


Abstract
HEK 293 cells stably expressing the human serotonin transporter (hSERT) were grown on coverslips, preincubated with [3H]5-hydroxytryptamine (5-HT), and superfused. Substrates of the hSERT [e.g., p-chloroamphetamine (PCA)], increased the basal efflux of [3H]5-HT in a concentration-dependent manner. 5-HT reuptake blockers (e.g., imipramine, paroxetine) also raised [3H]5-HT efflux, reaching approximately one-third of the maximal effect of the hSERT substrates. In uptake experiments, both groups of substances inhibited [3H]5-HT uptake. Using the low-affinity substrate [3H]N-methyl-4-phenylpyridinium (MPP+) to label the cells in superfusion experiments, reuptake inhibitors failed to enhance efflux. Similar results were obtained using human placental choriocarcinoma (JAR) cells that constitutively express the hSERT at a low level. By contrast, PCA raised [3H]MPP+ efflux in both types of cells, and its effect was inhibited by paroxetine. The addition of the Na+,K+-ATPase inhibitor ouabain (100 μM) to the superfusion buffer enhanced basal efflux of [3H]5-HT-loaded hSERT cells by approximately 2-fold; the effect of PCA (10 μM) was strongly augmented by ouabain, whereas the effect of imipramine was not. The Na+/H+ionophore monensin (10 μM) also augmented the effect of PCA on efflux of [3H]5-HT as well as on efflux of [3H]MPP+. In [3H]5-HT-labeled cells, the combination of imipramine and monensin raised [3H]5-HT efflux to a greater extent than either of the two substances alone. In [3H]MPP+-labeled cells, imipramine had no effect on its own and fully reversed the effect of monensin. The results suggest that the [3H]5-HT efflux caused by uptake inhibitors is entirely due to interrupted high-affinity reuptake, which is ongoing even under superfusion conditions.
The action of serotonin [5-hydroxytryptamine (5-HT)] released at nerve terminals is ended by high-affinity reuptake of the neurotransmitter from the synaptic cleft. This process is mediated by a selective Na+- and Cl−-dependent serotonin transporter (SERT). Moreover, the SERT is responsible not only for inward transport of released transmitter but also for outward movement of transmitter under certain pharmacological, physiological, and pathophysiological conditions (Levi and Raiteri, 1993).
Many drugs of therapeutic interest as well as drugs of abuse exert their action via this transmembrane protein. Antidepressants such as the tricyclics (e.g, imipramine, clomipramine) or the selective serotonin reuptake inhibitors (e.g., fluoxetine, citalopram) bind to the transporter and prevent substrate translocation. The subsequent rise of the 5-HT concentration in the vicinity of the postsynaptic receptors is believed to be the initial step in the therapeutic action of these drugs. On the other hand, the anorectic agent fenfluramine or the amphetamine derivative p-chloroamphetamine (PCA) competes with 5-HT for translocation and thereby promotes release of 5-HT both in vivo (Fuller et al., 1965) and in vitro (Rudnick and Wall, 1992; Gobbi et al., 1993) by a reversed transport mechanism.
Classic experimental models to study carrier-mediated serotonin transport are synaptosomes or blood platelets (for a review, seeRudnick, 1997). However, there are some disadvantages to these systems. In synaptosomes, for instance, the characterization of the neurochemical events associated with the uptake and release of a specific neurotransmitter is complicated by their metastable structure and great heterogeneity. Thrombocytes as well as synaptosomes not only transport 5-HT across their plasmalemmal membrane via the SERT but also concentrate 5-HT intracellularly in vesicles via the vesicular monoamine transporter. Therefore, cellular systems containing only the SERT have met with increased interest in studies on the function and regulation of this protein. In particular, an analysis of carrier-mediated processes on a molecular level has been allowed by the cloning and heterologous expression of high numbers of the human SERT (hSERT) in mammalian cells (Ramamoorthy et al., 1993; for a review, seeBlakely et al., 1997). Typically in these studies, the cells are grown in wells and incubated with radiolabeled transmitter in the absence and presence of drugs. For measurements of transporter-mediated efflux, the cells are first loaded with radiolabel, and then the release of radioactivity is monitored by pipetting medium from the well or by measuring the amount of radiolabel in the cells. This approach, however, has methodological limitations, because it detects only changes in net movement of neurotransmitter molecules, which may result from influences on inward or outward transport across the membrane. It also does not allow a satisfactory time resolution of the process under study. To circumvent these disadvantages, we applied a method that allows the superfusion of transfected cells after they have been loaded with radiolabeled neurotransmitter (Pifl et al., 1995). Superfusion is expected to quickly remove released radiolabel and thus to prevent reuptake (Raiteri et al., 1974).
Here, we present uptake and superfusion experiments in HEK 293 cells stably expressing the hSERT and in human placental choriocarcinoma (JAR) cells, which constitutively express the hSERT (Prasad et al., 1996). By using [3H]5-HT and [3H]N-methyl-4-phenylpyridinium (MPP+), two substrates of different affinity for the hSERT and of different transmembrane diffusion properties, as well as by experimental alterations of the transmembrane ion gradients with ouabain or monensin, we were able to distinguish drug effects on carrier-mediated inward and outward transport and to discern active reverse transport from diffusion of [3H]5-HT out of the cell.
Materials and Methods
Cell Line Transfection.
The plasmalemmal hSERT was originally cloned in pBluescript II KS−(Ramamoorthy et al., 1993). The hSERT cDNA was a generous gift of Dr. R. D. Blakely (Department of Pharmacology and Center for Molecular Neuroscience, School of Medicine, Vanderbilt University, Nashville, TN). The cDNA was excised with XbaI and HindIII and ligated into pEGFP-C1 (Clontech, Palo Alto, CA), which had been opened with NheI and HindIII and dephosphorylated. The GFP-tag is excised using these two enzymes, and therefore the cDNA is placed after the promoter without being modified. For stable expression into HEK 293 cells, the same method was used as described previously (Pifl et al., 1996). The stable transfectants (hSERT cells) were grown in minimal essential medium with Earle's salts andl-alanyl-l-glutamine (l-GlutaMAX I; Gibco Life Technologies, Grand Island, NY), 10% heat-inactivated fetal bovine serum, 50 mg/l gentamicin, and 500 μg/ml geneticin (G418) on 100-mm-diameter cell culture dishes at 37°C in an atmosphere of 5% CO2, 95% air.
JAR Cells.
JAR cells were a generous gift of Dr. H. Bönisch (Institute of Pharmacology and Toxicology, University of Bonn, Bonn, Germany). JAR cells were grown in RPMI-1640 medium, 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in an atmosphere of 5% CO2, 95% air.
Uptake Experiments.
The experiments were performed in poly(d-lysine)-coated 24-well plates (1 day after plating; 5 × 104 or 105cells/well for hSERT or JAR cells, respectively). Each well was washed with 0.5 ml of KRH buffer (Krebs-Ringer-HEPES buffer containing 10 mM HEPES, 120 mM NaCl, 3 mM KCl, 2 mM CaCl2, 2 mM MgCl2, final pH 7.3) and incubated with 0.05 ml of buffer containing 0.1 μCi of [3H]5-HT or [3H]MPP+ (27.5 Ci/mmol [3H]5-HT and 60 Ci/mmol [3H]MPP+, respectively) and various concentrations of unlabeled 5-HT, MPP+, or drugs in a total volume of 0.5 ml. After 2.5 min at room temperature, the uptake buffer was rapidly aspirated, and the cells were washed twice with 1 ml of ice-cold buffer. Cells were lysed with 0.5 ml of 1% SDS and transferred into scintillation vials for liquid scintillation counting. Nonspecific uptake was defined as uptake in the presence of 30 μM clomipramine and amounted to less than 2% of the total uptake in the presence of 120 mM NaCl. In experiments in which the effect of competing drugs was tested, the substances of interest were added 5 min before the addition of [3H]5-HT using at least six different concentrations.
Vmax,Km, EC50, and IC50 values were calculated using nonlinear regression fits performed with Prism (GraphPad, San Diego, CA). The equation used to estimate Km andVmax values was Y =Vmax ×X/(Km + X), where Y = V (picomoles per 106 cells per minute) and X = substrate concentration (moles per liter).
Uptake experiments on parental HEK 293 cells were performed in poly(d-lysine)-coated 12-well plates (1 day after plating; 2 × 105 cells in 500 μl of medium/well). The final concentrations of 5-HT and MPP+ were 10 μM; 20 μl of unlabeled 5-HT (260 μM) was added for HPLC experiments; for radiochemical experiments 2 μCi [3H]5-HT was added additionally. To study MPP+ accumulation, 260 μM unlabeled MPP+ and 2 μCi of [3H]MPP+ were used. After several time points of incubation at 37°C, the medium was removed quickly, and each well was washed twice with 2 ml of ice-cold KRH buffer. To perform HPLC detection, cells were lysed with 500 μl of 0.1 M perchloric acid and transferred to a 1.5-ml tube. The wells were washed with an additional 500 μl of 0.1 M perchloric acid. The cells were sonicated and centrifugated at 15,000g for 10 min at 4°C. The supernatant was transferred onto the HPLC column. For radiochemical detection, cells were lysed with 1 ml of 1% SDS and transferred into scintillation vials for liquid scintillation counting.
Superfusion Experiments.
The cells were grown overnight onto round glass coverslips (diameter, 5 mm) coated with poly(d-lysine) (25 μg/ml) at 2 × 104 cells/well for hSERT cells, 105 cells/well for JAR cells, and 4 × 104 cells/well for parental HEK 293 cells. For preincubation with labeled substrates, the following conditions applied: hSERT cells were incubated with [3H]5-HT (5 μM, 0.38 μCi/well) or [3H]MPP+ (10 μM, 1.54 μCi/well) for 20 min at 37°C in a final volume of 0.225 ml of culture medium. JAR cells were incubated with [3H]5-HT (1.7 μM, 2.3 μCi/well) or [3H]MPP+ (17 μM, 1.88 μCi/well) for 30 min at 37°C in a final volume of 0.125 ml of culture medium. Parental HEK 293 cells were incubated with [3H]5-HT (3.8 μM, 10 μCi/well) for 60 min at 37°C in a final volume of 0.11 ml of culture medium. Coverslips were then transferred to small superfusion chambers (0.2 ml) and superfused with KRH buffer (0.7 ml/min) as described recently (Pifl et al., 1995). Because of the very rapid loss of [3H]5-HT at 37°C, which seriously interfered with the observation of drug effects, a superfusion temperature of 25°C was chosen. A similar observation was made by Johnson et al. (1998) in hSERT transfected C6 glioma cells. After a washout period of 45 min to establish a stable efflux of radioactivity, the experiment was started with the collection of fractions (4-min duration). At the end of the experiment, cells were lysed in 1% SDS. Tritium in the superfusate fractions and in the SDS lysates was determined by liquid scintillation counting. The release of 3H was expressed as fractional rate (i.e., the radioactivity released during a fraction was expressed as a percentage of the total radioactivity present in the cells at the beginning of that fraction). Drug-induced release was calculated by subtracting the estimated basal release from total release during the first 8 min of drug exposure and is expressed as a percentage of radioactivity in the cell at the beginning of drug exposure.
HPLC Experiments.
The chromatography system consisted of a Jasco 980 HPLC pump (0.8 ml/min; Jasco International, Tokyo, Japan), a Rheodyne 7125 valve (100-μl loop; Rheodyne, Cotati, CA), a reversed phase column (250 × 4.6 mm; Hi-Pore RP Column; Bio-Rad Laboratories, Richmond, CA), and an LC-4C electrochemical detector (Bioanalytical Systems, West Lafayette, IN). The detector potential was set at 600 mV versus an Ag/AgCl reference electrode [mobile phase: 75 mM NaH2PO4, 0.11 mM EDTA, 0.2 mM sodium octane sulfonate, and 12% (v/v) methanol, with a final pH of 4.05].
Chemicals.
Tissue culture reagents were from Gibco Life Technologies. RPMI-1640 medium was obtained from Sigma-Aldrich Handels GmbH (Vienna, Austria). [3H]MPP+ and [3H]5-HT were purchased from New England Nuclear Life Sciences Products (Boston, MA). Paroxetine was kindly donated by SmithKline Beecham (Worthing, UK). MPP+ and fluoxetine were obtained from Research Biochemicals International (Natick, MA). Monensin,d-fenfluramine, PCA, imipramine, and serotonin were obtained from Sigma-Aldrich Handels GmbH. All other chemicals were obtained from commercial sources.
Statistical Analysis.
All results were expressed as mean ± S.E. values. Statistical analysis consisted of Spearman's correlation and repeated measures ANOVA followed by t test for paired or independent observations, as appropriate. A value ofP < .05 was considered to be the level of statistical significance.
Results
HEK 293 cells permanently expressing the hSERT exhibited a clomipramine-sensitive and time-, temperature-, and concentration-dependent accumulation of [3H]5-HT. Saturation analysis of initial uptake rates revealed a Vmax value of 141 ± 17 pmol/min/106 cells and aKm value of 0.396 ± 0.034 μM (mean ± S.E. of eight independent determinations). When [3H]MPP+ was used as substrate, a Vmax value of 196 ± 4 pmol/min/106 cells and aKm value of 13.68 ± 3.6 μM (mean ± S.E. of three independent determinations) were obtained.
Effects of hSERT Substrates and 5-HT Reuptake Inhibitors on Release and Uptake of [3H]5-HT.
Basal efflux of radioactivity from hSERT cells preincubated with [3H]5-HT amounted to 1.32 ± 0.07% during min 8 to 12, corresponding to 65.95 ± 5.43 pCi (n= 25; mean ± S.E.). HPLC analysis revealed that more than 95% of the radioactivity in superfusates and cells coeluted with authentic [3H]5-HT (data not shown). The addition of substrates of hSERT such as PCA or d-fenfluramine or inhibitors of 5-HT reuptake such as imipramine, paroxetine, or fluoxetine to the superfusion buffer caused a concentration- and time-dependent increase in [3H]5-HT efflux. Representative, the effects of PCA and imipramine are shown in Fig.1. Both drugs caused a concentration-dependent increase in efflux that reached a plateau after 8 min of drug exposure, with imipramine being approximately one-fourth as effective as PCA. Figure 2A shows concentration-response curves for various hSERT substrates and 5-HT reuptake inhibitors, and the resulting EC50values are given in Table 1. The maximum effects on release expressed as a percentage of radioactivity released during the first 8 min of drug exposure were 13 to 16% for the hSERT substrates and approximately 4% for the reuptake inhibitors. All drugs were also tested for inhibition of [3H]5-HT uptake. The concentration-response relationships are shown in Fig. 2B, and the corresponding IC50 values are given in Table 1. There was a good correlation between the log IC50 values obtained from uptake experiments with the log EC50 values obtained from superfusion experiments (r = 0.933, P < .01).

Time course of the effects of PCA and imipramine on the release of [3H]5-HT from HEK 293 cells expressing the hSERT. HEK 293 cells stably transfected with the hSERT were loaded with [3H]5-HT and superfused, and 4-min fractions were collected. After three fractions (12 min) of basal efflux, the buffer was switched to a buffer containing various concentrations of PCA (A) or imipramine (B). Data are presented as fractional release (i.e., each fraction is expressed as percentage of radioactivity present in the cells at the beginning of that fraction). Symbols represent mean ± S.E. of six observations (one observation = one superfusion chamber; triplicate determinations from two separate experiments).

Effect of hSERT substrates and of 5-HT reuptake inhibitors on uptake and release of [3H]5-HT in HEK 293 cells expressing the hSERT. A, concentration-response curves of hSERT substrates and 5-HT reuptake inhibitors for their effects on [3H]5-HT release. For experimental details, see Fig. 1. Drug-induced release was calculated from the total radioactivity released during the first 8 min of drug exposure minus estimated baseline release and expressed as a percentage of radioactivity present in the cells at the beginning of drug addition. Data are presented as fractional release (i.e., each fraction is expressed as a percentage of radioactivity present in the cells at the beginning of that fraction). Symbols represent mean ± S.E. of six to nine observations (one observation = one superfusion chamber; triplicate determinations from two or three independent experiments). The data were fitted by nonlinear regression. EC50 values are given in Table 1. B, concentration-inhibition curves of hSERT substrates or 5-HT reuptake inhibitors for their effects on [3H]5-HT uptake. The cells were incubated in 24-well plates with 0.1 μM [3H]5-HT for 2.5 min at room temperature as described inMaterials and Methods. Test drugs were added 5 min before [3H]5-HT using at least six different concentrations. Each well contained 105 cells. Nonspecific uptake was measured in the presence of 30 μM clomipramine. Symbols represent mean ± S.E. of 10 to 12 observations (duplicate determinations from five or six independent experiments). The data were fitted by nonlinear regression. IC50 values are given in Table 1. ▪, PCA; ▾, d-fenfluramine (d-FEN); ●, 5-HT; ▵, paroxetine; ■, fluoxetine; ○, imipramine.
IC50 and EC50 values of hSERT substrates and 5-HT reuptake inhibitors for inhibiting 5-HT uptake and inducing 5-HT release in HEK 293 cells expressing the hSERT
Interaction between hSERT Substrates and hSERT Blockers.
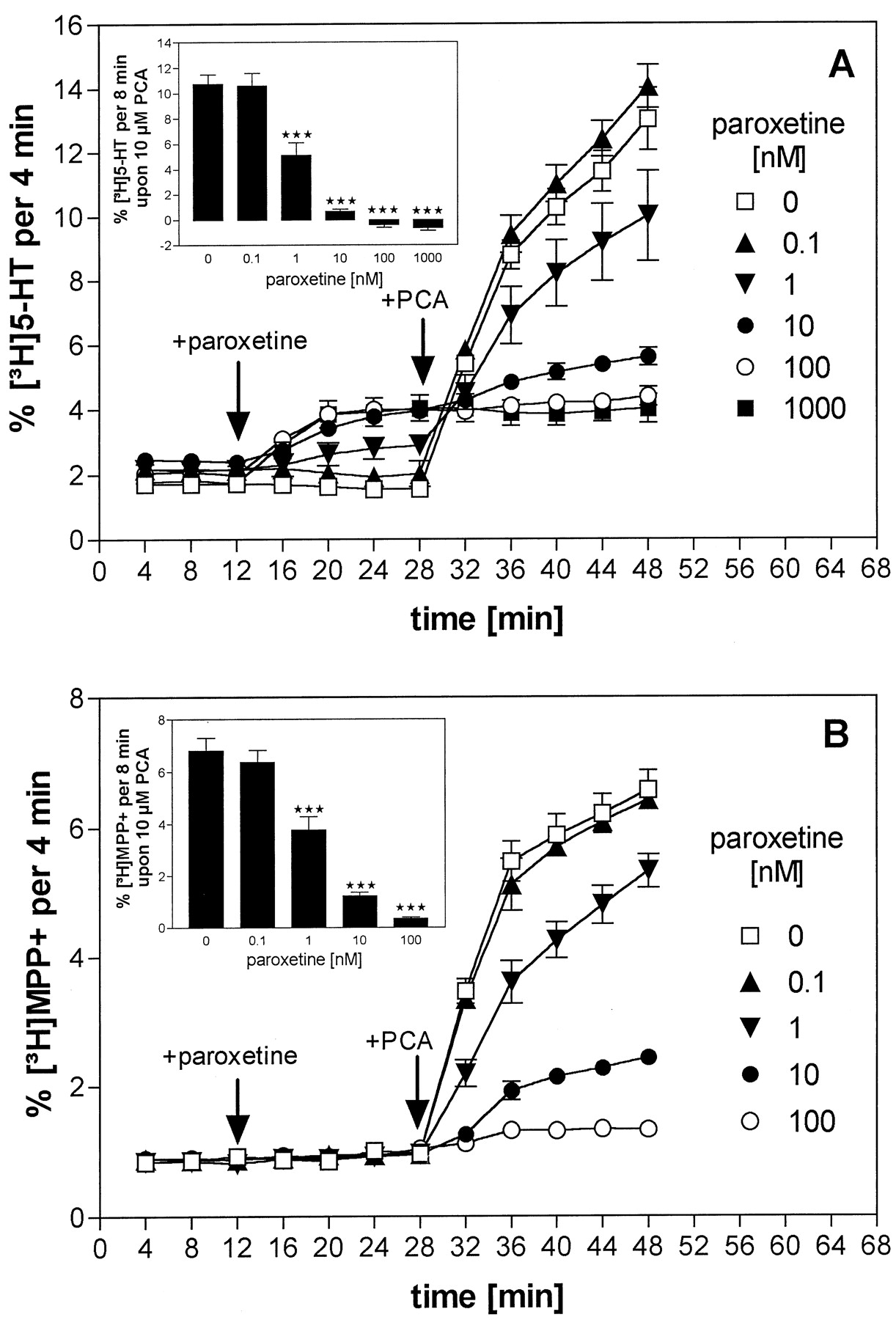
The effect of the potent uptake inhibitor paroxetine on PCA-induced [3H]5-HT release was analyzed. After a 16-min exposure to different concentrations of paroxetine (0.1 nM to 1 μM), PCA (10 μM) was added to the superfusion buffer for an additional 20 min. As shown in Fig. 3A, paroxetine alone increased [3H]5-HT efflux in a concentration-dependent manner but likewise inhibited further efflux induced by PCA. At paroxetine concentrations of more than 10 nM, the release-enhancing action of PCA was abolished. The same experiment was carried out using [3H]MPP+ as substrate instead of [3H]5-HT. Under these conditions, paroxetine did not increase [3H]MPP+ efflux but inhibited the effect of PCA, causing complete suppression at 100 nM (Fig. 3B).

Interaction of PCA and paroxetine on the release of [3H]5-HT or [3H]MPP+ from HEK 293 cells expressing the hSERT. HEK 293 cells stably transfected with the hSERT were loaded with [3H]5-HT (A) or with [3H]MPP+ (B) and superfused, and 4-min fractions were collected. After three fractions (12 min) of basal efflux, cells were exposed to buffers containing different concentrations of paroxetine (0.1 nM, ▴; 1 nM, ▾; 10 nM, ●; 100 nM, ○; 1 μM, ▪) or left at control conditions (■). At 16 min later, PCA (10 μM) was added to all superfusion channels. Data are presented as fractional release (i.e., each fraction is expressed as a percentage of radioactivity present in the cells at the beginning of that fraction). Symbols represent mean ± S.E. of six observations (one observation = one superfusion chamber; triplicate determinations from two separate experiments). Inset, drug-induced substrate-release during the first 8 min after PCA addition. ***P < .001 versus control conditions.
Parental HEK 293 Cells.
To investigate a possible role of diffusion in the efflux of [3H]5-HT or [3H]MPP+ from cells expressing the hSERT, parental HEK 293 cells were studied. The cells were incubated with the radiolabeled substrates (10 μM), and the amount of radioactivity entering the cells was measured. [3H]5-HT easily entered the cells during the 60-min incubation, reaching approximately 0.2 nmol/106 cells. By contrast, [3H]MPP+ displayed considerably lower intracellular accumulation (Fig.4A).

Uptake and superfusion experiments in parental HEK 293 cells. A, parental HEK 293 cells were incubated in 12-well plates with 10 μM [3H]5-HT or 10 μM [3H]MPP+ at 37°C for the times indicated. Each well contained 2 × 105 cells. Open columns ([3H]5-HT) and closed columns ([3H]MPP+) represent mean ± S.E. of 12 observations (performed in duplicate or triplicate determinations). B, parental HEK 293 cells were passively loaded with [3H]5-HT as described in Materials and Methods and superfused, and 4-min fractions were collected. After three fractions (12 min) of basal efflux, cells were either exposed to a buffer containing 10 μM PCA (▪) or 10 μM imipramine (●) or they were held at control conditions (○). Data are presented as fractional release (i.e., each fraction is expressed as a percentage of radioactivity present in the cells at the beginning of that fraction). Symbols represent mean ± S.E. of six observations (one observation = one superfusion chamber; triplicate determinations from two separate experiments).
In superfusion experiments with parental HEK 293 cells preloaded with [3H]5-HT (4 μM, 1 h), a very high fractional efflux rate of approximately 6% was observed. The addition of PCA (10 μM) or imipramine (10 μM) did not change the rate of efflux (Fig. 4B).
JAR Cells.
JAR cells, which constitutively express the hSERT at a low density, were studied in uptake and superfusion experiments. The cells concentration and time dependently accumulated [3H]5-HT. Saturation analysis of initial rates of [3H]5-HT uptake yielded aVmax value of 2.44 ± 0.3 pmol/min/106 cells and aKm value of 0.58 ± 0.1 μM (n = 4, with the latter value being similar to that found in hSERT cells).
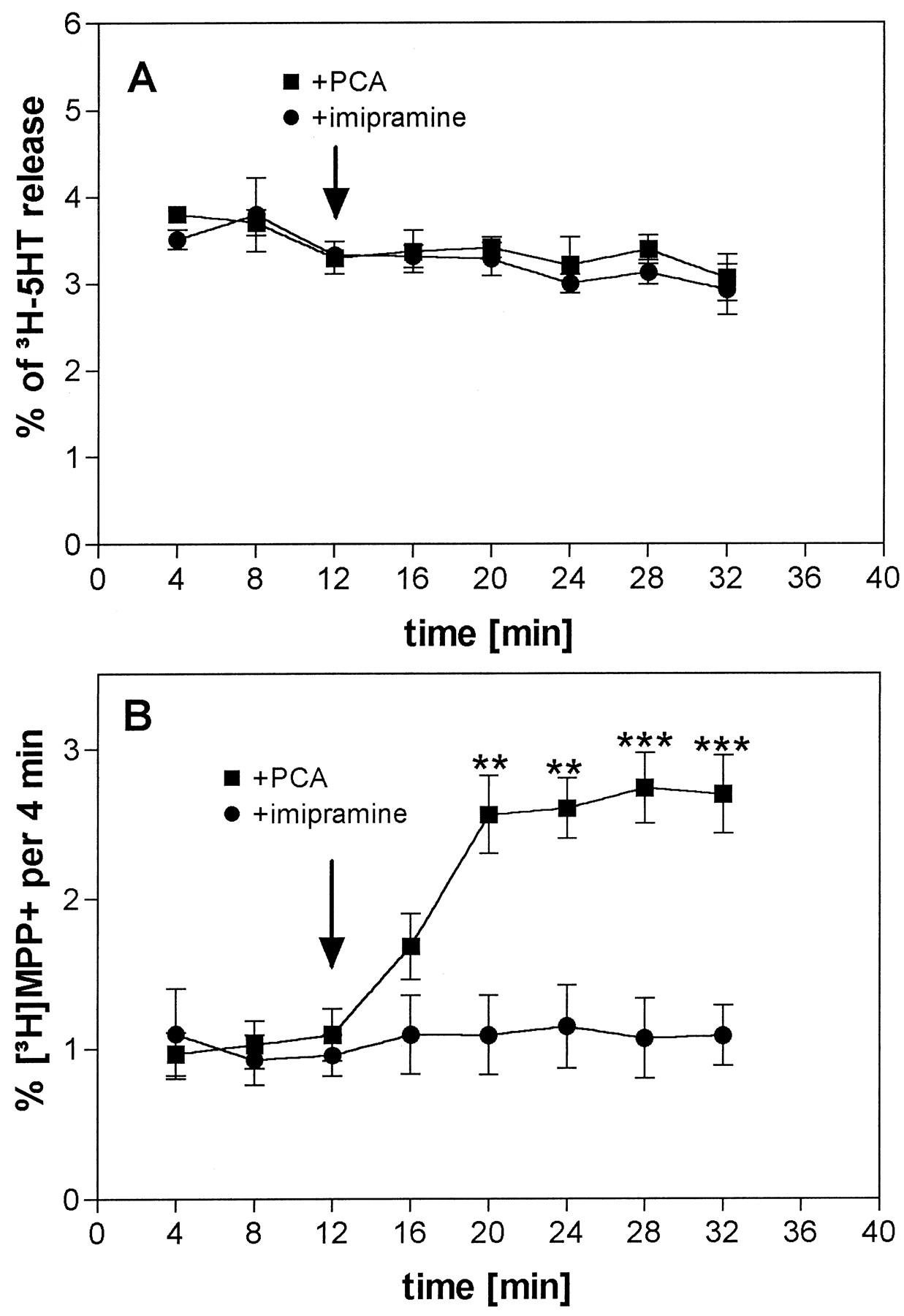
On superfusion, a high fractional rate of efflux of approximately 4% was observed (Fig. 5A). This was 2 to 4 times higher than the rate in hSERT cells superfused in parallel (not shown). The addition of PCA (10 μM) or imipramine (10 μM) had no significant effect on efflux (Fig. 5A). By contrast, the use of [3H]MPP+ as loading substrate resulted in a basal efflux of approximately 1%, comparable to hSERT cells. On the addition of PCA (10 μM), a significant increase in [3H]MPP+release was observed, whereas the addition of imipramine (10 μM) did not alter [3H]MPP+release (Fig. 5B).

Time course of the effects of PCA and imipramine on the release of [3H]5-HT or [3H]MPP+ from human placental choriocarcinoma (JAR) cells. JAR cells were loaded with [3H]5-HT (A) or with [3H]MPP+ (B) and superfused, and 4-min fractions were collected. After three fractions (12 min) of basal efflux, the buffer was switched to a buffer containing 10 μM PCA (▪) or 10 μM imipramine (●). Data are presented as fractional release (i.e., each fraction is expressed as a percentage of radioactivity present in the cells at the beginning of that fraction). Symbols represent mean ± S.E. of nine observations (one observation = one superfusion chamber; triplicate determinations from three separate experiments). **P < .01, ***P < .001 versus control condition.
Effects of Ouabain and Monensin on Efflux of [3H]5-HT and [3H]MPP+.
To investigate in more detail the unexpected finding that reuptake inhibitors caused an increase in release of [3H]5-HT, a series of experiments were performed under the conditions of an increased intracellular sodium concentration, which is expected to facilitate outward transport of substrate. For this purpose, the Na+,K+-ATPase inhibitor ouabain and the Na+/H+-selective ionophore monensin were used.
Ouabain (100 μM) led to a slow increase of [3H]5-HT efflux. When it was included in the superfusion buffer during the 45-min washout period, the efflux rate at the beginning of superfusate collection was more than twice as high as that under ouabain-free conditions (3.43 ± 0.37%,n = 9, versus 1.19 ± 0.11%, n = 9; P < .001: pooled values from Fig.6B). On the addition of PCA (10 μM) to the ouabain-containing buffer (cells were superfused for 57 min with ouabain at this point), efflux of [3H]5-HT was massively enhanced to a rate of more than 37% (Fig. 6A). Likewise, the subsequent addition of ouabain to PCA caused a pronounced enhancement of [3H]5-HT efflux (Fig. 6A).

Influence of Na+,K+-ATPase inhibition on the effects of PCA and imipramine on the release of [3H]5-HT from HEK 293 cells expressing the hSERT. HEK 293 cells stably transfected with the hSERT were loaded with [3H]5-HT and superfused, and 4-min fractions were collected. A, after three fractions (12 min) of basal efflux, the cells were exposed to a buffer containing 10 μM PCA (▪, ▵; arrow) or left at control conditions (▴). At 12 min later, an additional switch was performed, creating three experimental conditions: 100 μM ouabain alone (▴), 10 μM PCA alone (▪), and 10 μM PCA plus 100 μM ouabain (▵). ▪, an additional experimental condition was introduced by superfusing cells for 60 min with 100 μM ouabain before the collection of 4-min samples. After 72 min of superfusion (min 12 in plot), 10 μM PCA was added (■, arrow). B, same experimental design as in A except that 10 μM imipramine was used instead of PCA. ▴, effect of 100 μM ouabain. ●, effect of 10 μM imipramine. ▿, effect of 100 μM ouabain added after 10 μM imipramine. ○, 10 μM imipramine added after 72 min of superfusion with 100 μM ouabain. Data are presented as fractional release (i.e., each fraction is expressed as a percentage of radioactivity present in the cells at the beginning of that fraction). Symbols represent mean ± S.E. of nine observations (one observation = one superfusion chamber; triplicate determinations from three separate experiments). Bar indicates fractional release rates significantly higher than the efflux rate during min 8 to 12 (**P < .01).
By contrast, when imipramine (10 μM) was used instead of PCA in otherwise identical experiments, no apparent interaction was observed. Imipramine and the combination of ouabain and imipramine enhanced [3H]5-HT efflux to comparable values of approximately 5% (Fig. 6B).
The Na+/H+-selective ionophore monensin (10 μM) caused a distinct increase in the fractional efflux rate of [3H]5-HT from basal values to approximately 7% within 8 min. The subsequent addition of PCA (10 μM) caused a further strong increase to more than 35%. A similar combined effect was obtained when PCA was added first followed by monensin (Fig. 7A). In identically designed experiments in which imipramine (10 μM) was used instead of PCA, a qualitatively similar result was obtained. The combination of monensin and imipramine, regardless of the order of addition to the buffer, caused an increase in [3H]5-HT efflux to rates of approximately 9%, with the combined effect being significantly higher than the effect of each drug alone (Fig. 7B).

Influence of monensin (MON) on the effects of PCA and imipramine (IMI) on the release [3H]5-HT from hSERT cells. HEK 293 cells stably transfected with the hSERT were loaded with [3H]5-HT and superfused, and 4-min fractions were collected. A, after three fractions (12 min) of basal efflux, the cells were exposed to a buffer either containing 10 μM monensin (⋄, ♦) or 10 μM PCA (■, ▪). At 16 min later, an additional switch was performed, creating three experimental conditions: a buffer containing 10 μM monensin and 10 μM PCA was superfused onto one group of cells (♦, ▪); monensin alone was superfused onto a second group (⋄), whereas the third group was treated with PCA alone (■). B, same experimental design as in A, except that 10 μM imipramine was used instead of PCA. ⋄, effect of 10 μM monensin. ○, effect of 10 μM imipramine. ▾, effect of 10 μM monensin added after 10 μM imipramine. ●, 10 μM imipramine added after 10 μM monensin. *P < .05, **P < .01 versus monensin 10 μM (⋄). Data are presented as fractional release (i.e., each fraction is expressed as a percentage of radioactivity present in the cells at the beginning of that fraction). Symbols represent mean ± S.E. of nine observations (one observation = one superfusion chamber; triplicate determinations from three separate experiments).
The same set of experiments was repeated in hSERT cells loaded with [3H]MPP+ instead of [3H]5-HT. Monensin (10 μM) alone caused only a slight enhancement of [3H]MPP+ efflux. The subsequent addition of PCA (10 μM) to the monensin-containing buffer led to a pronounced increase of [3H]MPP+ efflux up to a maximum of approximately 25% (Fig. 8A). A similar effect was seen when monensin was added after PCA. The administration of imipramine to superfused hSERT cells loaded with [3H]MPP+ did not alter the basal efflux and prevented the effect of subsequently added monensin (Fig. 8B). When imipramine was added after monensin, the monensin-induced [3H]MPP+efflux was reduced to basal levels (Fig. 8B).

Influence of monensin (MON) on the effects of PCA and imipramine (IMI) on the release [3H]MPP+ from hSERT cells. HEK 293 cells stably transfected with the hSERT were loaded with [3H]MPP+ and superfused, and 4-min fractions were collected. A, after three fractions (12 min) of basal efflux, the cells were exposed to a buffer either containing 10 μM monensin (⋄, ♦) or 10 μM PCA (■, ▪). At 16 min later, an additional switch was performed, creating three experimental conditions: a buffer containing 10 μM monensin and 10 μM PCA was superfused onto one group of cells (♦, ▪); monensin alone was superfused onto a second group (⋄), whereas the third group was treated with PCA alone (■). B, same experimental designs as in A, except that 10 μM imipramine was used instead of PCA. [[diao[, effect of 10 μM monensin. ○, effect of 10 μM imipramine. ▾, effect of 10 μM monensin added after 10 μM imipramine. ●, 10 μM imipramine added after 10 μM monensin. *P < .05, **P < .01 versus monensin 10 μM (⋄). Data are presented as fractional release (i.e., each fraction is expressed as a percentage of radioactivity present in the cells at the beginning of that fraction). Symbols represent mean ± S.E. of six observations (one observation = one superfusion chamber; triplicate determinations from two separate experiments).
To test the possibility that ouabain or monensin exerted transporter-independent effects on efflux from the hSERT cells, experiments with parental HEK 293 cells were performed. Although ouabain (100 μM) did not change the rate of [3H]5-HT efflux from cells passively loaded with [3H]5-HT, monensin (10 μM) caused a clear-cut and sustained increase (Fig. 9, A and B, respectively).

Influence of Na+,K+-ATPase inhibition or monensin on the release of [3H]5-HT from parental HEK 293 cells. HEK 293 cells were passively loaded with [3H]5-HT as described in Materials andMethods and superfused, and 4-min fractions were collected. A, after three fractions (12 min) of basal efflux, the cells were exposed to a buffer containing 100 μM ouabain (●) or left at control conditions (○). In an additional experimental condition, cells were superfused for 60 min with 100 μM ouabain before the collection of 4-min samples (▴). B, after three fractions (12 min) of basal efflux, the cells were exposed to a buffer containing 10 μM monensin (⋄) or left at control conditions (●). Data are presented as fractional release (i.e., each fraction is expressed as a percentage of radioactivity present in the cells at the beginning of that fraction). Symbols represent mean ± S.E. of six observations (one observation = one superfusion chamber; triplicate determinations from two separate experiments). *P < .05, **P < .01 versus control conditions.
Discussion
The aim of the present study was to characterize 5-HT transport in cells stably expressing hSERT. For this purpose, the effects of uptake inhibitors as well as hSERT substrates were studied in uptake and superfusion experiments. All drugs inhibited uptake with IC50 values between 4 nM and 1 μM, displaying a comparable rank order of potency as described recently (Barker et al., 1994) using the same clone expressed in a different cell line.
Superfusion experiments involved preincubation of the cells with [3H]5-HT and monitoring the subsequent efflux of radioactivity. A particular advantage of this technique is believed to be the avoidance of concomitant high-affinity reuptake of released transmitter (Raiteri et al., 1974). Superfusion rests on the premise that the transmitter leaving the cell is immediately diluted by the superfusion medium and therefore is not available for reuptake. By contrast, performing the experiment using a stationary incubation in culture dishes or wells will allow reuptake of released 5-HT, which constitutes a recognized confounding factor in this type of studies. Superfusion conditions should thus enable different mechanisms of substances acting on hSERT to be clearly distinguished. If releasing drugs such as the hSERT substrates PCA or d-fenfluramine are added to superfused cells, the rate of [3H]5-HT efflux should increase, but if uptake blockers such as imipramine or paroxetine are added, which bind to the transporter and block transport in both directions, the rate of [3H]5-HT release into the superfusate should not change or even decrease.
This expectation was fully borne out by the results regarding substrates of the hSERT (i.e., 5-HT, PCA, andd-fenfluramine), all of which increased the efflux of [3H]5-HT. However, uptake inhibitors did not display the expected effect but instead caused a distinct enhancement of release. Substrate efflux induced by uptake inhibitors from cell lines expressing the SERT was also described by Wall et al. (1995) andJohnson et al. (1998). The apparent releasing effect would best be explained by a block of ongoing reuptake of [3H]5-HT diffusing out of the cells. However, such an effect is typical for slice or isolated organ preparations but is not expected in superfused cells or synaptosomes. For example, the uptake-blocking drug cocaine did not enhance transmitter efflux from superfused COS-7 cells transfected with either the plasmalemmal dopamine or noradrenaline transporter (Pifl et al., 1995, 1999). Moreover, releasing effects of various reuptake inhibitors in superfused rat brain synaptosomes preloaded with [3H]5-HT were not observed (Collard et al., 1981; Raiteri et al., 1984). By contrast, the effects of the uptake-inhibiting drugs in the present experiments had EC50 values in the nanomolar range and exhibited clearly defined maxima. If their action was indeed a consequence of interrupted high-affinity reuptake, several conditions should apply: 1) the uptake of an hSERT substrate that is added to the superfusion buffer should be inhibited, 2) diffusion must play a quantitatively significant role in the efflux of [3H]5-HT, and 3) the affinity of the substrate that is used for loading the cells and 4) the density of transporters expressed at the surface of the cell should determine reuptake. All four premises were tested experimentally. First, the interaction of the reuptake inhibitor paroxetine and the hSERT substrate PCA was studied. Paroxetine by itself enhanced efflux concentration dependently but clearly inhibited in the same manner the effect of subsequently added PCA (Fig. 3A). Inhibition by uptake blockade of substrate-induced release of 5-HT has also been observed recently using d-fenfluramine as releasing agent (Gobbi et al., 1992; Cinquanta et al., 1997). Information pertinent to the question of diffusion and substrateKm was obtained when the experiment was repeated using the substrate [3H]MPP+ instead of [3H]5-HT. Under this condition, paroxetine still inhibited the effect of PCA but did not increase the efflux of [3H]MPP+ by itself (Fig.3B). The positively charged substrate [3H]MPP+ showed considerably less diffusion through the plasma membrane of parental HEK 293 cells compared with 5-HT (Fig. 4A) and has a more than 30-fold higher Km value for the hSERT (14 μM for [3H]MPP+ and 0.4 μM for [3H]5-HT). These properties can both be expected to reduce reuptake by the hSERT: there is less substrate leaving the cell, and it is more easily diluted to a concentration at which reuptake is negligible. Finally, as to the premise of an influence of transporter density on the effect of uptake inhibitors, experiments were performed on JAR cells. These cells express the hSERT constitutively but display an initial uptake rate of [3H]5-HT that is <1% of the value obtained in hSERT cells. Consequently, one would expect less reuptake during superfusion, resulting in a high basal efflux rate and small or no effects of uptake blockers on efflux. This was in fact the case. The hSERT substrate PCA was also without apparent effect, probably due to the high basal rate masking any small drug-induced effect. With use of the less diffusible [3H]MPP+ as substrate, basal efflux was lower, and PCA displayed a distinct release-enhancing effect, whereas imipramine again did not change efflux (Fig. 5B).
To further characterize the effects of uptake-inhibiting drugs and hSERT substrates, we conducted a series of experiments in which the intracellular sodium concentration was modified by the addition of ouabain or monensin. Although ouabain inhibits Na+,K+-ATPase, monensin acts as ion-selective ionophore, facilitating the transmembrane exchange of sodium ions for protons (Mollenhauer et al., 1990). It is known that a rise in the sodium concentration at the inside of the plasma membrane results in a decrease inKm value for outward transport (Raiteri et al., 1978; Liang and Rutledge, 1982; Bönisch, 1986). In the present experiments, both ouabain and monensin massively augmented PCA-induced release of [3H]5-HT. This result was reproduced when monensin and PCA were studied in [3H]MPP+-loaded cells (Fig. 8A). The interactions with the uptake inhibitor imipramine, however, revealed more complexity. The combination of ouabain and imipramine raised the efflux rate of [3H]5-HT to the same level as imipramine alone. The combination of monensin and imipramine, on the other hand, increased the efflux rate to a distinctly higher level than imipramine on its own. Adding the drugs in sequence showed quite clearly the effect of monensin on top of the imipramine-induced rise of efflux (Fig. 7B, filled circles). This action of monensin was most likely not related to active transport. Three observations strongly support this notion: 1) when the less-diffusible [3H]MPP+was used instead of [3H]5-HT to label the cells, monensin did not display any effect after imipramine (Fig. 8B, filled circles). 2) Imipramine was able to completely reverse a monensin-induced rise in efflux of [3H]MPP+ when the uptake inhibitor was added after the ionophore (Fig. 8B, filled triangles). 3) When parental HEK 293 cells passively loaded with [3H]5-HT were superfused, monensin increased efflux of radioactivity (Fig. 9). This demonstrates an hSERT-independent mechanism by which [3H]5-HT leaves the cell in the presence of monensin. A possible explanation could be monensin-induced alkalinization of the cell interior due to outward movement of protons (Mollenhauer et al., 1990); an increase in the intracellular pH value toward the pKa value of 5-HT would favor outward diffusion by enhancing the mole fraction of the more lipophilic neutral form of 5-HT (Rudnick et al., 1989). The effect of imipramine on top of monensin in [3H]5-HT-labeled cells (Fig. 7B, filled triangles) indicates ongoing reuptake in the presence of the ionophore. Taken together, the data point to a combined effect of monensin (i.e., imipramine-sensitive outward transport of [3H]5-HT together with an increase in outward diffusion of [3H]5-HT, with the latter effect being partially offset by concomitant reuptake).
The finding that raising the intracellular sodium concentration drastically increases the releasing effect of PCA but not that of uptake blockers strongly argues against the possibility that efflux induced by uptake inhibitors is a consequence of reversed transport. A similar conclusion was reached in a study of the effects of cocaine and ouabain on dopamine efflux from LLC cells expressing the human noradrenaline transporter (Chen et al., 1998). A point that has not been experimentally addressed in the present study is whether the observed differences between PCA and imipramine may be related to changes in membrane potential brought about by the changes in ion distribution. Many sodium-coupled transport systems are responsive to changes in membrane potential (Wilson et al., 1998).
The present results may directly relate to a well known clinical caveat: the concomitant use of a monoamine oxidase inhibitor and a tricyclic antidepressant or a selective serotonin reuptake inhibitor is contraindicated because of a severe adverse reaction known as serotonergic syndrome (for a review, see Hilton et al., 1997). Symptoms include alterations in cognition, behavior, autonomic system function, and neuromuscular activity. Because treatment with monoamine oxidase inhibitors raises cytoplasmic levels of 5-HT, a situation may be created in vivo that is comparable to our experimental conditions in vitro. According to our results, introducing an uptake inhibitor in the presence of a relevant cytoplasmic pool of 5-HT should in fact lead to a markedly enhanced availability of 5-HT in the extracellular space.
Footnotes
-
Send reprint requests to: Dr. Harald H. Sitte, Department of Pharmacology, University of Vienna, Waehringerstr. 13a, A-1090 Vienna, Austria. E-mail:harald.sitte{at}univie.ac.at
-
↵1 This work was supported by the Austrian Science Foundation, Project P13183. Part of the work was presented at the Summer Meeting of the British and German Pharmacological Societies at the University of Nottingham, UK, July 1999.
- Abbreviations:
- 5-HT
- 5-hydroxytryptamine
- hSERT
- human serotonin transporter
- SERT
- serotonin transporter
- PCA
- p-chloroamphetamine
- MPP+
- N-methyl-4-phenylpyridinium
- Received October 18, 1999.
- Accepted February 4, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}