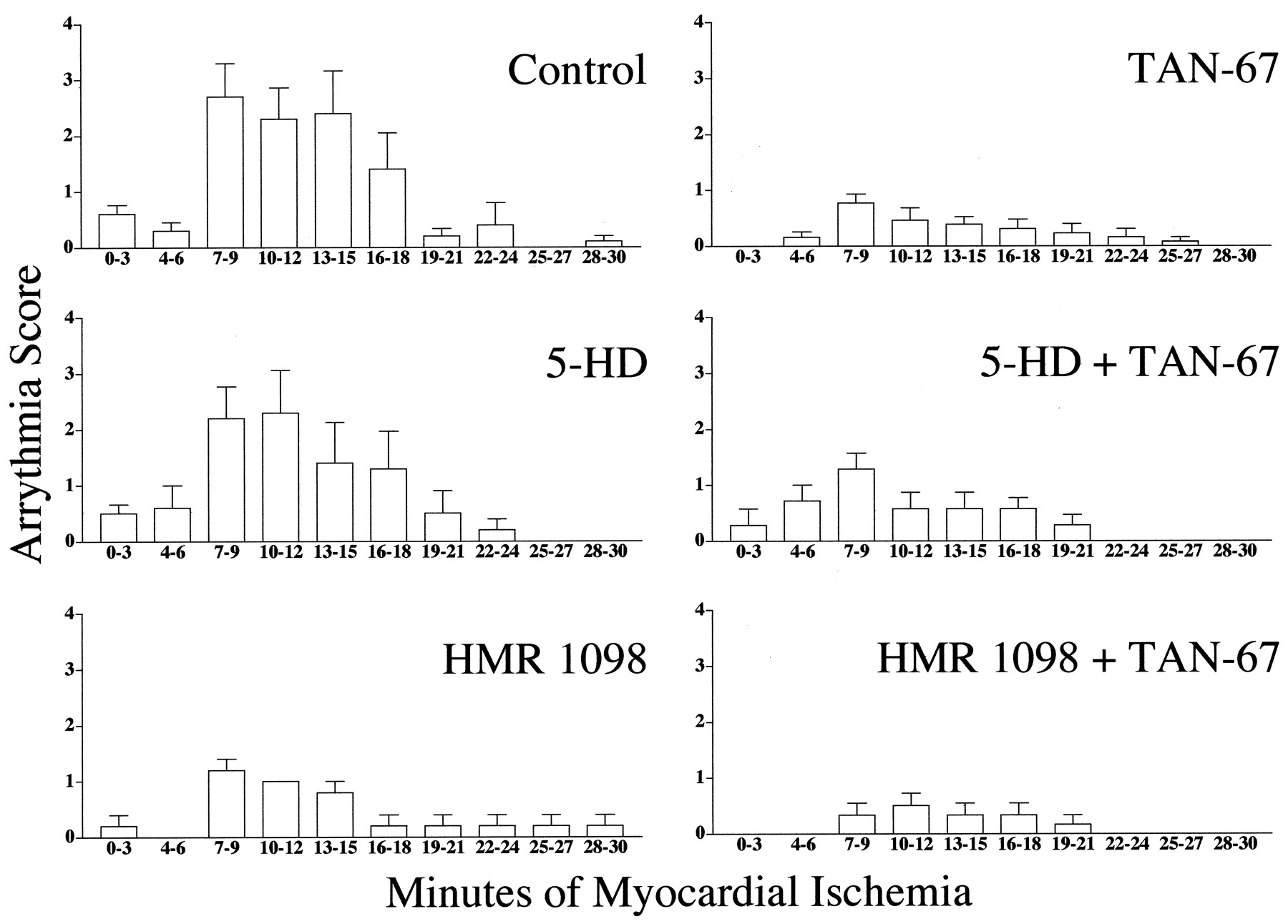
Incidence of cardiac arrhythmia during 3-min intervals in control animals and animals treated with the δ1-opioid agonist TAN-67 in the presence or absence of 5-HD or HMR 1098.

Total arrhythmias during 30 min of coronary artery occlusion in control (Con) animals and animals treated with the δ1-opioid agonist TAN-67 in the presence or absence of 5-HD or HMR 1098. *P < .05 versus control.
Arrythmia score
Discussion
These experiments demonstrate that inhibition of the mitochondrial, but not the sarcolemmal, KATPchannel can completely abolish cardioprotection induced by TAN-67, an agonist of the δ1-opioid receptor. We suggest that TAN-67-induced cardioprotection is mediated via activation of the mitochondrial KATP channel in the in vivo rat heart because the mitochondrial-selective KATPchannel inhibitor 5-HD, but not the sarcolemmal-selective KATP channel inhibitor HMR 1098, abolished cardioprotection. We have previously demonstrated that TAN-67-induced cardioprotection is mediated via a δ1-opioid receptor/Gi/o protein-coupled event that could be inhibited by pertussis toxin and the δ1-opioid receptor antagonist 7-benzylidenenaltrexone (BNTX) (Schultz et al., 1998b).
Because IS/AAR was not affected by pretreatment with HMR 1098, these experiments suggest that the sarcolemmal KATPchannel is not the main effector of cardioprotection, or that it may play only a minimal, supplementary role to the mitochondrial KATP channel. However, the potent antiarrhyhtmic and antifibrillatory effects of HMR 1098 were evident in this study. This is in agreement with Billman et al. (1998) who previously demonstrated the antifibrillatory effects of HMR 1883 induced by myocardial ischemia in conscious dogs. Their research demonstrates that HMR 1883 can prevent ischemia-induced reductions in the refractory period and incidence of VF. No animals treated with HMR 1098 in the presence or absence of TAN-67 exhibited VF. However, 60 and 40% of controls or animals administered 5-HD during a control protocol, respectively, exhibited VF. Similarly, we demonstrate that TAN-67 is antiarrhythmic, as determined by arrhythmia score, in the presence of HMR 1098. Although statistically insignificant, it appears that TAN-67 retains some antiarrhythmic properties in the presence of 5-HD and that HMR 1098 in the absence of TAN-67 may reduce arrythmia score. Although some research suggests that opioid agonists have no effect (Wong et al., 1990) or may be proarrhythmic (Lee et al., 1992; Wu et al., 1993), we demonstrate that TAN-67 induces a potent antiarrhythmic and antifibrillatory effect. Maslov et al. (1996) recently demonstrated that rats adapted to stress have an increased level of opioid peptides and show a decreased severity and incidence of cardiac arrhythmias induced by epinephrine that could be abolished with naloxone. In this study, the antiarrhythmic effects of TAN-67 could be blunted by the mitochondrial KATP channel antagonist 5-HD. These data are consistent with our observation that 5-HD, but not HMR 1098, could abolish opioid-induced cardioprotection as analyzed by IS and are in agreement with Kita et al. (1998) who demonstrated that 5-HD and naloxone could inhibit IPC-induced suppression of reperfusion arrhythmias in the rat.
These data are also in agreement with our previous observation that an opioid-induced “second window” of cardioprotection is mediated by mitochondrial KATP channels (Fryer et al., 1999a). Intraperitoneal injection of TAN-67 48 h before 30 min of ischemia and 2 h of reperfusion reduced IS compared with control animals administered saline. This response could be abolished by the δ1-opioid receptor antagonist BNTX and 5-HD, suggesting a δ1-opioid receptor-mediated event and the downstream involvement of the mitochondrial KATP channel.
The involvement of the KATP channel in opioid-induced cardioprotection has been previously identified in our laboratory with glibenclaminde, a nonselective inhibitor of the sarcolemmal and mitochondrial KATP channel (Schultz et al., 1996, 1998b). However, the advent of sarcolemmal or mitochondrial selective inhibitors of these channels has proved to be useful in dissecting out opioid-induced signaling pathways. Hoechst-Marion-Roussel recently synthesized the cardiac selective antifibrillatory drug HMR 1883 and its accompanying sodium salt HMR 1098 (Billman et al., 1998; Gogelein et al., 1998). Similarly, Marban's laboratory recently suggested that HMR 1883 and 1098 are sarcolemmal-selective inhibitors of the KATPchannel in rabbit myocytes (Sato et al., 2000). Marban's group also has demonstrated that 5-HD is selective for the mitochondrial KATP channel in rabbit cardiomyocytes (Sato et al., 1998). We have previously demonstrated that 5-HD exhibited a time-dependent inhibition of the mitochondrial KATP channel in Wistar rats (Fryer et al., 2000). We demonstrated that IPC-induced cardioprotection could be markedly attenuated with a 5-min pretreatment with 5-HD; however, when pretreatment was extended to either 10 or 30 min before IPC, 5-HD did not abolish cardioprotection. These data suggest that 5-HD is rapidly metabolized in Wistar rats and suggest that use of 5-HD as a selective inhibitor of the mitochondrial KATP channel in these rats requires time considerations similar to those previously shown in our laboratory for glibenclamide (Schultz et al., 1997b). However, we have found that 5-HD, administered 15 min before IPC in Sprague-Dawley rats (Schultz et al., 1997a), could still abolish the cardioprotective effects of IPC. Similar results have been found when 5-HD is administered 15 min before ischemia in rabbits (Janin et al., 1998; Bernardo et al., 1999). Therefore, Wistar rats may possess the unique ability to rapidly metabolize this drug. Similarly, because the dose of HMR 1098 in this study was antifibrillatory, the dose and time of HMR 1098 administration was adequate to block sarcolemmal KATP channels.
The reported proarrhythmic effects of potassium channel openers during myocardial ischemia may result in VF and sudden cardiac death. This is based on the potential of potassium channel-opening drugs to exacerbate ischemia-induced shortening of the action potential via rapid efflux of potassium through the sarcolemmal membrane (Wilde, 1994). Therefore, inhibition of this channel with HMR 1098 may be expected to limit the arrhythmia score in this investigation. However, we did not find a reduction in PVCs, but found a complete abolishment of the incidence of VF. Additionally, we demonstrated that TAN-67 maintains antiarrhythmic activity in the presence of HMR 1098. This may be due to nonopiate receptor-mediated effects, possibly via inhibition of Ca2+ current in the sarcolemmal membrane (Utz et al., 1995). Indeed, it has been previously demonstrated that stimulation of the κ-opioid receptor with 10−6mol/l U50,488H can abolish β-adrenoceptor-induced arrhythmias and intracellular Ca2+ oscillations (Yu et al., 1999). However, reports from the same laboratory also have reported that U50,488H (80, 400, and 800 nmol) is proarrhythmic in a dose-dependent manner via a phopholipase C/inositol phosphate3/Ca2+ and pertussis toxin-sensitive pathway (Bian et al., 1998). It appears that nanomolar concentrations of U50,488H may be proarrhythmic, whereas micromolar concentrations may be antiarrhythmic. Lishmanov et al. (1999) also have recently suggested that the κ1- and κ2-opioid agonist U-62066 is antiarrhythmic against adrenaline-induced dysrhythmias in rats via the activation of peripheral opioid receptors. In this study, we have not determined whether the antiarrhythmic effect of TAN-67 is a receptor-mediated event.
Although we suggest that the mitochondrial KATPchannel may play a role in opioid-induced acute cardioprotection, the signal transduction mechanism leading to KATPchannel activation remains elusive. PKC may serve as an integral mediator of this protection (Miki et al., 1998). There is strong evidence for the involvement of PKC in ischemic preconditioning (Liu et al., 1994; Ytrehus et al., 1994; Fryer et al., 1999b). Similarly, Miki et al. (1998) recently identified PKC as a component of morphine-induced cardioprotection in rabbits. Although morphine is thought to act via stimulation of the μ-opioid receptor, its cardioprotective effect does not appear to be the result of activation of this receptor. In this regard, Liang and Gross (1999) demonstrated that morphine-induced cardioprotection of chick cardiac ventricular myocytes can be abolished by the selective δ1-opioid receptor antagonist BNTX. Similarly, preliminary evidence from our laboratory suggests that PKC is an integral component of δ1-opioid receptor-induced cardioprotection and suggests that specific PKC-isoforms translocate to the sarcolemmal membrane, intercalated disk, and mitochondria after TAN-67 administration. Similarly, preliminary evidence from our laboratory suggests that the MAP kinase extracellular signal-regulated kinase is activated to a greater extent during reperfusion when administered with TAN-67 versus control animals. Similarly, Gutstein et al. (1997) have demonstrated in COS-7 cells that μ- or δ-opioid receptor stimulation could induce potent activation of extracellular signal-regulated kinase, but had weak or no effects on the stress-activated MAP kinases.
We have recently provided the first evidence to link mitochondrial KATP channel activation and cardioprotection at the level of mitochondrial function (Fryer et al., 2000). We demonstrated that 5-HD significantly attenuated the protective effect of IPC to preserve mitochondrial ATP synthesis and reduce IS. 5-HD pretreatment 5 min before IPC decreased the rate of ATP synthesis to 63% of the rate measured in IPC-treated hearts and partially abolished the reduction in IS produced by IPC. In contrast, HMR 1098 had no effect on the protective effect of IPC on mitochondrial ATP synthesis nor did it abolish cardioprotection based on IS reduction. Based on assessment of both IS as an index of cardiac injury and ATP synthesis as an index of mitochondrial function, we suggested a link between IPC, mitochondrial KATP channel activation, and improved mitochondrial function in the intact rat heart. However, preliminary evidence from our laboratory suggests that administration of TAN-67 does not induce a recovery of mitochondrial function versus control animals based on ATP synthesis through complex I of the mitochondrial electron transport chain (our unpublished observation). Therefore, the preservation of mitochondrial function during IPC may be an added benefit of, but not necessarily a causal factor in cardioprotection from IPC, whereas the lesser cardioprotection from opioid receptor stimulation versus IPC may not be sufficient to improve mitochondrial bioenergetics.
In summary, aforementioned studies in combination with the current studies from our laboratory support a role for the mitochondrial KATP channel in a rat model of pharmacologically induced cardioprotection. These conclusions are supported by the demonstration that the mitochondrial KATPchannel-selective antagonist 5-HD, but not the sarcolemmal-selective KATP channel antagonist HMR 1098, can effectively abolish opioid-induced cardioprotection in the in vivo rat model. Additionally, we demonstrate that the potent antiarrhythmic and antifibrillatory properties of TAN-67 and demonstrate that inhibition of the mitochondrial but not sarcolemmal, KATPchannel can abolish these effects.
Footnotes
-
Send reprint requests to: Garrett J. Gross, Ph.D., Department of Pharmacology and Toxicology, Medical College of Wisconsin, 8701 Watertown Plank Rd., Milwaukee, WI 53226-3548.
-
↵1 This study was funded in part by a predoctoral research grant from the American Heart Association (to R.M.F.) and National Institutes of Health Grant HL08311 (to G.J.G.).
- Abbreviations:
- KATP
- ATP-sensitive potassium channel
- PKC
- protein kinase C
- IPC
- ischemic preconditioning
- MAP
- mitogen-activated protein
- 5-HD
- 5-hydroxydecanoic acid
- IS
- infarct size
- AAR
- area at risk
- TTC
- triphenyltetrazolium chloride
- IS/AAR
- IS expressed as a percentage of AAR
- PVC
- premature ventricular contraction
- VF
- ventricular fibrillation
- LV
- left ventricular weight
- MBP
- mean arterial blood pressure
- BTXN
- 7-benzylidenenaltrexone
- Received February 29, 2000.
- Accepted April 17, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}