


Abstract
Several recent electrophysiological studies have demonstrated that nicotinic agonists stimulate the release of γ-aminobutyric acid (GABA) from rodent brain tissue. Our studies used a neurochemical approach to characterize nicotinic receptor-stimulated [3H]-GABA release from mouse brain synaptosomes. Nicotine increased [3H]-GABA release from synaptosomes preloaded with [3H]-GABA in a concentration-dependent manner. This release appeared rapidly, was Ca++ dependent, and was partially (about 50%) blocked by 100 nM tetrodotoxin and totally blocked by mecamylamine and dihydro-β-erythroidine. α-Bungarotoxin had no effect. Twelve nicotinic agonists were compared for their effects on [3H]-GABA release. The agonists differed in potency (EC50) and efficacy (Emax). The EC50 and Emax values were significantly correlated (r = 0.95, P < .001 for EC50; r = 0.93, P < .01 for Emax) to values obtained for these same agonists when 86Rb+ efflux was determined. A significant correlation (r = 0.84, P < .01) was found when the EC50 values for agonist-stimulated [3H]-GABA release and IC50 values for agonist inhibition of [3H]-l-nicotine binding were compared. Differences in [3H]-GABA release were detected in 12 brain regions and maximal release was significantly correlated with [3H]-nicotine binding. The pharmacological and regional comparisons suggest that the nAChR that stimulates [3H]-GABA release is the one that binds [3H]-nicotine with high affinity (α4β2). Unequivocal evidence that the receptor that modulates nicotine-stimulated [3H]-GABA release contains a β2 subunit was obtained in a study using wild-type, heterozygous and homozygous β2 null mutant mice. [3H]-GABA release and [3H]-nicotine binding decreased along with the number of copies of the null mutant gene.
The broad array of behavioral and physiological effects produced by nicotine are presumably initiated by binding to nAChRs that are located throughout the peripheral and central nervous systems. The postsynaptic nAChR found on electric organ and skeletal muscle is the best described of all neurotransmitter receptors (Galzi and Changeux, 1995; Karlin and Akabas, 1995) but motor neurons also seem to have presynaptic autoreceptors that modulate ACh release (Riker et al., 1957). Activation of these autoreceptors decreases ACh release under some circumstances but increases release under other circumstances (Tian et al., 1994; Domet et al., 1995).
Presynaptic nAChRs in brain apparently modulate release of several neurotransmitters (see Wonnacott, 1997, for a recent review). Nicotinic agonists stimulate the release of dopamine (Grady et al., 1992, 1997; Marshall et al., 1996; Rowell et al., 1987; Wonnacott et al., 1989), ACh (Beani et al., 1985; Lapchak et al., 1989; Meyer et al., 1987), and norepinephrine (Clarke and Reuben, 1996) from brain slice and/or synaptosomal preparations. These processes are Ca++dependent and are blocked by nicotinic antagonists such as mecamylamine.
Several neurochemical studies suggest that nicotine also stimulates GABA release, but it is not clear whether this is a direct or indirect effect. Wonnacott et al. (1989) reported that nicotine directly stimulates [3H]-GABA release from rat hippocampal synaptosomes. This effect was blocked by the nAChR antagonist DHβE but not by α-BTX. In contrast, Kayadjanian et al. (1994) reported that nicotine produces a transient increase in [3H]-GABA release from slices obtained from rat globus pallidus and substantia nigra, but this effect was blocked by dopamine receptor antagonists, suggesting that GABA release is a secondary response that follows nicotine-induced dopamine release. Bianchiet al. (1995) also concluded, from a study done with guinea pig cortical slices, that nicotine stimulates GABA release but only as a consequence of stimulating serotonin release that then stimulates GABA release.
Several electrophysiological studies indicate that nicotine stimulates GABA release directly via activation of nAChRs found on, or near, GABA nerve terminals. Léna et al. (1993) concluded that nicotinic agonists stimulated GABA release by activating preterminal nAChRs, because pretreatment of rat interpeduncular nucleus slices with TTX, the Na+ channel blocker, blocked nicotinic agonist-evoked increases in postsynaptic GABAergic currents. In contrast, Léna and Changeux (1997) concluded that nicotine-stimulated GABA release from mouse thalamic slices occurs via activation of receptors found on the nerve terminal. This conclusion was drawn, in part, because GABA release was not blocked by TTX.
Direct evidence that supports a presynaptic localization for nAChRs that modulate GABA release comes from the studies of Alkondon et al. (1996) who measured the effects of focal application of ACh on whole cell currents recorded from cultured, dissociated hippocampal pyramidal and bipolar cells; the latter presumably make up the majority of hippocampal GABAergic interneurons. These investigators reported that ACh-induced increases in current density increased with distance from the center of the cell soma suggesting that the nAChRs are at or near the nerve ending.
Mammalian brain contains many nAChR subunits (α2–α7, β2–β4) (reviewed in Lindstrom, 1996) and, assuming that brain nAChRs are pentameric, many different types of receptors might exist. However,in situ hybridization studies have shown that the mRNAs for some of the receptor subtypes are found in only a few brain regions whereas others, such as the α4, β2 and α7 subunits, are widespread leading to the postulate that receptors made up of these subunits should be most frequently encountered (reviewed in Lindstrom, 1996). Studies done using expression systems, primarilyXenopus oocytes, have demonstrated that both α and β subunits affect rank order of agonist potency and efficacy (Luetje and Patrick, 1991; Wheeler et al., 1993) as well as sensitivity to antagonists (Harvey et al., 1996; Luetje et al., 1990). These findings suggest that pharmacological approaches may be useful in establishing the subunit composition of receptors that modulate nicotinic agonist-evoked neurotransmitter release.
Molecular genetic strategies might also be useful in determining the functional roles of nAChR subunits. In the last few years transgenic mice have been developed where the β2 nAChR gene has been successfully “knocked out” resulting in mice that lack high affinity nicotine binding sites (Picciotto et al., 1995,1998). Léna and Changeux (1997) argued, based in part on the observation that β2 null mutant mice do not show nicotinic agonist-induced changes in thalamic GABAergic miniature synaptic currents, that the nAChR that modulates GABA release in mouse thalamus is made up of α4 and β2 subunits.
Our studies comprise a pharmacological assessment of nicotinic agonist-induced [3H]-GABA release from mouse brain synaptosomes. The results suggest that the same receptor that binds [3H]-nicotine with high affinity (α4β2) may be critically involved in modulating GABA release from synaptosomes prepared from many, if not all, mouse brain regions.
Methods
Materials.
[3H]-GABA (84–90 Ci/mmol) was purchased from Amersham Corp., Arlington Heights, IL. DMPP was obtained from Aldrich Chemical Co., Milwaukee, WI. (+)-Anatoxin-a hydrochloride, methylcarbachol chloride, R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2.3.4.5-tetrahydro-1H-3-benzazepine hydrochloride, sulpiride, 3-tropanyl-3.5-dichlorobenzoate, 6-cyano-7-nitroquinoxaline-2.3-dione and DHβE were purchased from Research Biochemicals International, Natick, MA. Mecamylamine was a gift from the Merck Research Laboratories, Rahway, NJ. Sucrose and HEPES were obtained from Boehringer-Mannheim, Indianapolis, IN. The following compounds were products of Sigma Chemical Co., St. Louis, MO: nicotine hydrogen (−)-tartrate (l-nicotine), (+)-nicotine-(+)-di-p-toluoyltartrate (d-nicotine), ACh, cytisine, (±)-epibatidine-l-tartrate, carbachol iodide, tetramethylammonium iodide, atropine sulfate, α-BTX, (±)-anabasine, (±)nornicotine, aminooxy acetic acid, GABA, sodium chloride, potassium chloride, calcium chloride, magnesium sulfate, potassium dihydrogen phosphate, veratridine, TTX, d-(+)-glucose,dl-2-amino-5-phosphonopentanoic acid and DFP. Econo-safe scintillation cocktail was purchased from Research Products International Corp., Arlington Heights, IL.
Animals.
Female C57BL/6J and β2 null mutant (wild-type, heterozygotes and homozygous null mutant) mice were used in this study. Animals were 60 to 90 days old and were bred at the Institute for Behavioral Genetics, Boulder, CO. The β2 null mutants were originally derived from a C57BL/6-DBA hybrid (Picciotto et al., 1995). The animals used in this study had been backcrossed onto a C57BL/6J background for six generations. Mice were housed five per cage and were allowed free access to food and water. The animal colony room was maintained on a 12 hr light/12 hr dark cycle with lights on between 7:00 a.m. and 7:00 p.m. All procedures were in accordance with the NIH Guide for Care and Use of Laboratory Animals and were approved by the University of Colorado animal care committee.
Synaptosome preparation.
Crude synaptosomes were prepared by hand homogenization of the mouse brain tissue in 0.32 M sucrose buffered with 5 mM HEPES (pH 7.5) in a glass-Teflon homogenizer. The homogenate was centrifuged at 1000 × g for 10 min. The supernatant was then centrifuged at 12,000 × g for 20 min. The resulting P2 pellet was resuspended in the perfusion buffer (128 mM NaCl, 2.4 mM KCl, 3.2 CaCl2, 1.2 mM KH2PO4, 1.2 MgSO4.7H2O, 25 mM HEPES, pH 7.5, 10 mM glucose). The volume of perfusion buffer used for resuspending the synaptosomes varied between 0.2 to 8 ml, depending on the brain region being studied.
[3H]-GABA uptake.
The crude synaptosomes were incubated for 10 min at 37°C in perfusion buffer containing 1 mM aminooxyacetic acid, an inhibitor of GABA transaminase. [3H]-GABA and unlabeled GABA were then added to final concentrations of 0.1 and 0.25 μM, respectively, and the suspension was incubated for another 10 min. Aliquots (80 μl) were collected with gentle suction onto 6-mm diameter A/E glass-fiber filters (Gelman Science, Ann Arbor, MI) and washed twice with 0.5 ml perfusion buffer. These filters were then transferred to the perfusion apparatus. Samples to be used with ACh were incubated with 10 μM DFP, an irreversible cholinesterase inhibitor, during uptake.
Perfusion and release.
The perfusion apparatus and procedure have been described in detail previously (Grady et al., 1992). Briefly, each 6-mm filter containing the synaptosomes was placed on a 13-mm glass-fiber filter mounted on a polypropylene platform and perfused with the buffer containing 1 g/liter bovine serum albumin at a rate of 1.8 ml/min for 10 min before fraction collection was started. All fractions were collected for 12 sec. In most experiments, with the exception of the time course experiment, agonists were added to the perfusate for 12 sec. This time period was selected simply because the fractions were collected every 12 sec. Atropine (1 μM) was included in the perfusion buffer for experiments with ACh and carbachol.
Data analysis.
To correct for differences in total synaptosomal [3H]-GABA content within and between experiments, the amount of [3H]-GABA release induced by an agonist stimulation was normalized as follows. The fractions before and after the stimulation that represent basal release were identified and were then fit as the first-order process Et = Eo * e−kt, where Et is the actual data obtained at each time, t; Eo is the initial basal release and k is the rate of decrease of release. This calculation yielded the theoretical basal release for each fraction. The release of [3H]-GABA exceeding baseline, which represents the agonist-stimulated release, was then calculated by subtracting the theoretical basal release from the actual data and was finally divided by the average baseline underlying the peak. The data are expressed as “units” (U) of release where one unit represents a doubling of the release above baseline. Release traces were constructed using this normalization (e.g., fig. 1). Total release for any stimulation was the sum of the counts exceeding baseline for all fractions after agonist treatment. The inset to figure 1 provides a concentration response curve for total release expressed in units relative to baseline.

Release of [3H]-GABA by nicotine. Normalized [3H]-GABA release traces for various concentrations of l-nicotine are shown. Each trace contains 20 fractions of 12-sec sample collection and a 12-sec exposure ofl-nicotine to synaptosomes was active between fraction 7 and 8. The inset is the concentration-effect curve for stimulation of [3H]-GABA release by nicotine calculated as total release. This curve was obtained by fitting data to the Michaelis-Menten equation with a nonlinear least-squares algorithm. Each point represents the mean ± S.E.M. calculated from stimulation of four to five separate [3H]-GABA-loaded synaptosomal preparations. Each synaptosomal preparation was assayed in duplicate.
Results for each agonist were initially calculated by fitting data to the Hill equation: E = (A * Sn)/(kn + Sn), where S is the agonist concentration, E is the observed response, A is the maximum release, k is EC50 and n is the Hill coefficient. Inasmuch as no Hill coefficient was significantly different from 1, the EC50 and Emax (the maximum stimulation of release) values for each agonist were estimated by fitting data to the Michaelis-Menten equation. An estimate of the IC50 value for antagonist inhibition of release stimulated by 30 μM nicotine was calculated by fitting the data to the following equation: Ec = Eo/(1 + C/K), where Ec is the response to 30 μM nicotine in the presence of the concentration, C, of an antagonist, Eo is the response to 30 μM nicotine in the absence of an antagonist and K is the IC50value. The nonlinear curve-fitting algorithm in SigmaPlot 5.0 (Jandel Scientific, San Rafael, CA), was used for all of the calculations described above.
[3H]-Nicotine binding.
Crude synaptosomes were prepared from wild-type, heterozygotes and homozygous β2 null mutant mice by the method of Romano and Goldstein (1980) as described previously (Marks et al., 1986, 1996). The incubations were conducted in 96-well polystyrene culture plates with 100 μl of the same buffer that was used to load the synaptosomes with [3H]-GABA (NaCl, 140 mM; CaCl2, 2 mM; KCl, 1.5 mM; MgSO4, 1 mM; HEPES, 25 mM; pH = 7.5). The concentration of [3H]-nicotine used for these measurements was 12 nM [Kd ∼ 2 nM (Markset al., 1996)]. Blanks were established by including 10 μM unlabeled l-nicotine in the incubations. Samples were incubated at 22°C for 30 min. The binding reaction was terminated by filtration onto glass fiber filters that had been soaked in perfusion buffer containing 0.5% polyethylenimine. Two different glass fiber filters were used: the top filter was grade GB100 (Microfiltration Systems, Dublin, CA) and the bottom filter was type A/E (Gelman Sciences, Ann Arbor, MI). Samples were washed six times after filtration. All filtration and wash steps were conducted in a cold room (4°C) with a precooled cell harvester equipped with a 96-place manifold (Inotech Biosystems, Lansing, MI) and cold wash buffer (NaCl, 140 mM; KCl, 1.5 mM; CaCl2, 2 mM; MgSO4, 1 mM; HEPES, 10 mM; pH −7.5).
Statistical analyses.
Student’s t tests were used to evaluate the TTX and Ca++ data. One-way analysis of variance was used to compare the EC50 and Emaxvalues obtained for the 12 agonists tested using whole brain synaptosomes, for analyses of brain regional differences, for analyzing effects of nonnicotine antagonists on [3H]-GABA release and for analyzing effects of genotypes of β2 null mutant on [3H]-GABA release and [3H]-nicotine binding. Post hoc comparisons were done using Tukey’s test with the significance level set at 0.05. Values of nreported in the figure legends represent the number of synaptosomal preparations each of which was derived from a different animal.
Results
Characterization of nicotine-stimulated [3H] GABA release.
Initial studies assessed the effects of nicotine on [3H]-GABA release from thalamic synaptosomes. This brain region was chosen because it has the highest levels of [3H]-nicotine binding of any of 12 brain regions that we routinely study (Marks et al., 1996). A concentration-dependent release of [3H]-GABA was observed following a 12-sec stimulation period (fig.1). The EC50 for nicotine-stimulated [3H]-GABA release calculated from these data is 5.60 ± 1.97 μM with a Hill coefficient of 0.88 ± 0.26. This Hill coefficient is not significantly different from 1.0.
Nicotine produced a rapid increase in [3H]-GABA release (fig. 2a), but [3H]-GABA release did not persist in the continued presence of agonist (fig. 2b). The total amount of [3H]-GABA release increased with the time of exposure to nicotine, but the rate of release decreased with exposure time (k = 0.062 ± 0.021 sec−1,t½ = 11.5 sec). The rate constant of the declining phase of release was k = 0.02 ± 0.006 sec−1 which yields a t½ of 34 sec. This decrease in response presumably arose because of receptor desensitization.

Time course of nicotine stimulation of [3H]-GABA release. Synaptosomes were exposed to 30 μMl-nicotine for the times indicated. Figure 2a presents normalized [3H]-GABA release traces for various exposure times of nicotine. Each point is the mean of normalized data for each 12-sec sample collection calculated from four to seven separate stimulations among four to six animals. The error bars represent S.E.M. Figure 2b is the time-course response curve for 30 μM nicotine-stimulated [3H]-GABA release. The curve was generated by fitting the data as the first order process: At = Ao * (1 − e−kt), At is total release at time, t; Ao is the maximal release; k is the rate of activation of release.
Effects of Ca++ and TTX on [3H]-GABA release.
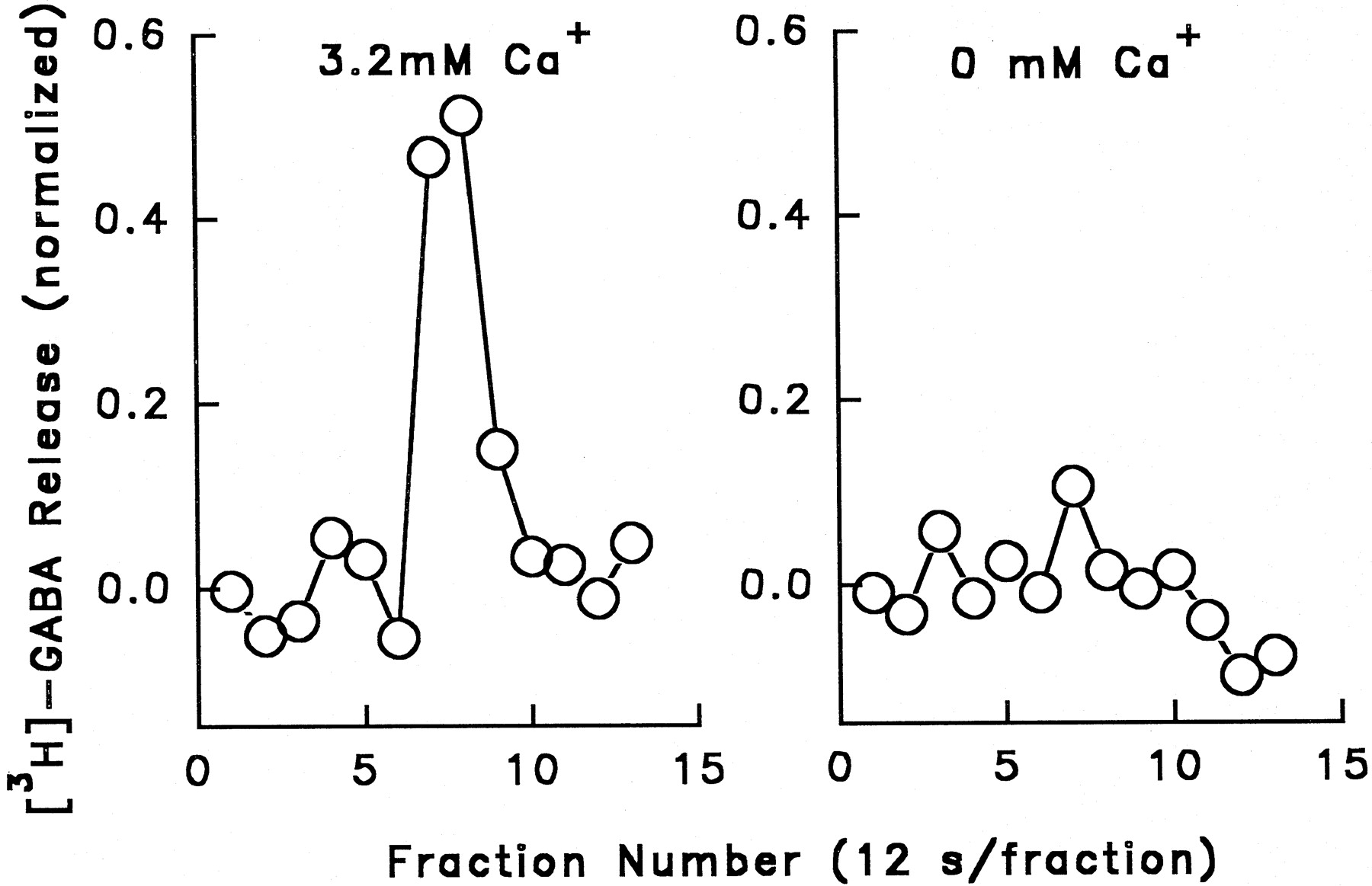
The Ca++ dependence of the release process was determined by measuring [3H]-GABA release stimulated by 30 μM nicotine added to perfusion buffer containing 3.2 mM Ca++ or nominally calcium-free buffer (4.8 mM NaCl was substituted for the 3.2 mM CaCl2). Results of a typical experiment are shown in figure 3. No release (0.06 ± 0.03 U) was seen in calcium-free media.

Ca++ dependence of nicotine-stimulated [3H]-GABA release. Whole brain synaptosomes were preloaded with [3H]-GABA and perfused with buffer containing 3.2 mM Ca++ or buffer containing no added Ca++ (0 mM Ca++). Nicotine (30 μM) stimulated [3H]-GABA release in Ca++-containing buffer but was ineffective in stimulating release in 0 mM Ca++buffer.
Inasmuch as TTX has been reported to partially inhibit nicotine-induced86Rb+ efflux (Marks et al., 1993) and [3H]-dopamine release (Marshall et al., 1996) from brain synaptosomes, the effects of TTX (100 nM) on [3H]-GABA release stimulated by nicotine, veratridine and K+ were measured (table 1). TTX treatment completely inhibited veratridine-stimulated release, but had no effect on K+-stimulated release, indicating that this concentration of TTX was adequate to block Na+channels but did not directly affect the release mechanism. TTX treatment inhibited nicotine-induced [3H]-GABA release by 50%.
Effects of TTX on nicotine, potassium, and veratridine-evoked [3H]-GABA release (% total synaptosomal [3H]-GABA content)
Effects of nicotinic receptor antagonists on nicotine-stimulated [3H]-GABA release.
The effects of three nAChR antagonists (DHβE, mecamylamine, α-bungarotoxin) on nicotine-stimulated [3H]-GABA release were examined using whole brain synaptosomes (fig. 4). DHβE and mecamylamine produced concentration-dependent inhibition of nicotine-evoked [3H]-GABA release with IC50values of 0.34 and 1.23 μM, respectively. α-BTX did not affect [3H]-GABA release even at the highest concentration tested (1 μM). This concentration of α-bungarotoxin completely inhibits the binding of [125I]-α-bungarotoxin to membranes prepared from mouse brain (Marks and Collins, 1982).

Concentration-response curves for inhibition of nicotine-stimulated [3H]-GABA release by DHβE, mecamylamine and α-BTX. Synaptosomes from whole brain were used and stimulated with 30 μM nicotine for 12 sec in the presence of various concentrations of each antagonist. DHβE and mecamylamine were only present during the 12-sec exposure to nicotine. α-BTX was incubated with synaptosomes for 60 min at 37°C before perfusion was begun and was not present during the perfusion (10 min). Each point represents the mean ± S.E.M. of four to six separate stimulations of synaptosomes. Open circles are 30 μM nicotine in the absence of antagonists. Lines are theoretical curve fit for inhibition (see “Methods” for equation).
Effects of neurotransmitter antagonists on nicotine-stimulated [3H]-GABA release.
Bianchi et al. (1995)noted that the 5-HT3 receptor antagonist, MDL-72222, inhibited nicotine-stimulated GABA release from guinea pig cortical slices, and Kayadjanian et al. (1994) reported that the D1 antagonist, (+)-SCH-23390, blocked [3H]-GABA release from rat substantia nigra slices. Consequently, the potential effects of MDL-72222 and (+)-SCH-23390 treatment on [3H]-GABA release were measured using synaptosomes prepared from whole brain. Potential effects of atropine (muscarinic antagonist), sulpiride (D2 antagonist), 6-cyano-7-nitroquinoxaline-2.3-dione (glutamate antagonist) and AP5 (NMDA antagonist) were also determined and compared with the effects produced by DHβE. All seven antagonists were present during the 10-min prewash period as well as during and after the 12-sec stimulation of 30 μM nicotine. The results of these experiments are depicted in figure 5. None of the antagonists, with the exception of DHβE, altered the [3H]-GABA release elicited by 30 μM nicotine. This suggests that [3H]-GABA release is a direct consequence of nAChR activation.

Effects of (+)-SCH-23390, sulpiride, MDL-72222, CNQX, AP-5, atropine and DHβE on [3H]-GABA release stimulated by 30 μM nicotine. Whole brain synaptosomes were exposed to the antagonists for 10 min before 12-sec stimulation of nicotine and antagonists continuously presented during and after stimulations of nicotine. Data are the mean ± S.E.M. calculated from at least four separate stimulations of synaptosomes. * P < .05vs. control. Analysis of variance with post hocmultiple comparison (Tukey’s test) was performed.
Effects of nicotinic agonists on [3H] GABA release.
The effects of perfusion for 12 sec with varying concentrations of 12 agonists on [3H]-GABA release from whole brain synaptosomes are illustrated in figure6. All 12 agonists stimulated a concentration-dependent increase in [3H]-GABA release. The Hill coefficients calculated from these data were not significantly different from 1.0 for any of the agonists. Therefore, the Michaelis-Menten equation was used to calculate EC50 and Emax values (table 2). Significant differences in agonist potency (F11,72 = 14.88, P < .01) were observed. The response showed stereoselectivity since the naturally occurring isomer, l-nicotine, was more potent than d-nicotine (EC50 = 1.6 and 12.8 μM, respectively).

Concentration-response curves for release of [3H]-GABA by 12 agonists. Whole brain synaptosomes were used and stimulated with various concentrations of each agonist for 12 seconds. Points represent the mean ± S.E.M. calculated from at least four separate stimulations of synaptosomes. Curves are theoretical nonlinear least-squares curve fits of data to the Michaelis-Menten equation.
Agonist-stimulated [3H]-GABA release from whole brain synaptosomes
The maximal [3H]-GABA release (Emax) stimulated by the 12 agonists also differed significantly (F11,72 = 45.4, P < .001). Not only wasl-nicotine more potent, it was also more efficacious thand-nicotine; the maximal [3H]-GABA release stimulated by d-nicotine was 60% of that elicited byl-nicotine. All of the quarternary ammonium compounds (acetylcholine, DMPP, carbachol, methylcarbachol and TMA) appeared to have similar, high maximal release (1.16–1.37 U). Epibatidine also elicited a high maximal GABA release (1.35 U). With the exception of l-nicotine (0.83 U) and epibatidine, the other nonquaternary compounds (d-nicotine, cytisine, (+)-anatoxin-a, nornicotine and (±)-anabasine) produced low maximal GABA release (Emax values ranged from 0.27–0.5 U).
Studies using β2 null mutants.
Figure7 presents the results of experiments where nicotine- and K+-stimulated [3H]-GABA release were measured in synaptosomes (whole brain) prepared from homozygous wild type (+/+), heterozygote (+/−) and homozygous null (−/−) β2 mutant mice (Picciotto et al., 1995, 1998). Figure 7a shows release traces obtained following stimulation for 12 sec with 30 μM nicotine for one mouse of each genotype. Figure 7b provides the overall results: Genotype exerted a significant overall effect on nicotine-stimulated [3H]-GABA release (F2,14 = 11.55, P < .01). There was virtually no release obtained in synaptosomes prepared from the homozygous β2 null mutants. The [3H]-GABA release seen in heterozygotes was intermediate between the wild-type controls and the homozygous null mutants. No differences were seen among the three genotypes after 20 mM K+ stimulation (fig. 7c) indicating that the [3H]-GABA release mechanism was not disrupted by the null mutation. [3H]-Nicotine binding was also measured in membrane fractions prepared from the whole brain synaptosomes (fig.7d). A significant effect of genotype on [3H]-nicotine binding was observed (F2,14 = 43.72, P < .001). As was the case for the [3H]-GABA release data, a gene dose effect was seen: [3H]-nicotine binding was virtually absent in the β2 null mutants and the heterozygotes were midway between the mutant and wild-type. [3H]-Nicotine binding and nicotine-stimulated [3H]-GABA release were significantly correlated (r = 0.76, P < .001) across the three genotypes (+/+, +/− and −/−).

Comparison of [3H]-GABA release and [3H]-nicotine binding in β2 nAChR receptor null mutant mice. The effects of 30 μM nicotine and 20 mM K+ on simulation of [3H]-GABA release, and [3H]-l-nicotine binding, were examined in synaptosomes (whole brain) prepared from wild type (control +/+,n = 4), heterozygote (+/−, n = 7) and homozygous β2 null mutant (−/−, n = 5) mice. Panel a provides representative normalized [3H]-GABA release traces for 30 μM nicotine. Figure 7b provides total [3H]-GABA release stimulated by 30 μM nicotine. Figure7c presents the [3H]-GABA release elicited by 20 mM K+. Figure 7d presents [3H]-l-nicotine binding from these same animals. Data are mean ± S.E.M. * P < .05 vs.wild-type.
Regional comparison of nicotine-stimulated [3H] GABA release.
Figure 8 illustrates the results of experiments where concentration-effect curves for nicotine-stimulated GABA release were determined in 11 brain regions of C57BL/6 mice; whole brain data are included for comparison. Nicotine stimulated concentration-dependent increases in [3H]-GABA release in every brain region studied. Hill coefficients calculated for each of these curves were not significantly different from 1.0. The EC50 and Emax values were calculated using the Michaelis-Menten equation and are presented in table3. The EC50 values for nicotine-stimulated release ranged between (1.43–19.9 μM). The EC50 value in cerebellum is significantly different from those obtained in any other regions (F10,58 = 2.53, P < .05). The Emax values differed significantly among the brain regions (F10,58 = 21.22, P < .001) with the release observed in striatum being the highest (1.41 U) and the release observed in olfactory bulb being the lowest (0.34 U).
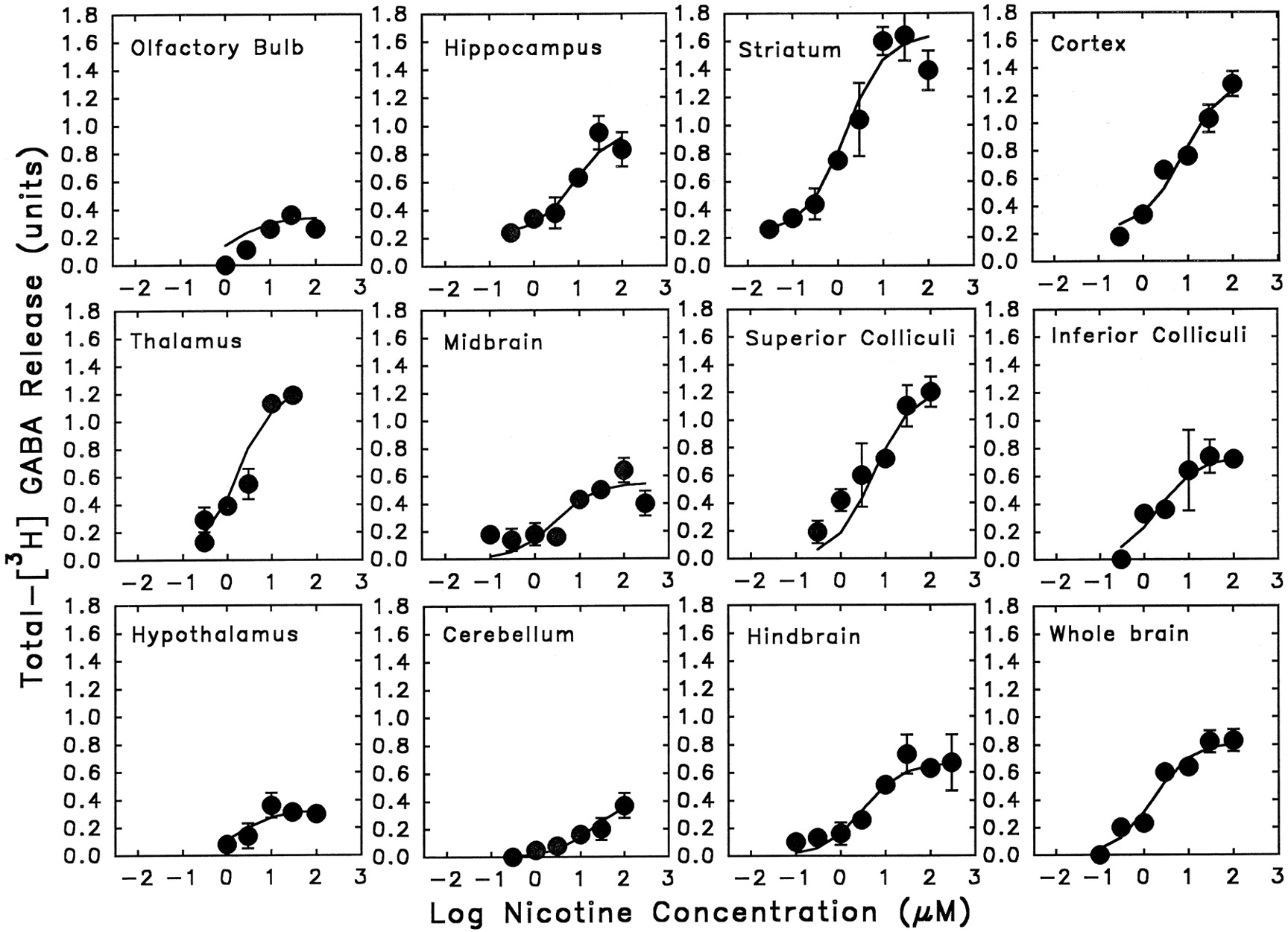
A regional comparison of nicotine-stimulated [3H]-GABA release from synaptosomes. Synaptosomes were stimulated for [3H]-GABA release by 12-sec exposure of various concentrations of nicotine. Each point represents the mean ± S.E.M. calculated from four to eight separate stimulations of synaptosomes. The curves were obtained by fitting the data to the Michaelis-Menten equation with a nonlinear least-squares algorithm.
EC50 values and Emax values for nicotine-stimulated [3H]-GABA release from synaptosomes obtained from 11 brain regions of C57BL/6 mice
The pattern of regional stimulation of maximal [3H]-GABA release was compared to l-[3H]-nicotine binding (Marks et al., 1992; Marks, unpublished data) and analyzed by correlation analysis (fig.9). Maximal [3H]-GABA release induced by nicotine was significantly correlated with the regional distribution of nicotine binding (r = 0.69, P < .01).

Correlation between maximal [3H]-GABA release and maximal [3H]-nicotine binding in 11 brain regions. The abbreviations for brain regions are as following, OB, olfactory bulb; HT, hypothalamus; CB, cerebellum; MB, midbrain; HB, hindbrain; HP, hippocampus; IC, inferior colliculi; SC, superior colliculi; CX, cortex; TH, thalamus; and ST, striatum. The Emax (maximum of [3H]-GABA release) values are summarized in table 3 and the [3H]-nicotine binding (Bmax) data were obtained from P2 synaptosomal preparation (Markset al., 1993).
Discussion
Our data demonstrate that application of nicotinic agonists to synaptosomes that have been preloaded with [3H]-GABA results in a concentration-dependent release of [3H]-GABA. GABA release occurs rapidly after agonist application and decreases to zero in the continued presence of agonist, suggesting that desensitization occurs. Agonist-stimulated release is Ca++ dependent and is blocked by classical nAChR antagonists. Nicotine stimulated the release of [3H]-GABA from synaptosomes prepared from every brain region tested. This finding argues that nicotine-evoked GABA release may play a major role in regulating behavioral and centrally mediated physiological responses to nicotine.
Nicotinic-receptor-mediated GABA release has previously been observed using electrophysiological and biochemical techniques. Electrophysiological studies with slices prepared from rat interpeduncular nucleus (Léna et al., 1993), mouse thalamus (Léna and Changeux, 1997), and rat hippocampus (Alkondonet al., 1997) have detected a nicotine-stimulated increase in miniature inhibitory postsynaptic currents that were blocked by GABA antagonists. Nicotine also promotes the release of GABA in vivo. Iontophoretic application of nicotine to the rat medial septum results in a decrease in neuronal firing rate which seems to be due to GABA release (Yang et al., 1996), and nicotine promotes the release of GABA in the rat dorsal motor nucleus of the vagus (Bertolino et al., 1997). Using biochemical methods,Wonnacott et al. (1989) observed nicotine stimulated release of [3H]-GABA from rat hippocampal synaptosomes, andKyadjanian et al. (1994) and Bianchi et al.(1995) reported nicotine stimulated release from tissue slices. On the basis of pharmacological data, the latter two studies suggested that GABA release may be secondary to nicotine-stimulated dopamine or serotonin release, respectively. However, such secondary effects seem unlikely in our study because synaptomes were used, a rapid response was observed and the sample perfusion rate was rapid, reducing neurotransmitter accumulation. Furthermore, antagonists of dopamine, serotonin, glutamate and muscarinic receptors had no effect on nicotine-stimulated GABA release, indicating that activation of these receptors was not involved in the release. Consequently, it is likely that the GABA release that was measured in our studies occurs because of a direct activation of nAChRs found on GABAergic neurons. It is possible, however, that other neurotransmitters modulate nicotinic activation-induced GABA release in vivo.
Nicotinic-receptor-mediated GABA release has been reported to occur at either the nerve terminal or preterminal depending on the nerve pathways involved. Léna et al. (1993) argued that the nAChR that modulates GABA release from rat interpeduncular nucleus is preterminal based on the observation that nicotine’s actions are blocked by the Na+ channel blocker, TTX. In contrast, TTX did not block nicotinic agonist-evoked GABA release when measured in two areas (ventrobasal complex, lateral geniculate) of the mouse thalamus (Léna and Changeux, 1997) suggesting that the thalamic nAChRs are found at the nerve terminal. Similarly, TTX did not affect nicotine-activated GABA release in the rat dorsal motor nucleus of the vagus (Bertolino et al., 1997). We detected a significant (approximately 50%) inhibition of nicotine-induced [3H]-GABA release from mouse brain synaptosomes by TTX. In contrast, veratridine-stimulated release was completely inhibited by the same concentration of TTX. These findings suggest that approximately half of the total release that we measured resulted from a cascade where nAChR stimulation produced enough voltage change to activate TTX-sensitive Na+ channels that, in turn, generated enough voltage change so that voltage-gated Ca++channels were activated leading to transmitter release. This might occur if a significant fraction of the synaptosomes contained preterminal elements where the nAChRs were not in close proximity to the relevant Ca++ channels. However, the finding that TTX did not block approximately 50% of the nicotine-evoked [3H]-GABA release suggests either that the Ca++ permeability of the relevant nAChRs is sufficient to stimulate the release process directly or the nAChRs are in close proximity to voltage-gated Ca++ channels that are activated by the voltage change produced by nAChR activation. This might occur if approximately half of the synaptosomes were derived from neurons where the nAChRs are directly associated with the terminal.
Although definitive assignment of a distinct nicotinic receptor as the mediator of nAChR-stimulated GABA release is not yet possible, the abolition of the response in β2 null mutants indicates that this subunit is present in the nAChR subtype that mediates GABA release from mouse brain synaptosomes. An identical result has been described byLéna and Changeux (1997) for GABA release in the ventrobasal complex and the dorsolateral geniculate nucleus of mouse thalamus. Consistent with this observation, Alkondon et al. (1997)postulated that the α4β2-nAChR subtype modulates GABA release from rat hippocampal interneurons and is the basis of the type II current observed in hippocampal cells.
The α4β2-nAChR subtype has also been postulated to mediate agonist-stimulated 86Rb+ efflux from mouse synaptosomes (Marks et al., 1993). Inasmuch as the86Rb+ efflux assay uses methods nearly identical to those used for GABA release, a direct comparison of the results for these two responses is possible. Figure10a presents dose-response curves for the effects of four agonists (ACh, cytisine, DMPP, nicotine) on [3H]-GABA and 86Rb+ efflux from mouse brain synaptosomes. As is readily evident from the concentration effect curves shown for the four agonists, virtually identical EC50 and Emax values were obtained for the four agonists in the two assays. Figure 10b provides a direct comparison of the EC50 and Emax values for all 12 of the agonists. Significant correlations for the EC50 (r = 0.95, P < .001) and Emax (r = 0.93, P < .01) values were obtained when agonist effects on the two assays were compared. This finding suggests that the two assays are measuring the same receptor(s).

Comparisons of nicotinic agonist-stimulated [3H]-GABA release with agonist-evoked86Rb+ efflux. Figure 10a shows the concentration-responses curves of acetylcholine, nicotine, DMPP and cytisine for stimulation of [3H]-GABA release (open symbols) and stimulations of 86Rb+ efflux (closed symbols). In figure 10b, upper panel provides a comparison of EC50 values of 12 agonists for stimulation of [3H]-GABA release (table 2) and EC50 values of these same agonists for stimulation of 86Rb+efflux (Marks et al., 1996). Lower panel of figure 10b provides a comparison of maximal release of [3H]-GABA and maximal 86Rb+ efflux stimulated by the 12 agonists. Points represent the mean ± S.E.M. for the 12 agonists.
Figure 11 presents a comparison of the relationship between the EC50 values for agonist-stimulated release of [3H]-GABA and the IC50 values of these same agonists for inhibition of [3H]-l-nicotine binding. The binding data are those presented in Marks et al. (1993 and 1996). The potency of agonist-stimulated [3H]-GABA release was highly correlated to the IC50 values of agonist inhibition [3H]-nicotine binding (r = 0.84, P < .01).

Relationship between EC50 values of the 12 agonists for stimulations of [3H]-GABA release and IC50 values of these same agonists for inhibition ofl-[3H]-nicotine binding. The binding data are those presented in Marks et al. (1996 and 1993). The correlation coefficient is significant (P < .01).
The potencies of agonist stimulation of [3H]-GABA release (EC50 values) were highly correlated with the potencies of these 11 compounds with respect to stimulation of86Rb+ efflux and inhibition of [3H]-nicotine binding (IC50 values). These findings argue that the receptor that modulates nicotine-evoked GABA release is very similar, if not identical, to the receptor that modulates 86Rb+ release at low μM concentrations and binds [3H]-nicotine with high affinity. Immunological evidence (Whiting and Lindstrom, 1988) and evidence obtained with the β2 null mutants (Picciotto et al., 1995; our data) argue that the high affinity [3H]-nicotine binding site includes a β2 subunit. Because antibodies directed against the α4 subunit precipitate more than 90% of rat brain high affinity nicotine binding sites (Floreset al., 1992), it seems highly likely that this binding site is made up of α4 and β2 subunits. These considerations suggest that α4β2-containing nAChRs account for a major percentage of the nAChRs that modulate GABA release. This conclusion agrees with the conclusions drawn by Alkondon et al. (1997) and Léna and Changeux (1997).
A significant association (r = 0.67, P < .01) was found between maximal [3H]-GABA release and [3H]-nicotine binding when these parameters were measured in 11 brain regions. This finding also supports the argument that the receptor that binds nicotine with high affinity is also the one that modulates GABA release. However, when we made a similar comparison for nicotine-stimulated 86Rb+ efflux a higher correlation (r = 0.93) was obtained between these measures across the same brain regions (Marks et al., 1993). One potential explanation for this difference is that α4β2-containing receptors modulate nicotine-induced GABA release in most brain regions, but in some regions another receptor, or additional receptors may modulate GABA release. In addition, it seems likely that the receptor that binds [3H]-nicotine with high affinity (α4β2) has functions in addition to modulating GABA release. These other functions may well vary across brain regions.
One potential explanation for regional differences in nicotine-stimulated [3H]-GABA release is brain regions clearly vary in numbers of GABA neurons. This, obviously, should result in regional variability in [3H]-GABA uptake into synaptosomes. This variability does not, however, explain the regional variability in nicotine-stimulated [3H]-GABA release as evidenced by the observation that striatum, thalamus, inferior and superior colliculli, cortex and hippocampus had nearly identical [3H]-GABA uptake although they showed a 2-fold difference in nicotine-stimulated [3H]-GABA release. In contrast, cerebellum, olfactory bulbs and hypothalamus showed a 2-fold difference in [3H]-GABA uptake although the nicotine-stimulated [3H]-GABA release of these three regions was virtually the same.
Alkondon et al. (1996, 1997) have obtained data that argue that nicotinic agonist-induced GABA release may be modulated by α7-containing nAChRs that bind α-bungarotoxin with high affinity. Our data do not support this argument because α-bungarotoxin did not block release. Similarly, α-bungarotoxin does not block [3H]-GABA release from rat hippocampal synaptosomes (Wonnacott et al., 1989). It is probably unwise, however, to conclude that α7-containing nAChRs do not modulate GABA release because α7-containing nAChRs desensitize very quickly (Couturieret al., 1990), and synaptosomal perfusion studies may not be capable of detecting [3H]-GABA release produced by a receptor that desensitizes quickly. Thus, α7-containing nAChRs may modulate some GABA release but it is highly unlikely that α7-modulated release contributes substantially to the release that we measured.
The results of the experiments reported here clearly demonstrate that activation of presynaptic nAChRs results in concentration-dependent release of GABA. Because this effect is not seen in synaptosomes prepared from whole brain of β2 null mutant mice, it seems very safe to conclude that the β2 subunit is a component of all of the receptors that modulate this response. The pharmacological approach, primarily the agonist studies, argue that an α4 subunit is also involved, at least in most brain regions.
Footnotes
-
Send reprint requests to: Dr. Allan C. Collins, Institute for Behavioral Genetics, Campus Box 447, University of Colorado, Boulder, CO 80309-0447.
-
↵1 This work was supported by Grants DA-03194 and DA-00197 from the United States National Institute on Drug Abuse and by the Collège de France, the Centre National de la Recherche Scientifique, the Association Française contre la Myopathie, the Council for Tobacco Research, a Biotech contract from the Commission of the European Communities.
-
↵2 Current address: Department of Psychiatry, Yale University School of Medicine, New Haven, CT 05508.
-
↵3 Current address: URA CNRS 1284, Neurobiologie Moleculaire, Institut Pasteur, Paris, France.
- Abbreviations:
- Ach
- acetylcholine
- nAChR
- nicotinic cholinergic receptors
- GABA
- γ-aminobutyric acid
- DHβE
- dihydro-β-erythroidine
- TTX
- tetrodotoxin
- DMPP
- dimethylphenyl piperazinium
- DFP
- diisopropyl flourophosphate
- α-BTX
- α-bungarotoxin
- HEPES
- N-[2-hydroxyethyl]-piperazine-N′-[2-ethanesulfonate] hemisodium salt
- CNQX
- 6-cyano-7-nitroquinoxaline-2.3-dione
- R(+)-SCH23390
- R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2.3.4.5-tetrahydro-1H-3-benzazepine hydrochloride, MDL-72222, 3-tropanyl-3.5-dichlorobenzoate
- AP-5
- DL-2-amino-5-phosphonopentanoic acid
- Received April 8, 1998.
- Accepted June 18, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}