


Abstract
The novel benzoindane S 18126 possessed > 100-fold higher affinity at cloned, human (h) D4 (Ki = 2.4 nM) vs. hD2 (738 nM), hD3(2840 nM), hD1 (> 3000 nM) and hD5 (> 3000 nM) receptors and about 50 other sites, except ς1receptors (1.6 nM). L 745,870 similarly showed selectivity for hD4 (2.5 nM) vs. hD2 (905 nM) and hD3 (> 3000 nM) receptors. In contrast, raclopride displayed low affinity at hD4 (> 3000 nM) vs.hD2 (1.1 nM) and hD3 receptors (1.4 nM). Stimulation of [35S]-GTPγS binding at hD4receptors by dopamine (DA) was blocked by S 18126 and L 745,870 withKb values of 2.2 and 1.0 nM, respectively, whereas raclopride (> 1000 nM) was inactive. In contrast, raclopride inhibited stimulation of [35S]-GTPγS binding at hD2 sites by DA with a Kb of 1.4 nM, whereas S 18126 (> 1000 nM) and L 745,870 (> 1000 nM) were inactive. As concerns presynaptic dopaminergic receptors, raclopride (0.01–0.05 mg/kg s.c.) markedly enhanced DA synthesis in mesocortical, mesolimbic and nigrostriatal dopaminergic pathways. In contrast, even high doses (2.5–40.0 mg/kg s.c.) of S 18126 and L 745,870 were only weakly active. Similarly, raclopride (0.016 mg/kg i.v.) abolished inhibition of the firing rate of ventrotegmental dopaminergic neurons by apomorphine, whereas even high doses (0.5 mg/kg i.v.) of S 18126 and L 745,870 were only weakly active. As regards postsynaptic dopaminergic receptors, raclopride potently (0.01–0.3 mg/kg s.c.) reduced rotation elicited by quinpirole in rats with unilateral lesions of the substantia nigra, antagonized induction of hypothermia by PD 128,907, blocked amphetamine-induced hyperlocomotion and was effective in six further models of potential antipsychotic activity. In contrast, S 18126 and L 745,870 were only weakly active in these models (5.0–> 40.0 mg/kg s.c.). In six models of extrapyramidal and motor symptoms, such as induction of catalepsy, raclopride was likewise potently active (0.01–2.0 mg/kg s.c.) whereas S 18126 and L 745,870 were only weakly active (10.0–80.0 mg/kg s.c.). In freely moving rats, raclopride (0.16 mg/kg s.c.) increased levels of DA by + 55% in dialysates of the frontal cortex. However, it also increased levels of DA in the accumbens and striatum by 70% and 75%, respectively. In contrast to raclopride, at a dose of 0.16 mg/kg s.c., neither S 18126 nor L 745,870 modified frontal cortex levels of DA. However, at a high dose (40.0 mg/kg s.c.), S 18126 increased dialysate levels of DA (+ 85%) and noradrenaline (+ 100%), but not serotonin (+ 10%), in frontal cortexwithout affecting DA levels in accumbens (+ 10%) and striatum (+ 10%). In conclusion, S 18126 and L 745,870 behave as potent and selective antagonists of cloned, hD4vs. other dopaminergic receptor types in vitro. However, their in vivo effects at high doses probably reflect residual antagonist actions at D2 (or D3) receptors. Selective blockade of D4receptors was thus associated neither with a modification of dopaminergic transmission nor with antipsychotic (antiproductive) or extrapyramidal properties. The functional effects of selective D4 receptor blockade remain to be established.
Dopamine receptors are currently classified into two families on the basis of their primary structures, their coupling to intracellular transduction mechanisms and their pharmacological profiles of ligand recognition: D1 and D5 receptors and D2, D3 and D4 receptors (Seeman, 1992; Sokoloff and Schwartz, 1995). The discovery of novel D3 and D4 receptors has raised the question of their respective roles in mediating the actions of DA and their pathophysiological significance in disorders reflecting a perturbation of dopaminergic transmission. Furthermore, because most ligands traditionally employed for the evaluation of actions at D2receptors possess comparable affinity for D2, D3 and D4 receptors (Chabert et al., 1994; Malmberg et al., 1993; Millan et al., 1995a; Newman-Tancredi et al., 1997a; Roth et al., 1995; Sokoloff et al., 1992, Van Tol et al., 1991), it appears necessary to reappraise the putative role and pathophysiological significance of D2 receptors. Indeed, apomorphine and other dopaminergic agonists employed for the treatment of Parkinson’s disease, as well as haloperidol and other antipsychotics utilized for the management of psychotic disorders, exhibit pronounced activity at D3 and D4receptors. As concerns the treatment of psychotic disorders, it is important to determine whether selective blockade of D3 or D4 receptors may control productive and/or deficit-cognitive symptoms of schizophrenia in the absence of undesirable, extrapyramidal side effects (Roth et al., 1995;Seeman, 1992; Sokoloff and Schwartz, 1995).
In this regard, D4 receptors have been the focus of particular attention for several reasons. First, neuroanatomical studies employing antibodies against the receptor protein (Arianoet al., 1997; Defagot et al., 1997; Harlanet al., 1996; Mauger et al., 1996) radiolabeled, selective antagonists (Primus et al., 1997; Tallman et al., 1997; Tarazi et al., 1997) and immunocytochemical localization of the corresponding mRNA (Matsumoto M. et al., 1996; Matsumoto et al., 1995; Meador-Woodruff et al., 1996; Wang et al., 1996) have suggested a preferential localization of D4 receptors in cortical and limbic structures involved in the regulation of mood and cognition, as well as in the etiology of schizophrenia. By contrast, their levels are comparatively low in the striatum and other structures involved in the modulation of motor behavior and in the induction of extrapyramidal symptoms. Second, elevated levels of D4 receptors have been documented in certain cerebral structures, including the striatum and nucleus accumbens, of schizophrenic patients (Murray et al., 1995; Seeman et al., 1993 and 1995; Seeman and Van Tol, 1995). Third, chronic treatment of rodents with antipsychotics alters cerebral levels of mRNA-encoding D4 receptors (Baldessariniet al., 1996; Schoots et al., 1995). Fourth, in contrast to haloperidol, clozapine was reported to possess a marked (10-fold) preference for cloned hD4 over hD2receptors (Murray et al., 1995; Seeman, 1992; Seemanet al., 1997; Van Tol et al., 1991). In fact, more recent studies suggest that the preference of clozapine for D4 over D2 sites is modest, on the order of 2- to 5-fold (Chabert et al., 1994; Newman-Tancredi et al., 1997a; Roth et al., 1995). Furthermore, the observation of a putative increase in levels of D4receptors in schizophrenic brain remains controversial (Lahti et al., 1996a and b; Mulcrone and Kerwin, 1996; Reynolds and Mason, 1995; Seeman and Van Tol, 1995). In addition, levels of D4receptors in cerebral tissue are low relative to those of D2 receptors—with the exception, however, of certain regions of the cerebral cortex (Lahti et al., 1996a;Matsumoto M. et al., 1995, 1996; Meador-Woodruff et al., 1996; Primus et al., 1997; Reynolds and Mason, 1995).
The utility of novel, antisense receptor “knockdown” and transgenic gene “knockout” approaches notwithstanding (Accili et al., 1996; Paulus et al., 1996; Tepper et al., 1997), the availability of chemically diverse, selective ligands remains essential for a broad-based experimental and clinical exploration of the pathophysiological significance of D4—as well as D2 and D3—receptors. Correspondingly, intensive efforts have been made to identify selective antagonists at D4 receptors, and recently, several structures have been presented in either preliminary (Hartman et al., 1996; Zorn et al., 1996) or more complete (Boyfield et al., 1996; Hidaka et al., 1996; Kulagowski et al., 1996; Merchant et al., 1996; Patel et al., 1996a; Rowley et al., 1996;Tallman, 1987, Tallman et al., 1997; Thurkauf et al., 1997) reports (see Hadley, 1996 for a review). However,in vivo functional data concerning the actions of selective D4 antagonists are very limited.
In this light, the present report describes the in vitro andin vivo properties of a novel, potent, competitive, selective and orally active D4 receptor antagonist S 18126 (fig. 1). Its actions were compared with those of the arylpiperazine D4 receptor antagonist L 745,870 (Bristow et al., 1997; Kulagowski et al., 1996; Patel et al., 1996b) and with those of the benzamide raclopride, which possesses negligible affinity at D4receptors but marked affinity at D2/D3receptors (Asghari et al., 1995; Millan et al., 1995a and b). In addition, for several key functional parametersin vivo, we extended these studies to a series of benzamides possessing differential affinity at hD2 and hD4receptors (Giuliani and Ferrari, 1997; Nasello et al., 1991;Rumigny et al., 1984; Steele et al., 1993) (see “Discussion”). Their utilization made possible a correlation analysis of the respective involvement of D4vs.D2 receptors in several functional paradigms. Thereby, doses of S 18126 and L 745,870 active at D2 receptorsin vivo could be determined, which, in turn, permitted the calculation of a theoretical dose range over which S 18126 and L 745,870 should act as selective D4 receptor antagonists. Thus, in addition to a characterizing S 18126 per se, we sought to determine more generally the putative functional significance of D4 receptor blockade in several models of antipsychotic and extrapyramidal properties. A preliminary account (in Abstract form) of some of the present data has been presented elsewhere (Millanet al., 1996).

Structure of S 18126.
Materials and Methods
Binding at hD4 and other dopaminergic receptor types.
Competition binding to CHO-D4.4 (Receptor Biology Inc., Beltsville, MD) and CHO-hD2S cell membranes was carried out as described in Newman-Tancredi et al.(1997a). Briefly, membranes (10–20 μg protein) were incubated with [3H]-spiperone at 25°C for 60 min in a buffer containing TRIS 50 mM (pH 7.4), NaCl 120 mM, KCl 5 mM, EDTA 1 mM and MgCl2 5 mM. Nonspecific binding was defined with haloperidol (1 μM). S 18126 was tested in competition binding experiments at a range of other recombinant and native brain binding sites. Experiments were carried out “in house” or by the screening company, CEREP (Celle L’Evescault, France). Isotherms were analyzed by nonlinear regression, using the program PRISM (Graphpad Software Inc., San Diego, CA) to yield IC50 values. Inhibition constants (Ki values) were derived from IC50values according to the Cheng-Prusoff equation:Ki = IC50/(1 +L/Kd ); where L is the concentration of radioligand and Kd is the dissociation constant of the radioligand.
Measurement of agonist efficacy and antagonist potency at hD2 and hD4 receptors.
Receptor-linked G protein activation at hD2 and hD4 receptors was determined by measuring the stimulation of [35S]-GTPγS (1332 Ci/mmol; NEN, Les Ulis, France) binding as described inNewman-Tancredi et al. (1997a). Briefly, CHO-D4membranes (50 μg protein) were incubated (20 min, 22°C) with agonists and/or antagonists in a buffer containing HEPES 20 mM (pH 7.4), GDP 3 μM, MgCl2 3 mM, NaCl 100 mM and [35S]-GTPγS 0.1 nM. Nonspecific binding was defined with GTPγS (10 μM). Agonist efficacy was expressed relative to that of DA (= 100%), which was tested at a maximally effective concentration in each experiment. For antagonist tests, membranes were preincubated with agonist and a single concentration of antagonist for 30 min before the addition of [35S]GTPγS. For concentration-response curves of the inhibition of DA-stimulated [35S]GTPγS binding, Kb values were calculated as described in Newman-Tancredi et al.(1997a). Experiments were terminated by rapid filtration through Whatman GF/B filters (pretreated with 0.1% polyethyleneimine in the case of [3H]spiperone binding) using a Brandel cell harvester. Radioactivity retained on the filters was determined by liquid scintillation counting. Protein concentration was determined colorimetrically using a bicinchoninic acid assay kit (Sigma Chimie, St-Quentin-Fallavier, France). All results are expressed as means ± S.E.M. of ≥ 3 independent determinations.
In vivo studies.
Male Wistar rats (220–240 g b.wt.) and, in most studies, NMRI mice (22–25 g) (Iffa-Credo, L’Arbresle, France) were housed in sawdust-lined cages with free access to chow and water. Laboratory temperature was 21°C ± 1.0°C and humidity was 60% ± 5%. There was a 12 hr/12 hr light-dark cycle with lights on at 7:30. Male CD1 (ICR) BR mice (22–25 g) (Charles River, Saint-Aubin-les-Elbeuf, France) were used for the apomorphine-induced climbing and rotarod tests.
Influence on the electrical activity of dopaminergic neurons.
As previously described in detail (Lejeune and Millan, 1995), rats were anesthetized with chloral hydrate (400 mg/kg i.p.), the femoral vein was catheterized and they were placed in a sterotaxic apparatus. A tungsten electrode was lowered into the ventrotegmental area according to coordinates derived from Paxinos and Watson (1986): AP: −5.5 from bregma, L: 0.7 and H: 9.7/8.5 from the dura. Dopaminergic neurons were identified as before, according to their wave-form (Lejeune et al., 1997; Wang, 1981), and base-line recording was performed over 5 min. Drugs were dissolved in sterile water and injected i.v. in a volume of 0.5 ml/kg, followed by a 0.1-ml saline flush. Drugs were administered alone (dose-response curves) cumulatively i.v. at intervals of 2 to 5 min. In antagonist studies, they were administered (1 dose per experiment) 2 min after a single injection of apomorphine (63 μg/kg i.v.). Data acquisition was performed with Spike 2 software (C.E.D., Cambridge, England), and results are expressed as firing rate (60-sec bins at time of peak drug action) as a percentage of base-line, preinjection values.
Influence on DA turnover and PRL levels.
As described in detail previously (Gobert et al., 1995), the influence of drugs on DA turnover in rats was evaluated by measuring the levels of DA compared with its metabolite, DOPAC, in terminal regions of mesocortical (FCX), mesolimbic (accumbens and olfactory tubercles) and nigrostriatal (striatum) pathways 30 min after their s.c. injection. Tissues were homogenized in 500 μl of 0.1 M HClO4containing 0.5% Na2S2O5 and 0.5% EDTA and then were centrifuged at 15,000 × g for 15 minutes at 4°C. Supernatants were diluted in the mobile phase. HPLC analysis followed by electrochemical detection was employed for determination of tissue levels of DA and DOPAC. The column characteristics and elution phases were as follows: column, hypersil ODS 5 μm, C18, 150 × 4.6 mm maintained at 25°C; mobile phase, KH2PO4, 100 mM, EDTA, sodium octylsulphonate (0.5 mM) and methanol 5% adjusted to pH 3.15 with PO4H3. The flow rate was 1 ml/min. Electrochemical detection was performed using a Waters M460 detector with a working potential of 850 mV against an Ag/AgCl reference. Levels of DA and DOPAC were expressed as a function of the tissue content of protein. The mean levels of DA, DOPAC and DOPAC/DA ratios determined in animals treated with vehicle were considered control values (100%). The influence of drugs was expressed as a percentage thereof. Data were analyzed by ANOVA followed by Dunnett’s test, for which the level of significance was set at P < .05. PRL levels were determined in systemic plasma using a radioimmunoassay and a specific antibody against rat PRL (Amersham, Buckingham, England) as described previously (Millan et al., 1995a). Results were expressed as a percentage of values obtained in control, vehicle-treated animals. Data were analyzed by ANOVA followed by Dunnett’s test. For DA turnover, AD50 values plus 95% CL were calculated, and for PRL levels, drug potency was expressed in terms of the minimal effective dose (P < .05) derived from Dunnett’s test.
Inhibition of 7-OH-DPAT- and PD 128,907-induced hypothermia.
As detailed previously (Millan et al., 1995a), CT was determined in rats by use of thermistoprobe (Testoterm, Forbach, France) in loosely restrained rats over 30 sec. CT was determined and rats were injected with vehicle or drug, followed 30 min later by an injection of vehicle, 7-OH-DPAT (0.16 mg/kg s.c.) or PD 128,907 (0.63 mg/kg s.c.). After 30 min, CT was again measured and the difference in temperature to basal values was calculated. Data were analyzed by ANOVA followed by Dunnett’s test, and ID50 values plus 95% CLs were calculated. The percent inhibition was computed as follows:
Inhibition of rotation induced by quinpirole.
The procedure employed was described in detail previously (Millan et al., 1995b). Briefly, rats were anesthetized with pentobarbital (45 mg/kg i.p.) and placed in a stereotaxic apparatus. The left substantia nigra pars compacta was injected, over 4 min, with 4.0 μl of 6-hydroxydopamine (2 μg/μl). After 3 weeks of recovery, those rats that showed a pronounced contralateral turning response to apomorphine (0.04 mg/kg s.c.) were selected for further study. Rats were trained with quinpirole (0.02 mg/kg s.c.), and rotation was recorded over the 20- to 50-min period after its application. Rotation was monitored automatically via a harness coupled to a Rotacount 8 microcomputer (Columbus Instruments, Columbus, OH). Rats received vehicle and quinpirole in alternating sessions. Rotation was expressed as a percentage of the mean of the sessions that preceded and those that followed drug treatment. Drugs were given 25 min before quinpirole. Data were analyzed by a paired Student’s t test (P < .05) and ID50 values (95% CLs) were calculated to estimate drug potency.
Apomorphine-induced climbing.
As before (Millan et al., 1995b), mice were administered drug or vehicle and placed individually in upturned cylinders (14 cm in diameter, 14 cm high) with walls of vertical bars (2 mm in diameter, 1 cm apart). Thirty minutes later, they were injected with apomorphine (0.75 mg/kg s.c.) and placed again in the cylinders. Each animal was observed for climbing behaviour (total score: 0–4) at 10 and 20 min after the injection of apomorphine. Data (percentages of animals with total climbing score <2) were analyzed by Fisher’s exact probability test (P < .05) and ED50 values (95% CLs) were calculated to estimate drug potency.
Inhibition of amphetamine, cocaine-, dizocilpine- and PCP-induced locomotion.
The procedure employed was as described previously byMaurel-Remy et al. (1995). Rats were administered drug or vehicle and placed in individual transparent polycarbonate cages (45 × 30 × 20 cm). Thirty minutes later, they were injected with amphetamine (2.5 mg/kg i.p.), cocaine (20 mg/kg i.p.), dizocilpine (0.16 mg/kg s.c.) or PCP (20.0 mg/kg s.c.), and the cages were placed in activity chambers (Lablinc System, Coulbourn, Lehigh Valley, PA). These were equipped with two infrared beams 4 cm above the floor and 24 cm apart. The consecutive interruption of two beams within 3 sec was computed as a movement (locomotor activity). Activity was monitored over 60 min after injection of amphetamine, cocaine, dizocilpine or PCP. Data were analyzed by ANOVA followed by Dunnett’s test (P < .05), and ID50 values (95% CLs) were calculated to estimate drug potency.
Inhibition of DOI-induced head-twitches.
As described previously (Schreiber et al., 1995), rats were injected with DOI (2.5 mg/kg i.p.) and placed in transparent Plexiglas observation cages (33.5 × 23.5 × 19.0 cm) without a sawdust lining. Five minutes after the administration of DOI, the number of head-twitches was counted over 5 min. Drugs were given 30 min before DOI. Data were analyzed by ANOVA followed by Dunnett’s test (P < .05), and ID50 values (95% CLs) were calculated to estimate drug potency.
Conditioned avoidance paradigm.
Rats used for the study were trained to move from one compartment of a shuttle-box (Letica, Barcelona, Spain) to the other when a stimulus light was on, in order to avoid an electric shock through the gridfloor. They were subjected to a daily session of 10 trials separated by 30-sec intertrial intervals. Each trial consisted of a 10-sec period (maximal duration) with the stimulus light on, followed or not by a 5-sec period (maximal duration) with an electric shock (560 μA), depending on the response of the animal to the stimulus light. The trial terminated once the rat had moved into the other compartment, either during the “light on” period (conditioned avoidance response) or during the shock period (escape response). Data were the number of conditioned avoidance responses (maximal value: 10) per session. The animals were their own controls, the control (vehicle) session being performed on the day before the test (drug) session. Vehicle or drugs were injected 30 min before the session. Data were analyzed by a paired Wilcoxon signed-rank test (P < .05), and ID50 values (95% CLs) were calculated to estimate drug potency.
Determination of extracellular levels of DA, 5-HT and NAD in the FCX, accumbens and striatum.
The procedure employed has been described in detail elsewhere (Gobert et al., 1995 and1997). Under pentobarbital anesthesia (60 mg/kg i.p.), rats were placed in a stereotaxic apparatus, and a guide cannula was implanted in the FCX or in both the accumbens and the contralateral striatum. The coordinates, according to Paxinos and Watson (1986), were as follows. FCX (AP: +2.2, L: ±0.6, DV: −0.2); accumbens (AP: +1.8, L: +1.6, DV: −4.5) and striatum (AP: +0.5, L: −2.8, DV: −3.0). Five days later, a Cuprophan CMA/11 probe (4 mm (FCX and striatum) and 2 mm (accumbens), 0.24 mm outside diameter) was lowered into position and perfused at 1 μl/min with a phosphate-buffered Ringer solution (147.2 mM NaCl, 4 mM KCl and 2.3 mM CaCl2, pH 7.3). Dialysis commenced 2 hr later, and samples were taken every 20 min. Three basal samples were taken; then the drug was injected. Samples were taken for a further 3 hr. Levels of DA, NAD and 5-HT were simultaneously quantified in individual samples via HPLC and coulometric detection with the following conditions. First, 20-μl dialysate samples were diluted with 20 μl of mobile phase (NaH2PO4: 75 mM, EDTA: 20 μM, sodium decanesulphonate: 1 mM, methanol: 17.5%, triethylamine 0.01%, pH: 5.70). Therefrom, 33-μl samples were analyzed by HPLC with a column (hypersil ODS 5 pm, C18, 150 × 4.6 mm, particle size, 5 μm) maintained at 43°C for separation and a coulometric detector (ESA 5014, Coulochem II) for quantification. The first electrode of the detector was set at −90 mV (reduction) and the second at +280 mV (oxidation). The mobile phase was delivered at a flow rate of 2 ml/min. The assay sensitivity was between 0.1 and 0.2 pg per sample for DA, NAD and 5-HT. Drug effects were expressed as a percentage of basal values (= 100%). Data were analyzed by a factorial ANOVA with drug as the between-subjects factor.
Induction of catalepsy.
Catalepsy was measured as previously (Millan et al., 1995a). Rats were placed in a position wherein the left and right hind paws were placed over the ipsilateral forepaws. The time over which this position was maintained was determined, with a cutoff of 30 sec (100% effect). The mean of three measures, separated by 1-min intervals, was determined. Drugs were injected 30 min before testing. Data were analyzed by ANOVA followed by Dunnett’s test, and AD50 values (95% CLs) were calculated to estimate drug potency.
Inhibition of methylphenidate-induced gnawing.
As before (Millan et al., 1995b), rats were administered methylphenidate (40.0 mg/kg i.p.) and placed in transparent Plexiglas observation cages (33.5 × 23.5 × 19.0 cm) with a grid floor. After 30 min, the number of periods (out of 10) with gnawing was determined over 10 min (one 10-sec observation period/min). Under such conditions, methylphenidate yielded a maximal response of 10. Drugs were administered 30 min before methylphenidate. Data were analyzed by ANOVA followed by Dunnett’s test, and ID50 values (95% CLs) were calculated to estimate drug potency.
Rotarod test: induction of ataxia.
As before (Millanet al., 1995b), 30 min after drug or vehicle injection, mice were placed on the bar of the rotarod apparatus (Ugo Basile, Varese, Italy) rotating with a gradual acceleration from 4 to 40 rpm over a period of 300 sec. The latency of mice to fall was determined with a cutoff of 360 sec. For determination of drug potency, ID50values (95% CLs) were calculated with respect to values in vehicle-treated animals (defined as 100%).
Oral activity of S 18126.
In several procedures (table 8), the activity of S 18126 was evaluated after its administration p.o. Under these conditions, S 18126 was given 60 min (rotarod) or 30 min (other tests) of pretesting, and its activity was expressed as a ratio to that obtained upon s.c. administration.
Activity of S 18126 upon oral administration
Drugs.
All drugs were dissolved in sterile water with a few drops of lactic acid. The pH was adjusted to as close to neutrality as possible (> 5.0). Drugs were injected s.c. unless otherwise specified. In general, full dose-response curves were performed for all studies. However, in view of limitations of drug solubility, the highest doses of S 18126 and L 745,470 tested in rats/mice were 160.0/80.0 and 40.0/10.0 mg/kg s.c., respectively. Thus for both S 18126 and L 745,870, in all procedures employed, doses were used that may be assumed fully to occupy central D4 receptors (see “Discussion”). Drug sources and salts were as follows: d-amphetamine sulfate (Calaire Chimie, Calais, France), cocaine hydrochloride (Coopérative Pharmaceutique Française, Melun, France), (±) DOI, (1-[2,5-dimethoxy-4-iodophenyl]-2-aminopropane hydrochloride (Research Biochemicals International, Natick, MA), apomorphine hydrochloride (Sigma Chimie, St Quentin-Fallavier, France), dizocilpine hydrogen maleate and raclopride tartrate (Research Biochemicals International, Natick, MA), methylphenidate hydrochloride (Ciba-Geigy) and PCP hydrochloride (Sigma Chimie, St Quentin-Fallavier, France). S 18126 and L 745,870 were synthetized by J.-L. Peglion (I.d.R.S.).
Results
Selectivity of S 18126 for hD4 receptors.
The affinity of S 18126 at hD4.4 receptors (Ki = 2.4 nM) was similar to that of L 745,870 but > 2000-fold greater than that of raclopride (fig.2; table1). Like L 745,870, S 18126 was 100-fold more selective for hD4 receptors than for other dopaminergic receptors (fig. 2; table 2). Furthermore, S 18126 showed low affinity at adrenergic, serotonergic, histaminergic and muscarinic receptors (table3). S 18126 also displayed low affinity (> 1000 nM) for 5-HT, NAD, DA and choline reuptake sites; nicotinic, imidazoline I2, adenosine A1, adenosine A2, AMPA, neurokinin1, neurokinin2, bradykinin B2, l-type Ca++ channel, site 2 Na+ channel, μ-opioid, cannabinoid, GABAA, GABAB, central benzodiazepine, NMDA, neuropeptide Y, endothelin-A, estrogen, progesterone and testosterone binding sites and MAO A, MAO B and NO synthase. Indeed, S 18126 was > 100-fold selective for hD4 receptors over all sites tested except ς1 sites, which showed marked affinity for S 18126 (Ki = 1.6 ± 0.6 nM). L 745,870 showed modest affinity for ς1 sites (123 ± 48 nM), whereas the affinity of raclopride at ς1 sites was low (> 1000 nM).

Interaction of S 18126, L 745,870 and raclopride at DA hD4 compared with other DA receptor types. [3H]-Spiperone competition binding experiments were carried out at recombinant human hD4 (hD4.4), hD2 (hD2S) and hD3 receptors. Points shown are means of triplicate determinations from representative experiments repeated on at least three independent occasions. A) S 18126; B) L 745,870 and C) raclopride.
Binding affinities of S 18126, L 745,870 and raclopride at dopaminergic receptor subtypes
Binding affinity of S 18126 for various receptor subtypes
Actions of drugs in models of activity at postsynaptic dopaminergic receptors
Antagonism by S 18126, L 745,870 and raclopride of agonist-stimulated [35S]-GTPγS binding at hD4 and hD2 receptors.
At hD4receptors, S 18126 alone did not induce any stimulation of [35S]-GTPγS binding (fig.3A) but concentration-dependently inhibited the stimulation of [35S]-GTPγS binding induced by 1 μM DA (fig. 3). L 745,870 likewise inhibited the action of DA at hD4 sites (Kb = 1.0 ± 0.1 nM). S 18126 also antagonized the stimulation of [35S]-GTPγS binding at hD4 receptors induced by 100 μM NAD (fig. 3B). DA stimulated [35S]-GTPγS binding at CHO-hD4 membranes with an EC50 of 107 ± 17 nM. The DA stimulation curve for [35S]-GTPγS binding to CHO-hD4membranes was concentration-dependently shifted to the right in the presence of fixed concentrations of S 18126 (fig. 3C). A Schild plot of the data yielded a linear isotherm with a slope close to unity and a pA2 value of 8.90 ± 0.06 (fig. 3D). Compared with hD4 receptors, S 18126 and L 745,870 only weakly inhibited DA (3 μM)-stimulated [35S]-GTPγS binding at hD2 receptors (fig. 4). In contrast, raclopride potently inhibited [35S]-GTPγS binding at hD2 receptors (fig. 4). DA stimulated [35S]-GTPγS binding to CHO-hD2 membranes with an EC50 of 353 ± 52 nM. The stimulation isotherm was not markedly altered by the addition of 30 nM S 18126 or 30 nM L 745,870 but was shifted 10-fold to the right by raclopride (30 nM) (fig. 4).

Antagonist actions of S 18126 at dopamine hD4 receptors as defined by the binding of [35S]-GTPγS. [35S]-GTPγS binding experiments were carried out on membranes of CHO cells expressing hD4 (hD4.4) receptors. A) Absence of stimulation of [35S]-GTPγS binding by S 18126 alone and antagonism of 1 μM DA-stimulated [35S]-GTPγS binding by S 18126 (IC50 = 23.7 ± 3.8 nM;Kb = 2.17 ± 0.35 nM). B) Inhibition of 1 μM NAD-stimulated [35S]-GTPγS binding by S 18126 (IC50 = 42.7 ± 10.3 nM; Kb = 2.32 ± 0.56 nM). C) “Shift” of the DA stimulation curve by fixed concentrations of S 18126. D) Schild plot of data from panel C:r = 0.99 ± 0.01, slope = 1.08 ± 0.01 and pA2 = 8.90 ± 0.06.

Antagonist actions of S 18126, L 745,870 and raclopride at dopamine hD2 receptors as defined by the binding of [35S]-GTPγS. [35S]-GTPγS binding experiments were carried out on membranes of CHO cells expressing hD2 (hD2S) receptors. A) Absence of stimulation of [35S]-GTPγS binding by S 18126, L 745,870 and raclopride alone and inhibition of 3 μM DA-stimulated [35S]-GTPγS binding (IC50 = 1500 ± 630, 2780 ± 510 and 13.6 ± 0.4 nM;Kb for raclopride = 1.44 ± 0.03 nM). B) “Shift” of the DA stimulation curve by fixed concentrations (30 nM) of S 18126, L 745,870 and raclopride: EC50, DA alone = 353 ± 52 nM; DA, S 18126 (30 nM) = 637 ± 123 nM; DA, L 745,870 (30 nM) = 456 ± 49 nM and DA, raclopride (30 nM) = 3570 ± 470 nM. Kb for raclopride = 2.79 ± 0.34 nM.
Activity at presynaptic dopaminergic receptors: influence on the activity of dopaminergic neurons.
As shown in figure5, raclopride markedly increased DA turnover throughout the brain, whereas S 18126 exerted only minor effects even at high doses, and L 745,870 was ineffective. Similarly, whereas raclopride increased the firing rate of ventrotegmental area-localized dopaminergic neurons, their activity was little affected by either S 18126 or L 745,870 (fig. 6). Raclopride also potently blocked the inhibition of firing elicited by the dopaminergic agonist apomorphine, whereas only a high dose of S 18126 interfered (partially) with the action of apomorphine, and L 745,870 was inactive (fig. 6).

Influence of S 18126, as compared with L 745,870 and raclopride, on the turnover of DA in various cerebral tissues. Data are means ± S.E.M.; N = 4 to 6 per value. Values (ratio of DOPAC to DA levels) are expressed as a percentage of those in control, vehicle-treated animals (= 100%). Absolute levels, in nanograms per milligram of protein, were as follows. Frontal cortex, DA = 2.50 ± 0.23 and DOPAC = 1.26 ± 0.21; nucleus accumbens, DA = 94.69 ± 11.52 and DOPAC = 12.23 ± 1.01; olfactory tubercles; DA = 40.95 ± 2.85 and DOPAC = 7.74 ± 0.86; striatum, DA = 128.42 ± 9.18 and DOPAC = 12.91 ± 0.76. Results of one-way ANOVA were as follows. S 18126: frontal cortex, F(3,22) = 4.1, P < .05; nucleus accumbens, F(3,21) = 12.9, P < .01; olfactory tubercles, F(3,22) = 4.9, P < .01 and striatum, F(3,21) = 9.7, P < .01. L 745,870: frontal cortex, F(2,10) = 0.7, P > .05; nucleus accumbens,F(2,10) = .4, P > .05; olfactory tubercles,F(2,10) = 1.3, P > .05 and striatum,F(2,10) = 1.3, P > .05. Raclopride: frontal cortex,F(6,42) = 22.5, P < .01; nucleus accumbens,F(6,42) = 88.9, P < .01; olfactory tubercles,F(6,42) = 22.9, P < .01 and striatum,F(6,42) = 25.7, P < .01. Asterisks indicate significance of differences from vehicle values in Dunnett’s test. * P < .05.

Influence of S 18126, compared with L 745,870 and raclopride, on the firing rate of dopaminergic neurons in the ventrotegmental area. Data are means ± S.E.M.; N= 4 to 5 per value. Results of one-way ANOVA were as follows. S 18126 alone, F(2,24) = 0.5, P > .05 and vs.apomorphine, F(2,24) = 7.6, P < .05. L 745,870 alone,F(2,24) = 0.9, P > .05 and vs. apomorphine,F(2,24) = 0.6, P > .05. Raclopride alone,F(3,32) = 41.9, P < .001 and vs.apomorphine, F(3,32) = 12.9, P < .001. Asterisks indicate significance of differences from vehicle values in Dunnett’s test. * P < .05.
Activity at postsynaptic dopaminergic receptors: hypothermia and rotation.
The dopaminergic agonists 7-OH-DPAT and PD 128,907 elicited a hypothermia that was potently and dose-dependently inhibited by raclopride. In contrast, only high doses of S 18126 and L 745,870 inhibited the induction of hypothermia by these agonists (table 3; fig.7A and B). None of the antagonists modified CT when administered alone. In rats sustaining unilateral lesions of the substantia nigra pars compacta, the dopaminergic agonist quinpirole elicited contralateral rotation. This action was potently abolished by raclopride, whereas S 18126 and L 745,870 only weakly modified the effect of quinpirole even at high doses (table 3 and fig.7C). Administered alone, they did not elicit rotation (not shown).

Influence of S 18126, compared with L 745,870 and raclopride, on the induction of hypothermia by 7-OH-DPAT and PD 128,907 and on the induction of rotation by quinpirole in rats with unilateral lesions of the substantia nigra pars compacta. A) Influence of antagonists alone (open symbols) on core temperature and on the induction of hypothermia by 7-OH-DPAT (filled symbols). B) Influence of antagonists on the induction of hypothermia by PD 128,907. Panel C) Influence of antagonists on the induction of rotation by quinpirole. Data are means ± S.E.M.; N = 4 to 8 per value. Results of one-way ANOVA were as follows. Hypothermia: S 18126 alone,F(3,22) = 1.0, P > .05; vs. 7-OH-DPAT,F(4,26) = 11.2, P < .001; vs. PD 128,907,F(4,24) = 10.8, P < .01. L 745,870 alone,F(3,20) = 1.7, P > .05; vs. 7-OH-DPAT,F(3,21) = 3.3, P < .05; vs. PD 128,907,F(4,18) = 6.2, P < .01. Raclopride alone,F(3,37) = 2.9, P > .05; vs. 7-OH-DPAT,F(4,39) = 26.6, P < .001; vs. PD 128,907,F(5,26) = 20.6, P < .001. VEH = vehicle. Asterisks indicate significance of differences from vehicle values in Dunnett’s test (hypothermia) and in paired Student’s ttest (rotation). * P < .05.
Activity in models predictive of antipsychotic activity.
As shown in table 4 and figure8, raclopride was potently active in several models predictive of antipsychotic properties: inhibition of amphetamine-, dizolcipine-, cocaine- and PCP-induced locomotion in rats, inhibition of apomorphine-induced climbing in mice, reduction of conditioned avoidance responses in rats and inhibition of DOI-induced head-twitches in rats. S 18126 displayed only modest activity in several of these models, even over a much higher dose range than for raclopride. Over the dose range tested, L 745,870 was also weakly active in these models.
Actions of drugs in models predictive of antipsychotic activity

Influence of S 18126, compared with L 745,870 and raclopride, in several models predictive of antipsychotic (antiproductive) activity. A) Inhibition of locomotion elicited by amphetamine (2.5 mg/kg i.p.). B) Inhibition of locomotion elicited by cocaine (20 mg/kg i.p.). C) Inhibition of locomotion elicited by dizocilpine (0.16 mg/kg s.c.). D) Inhibition of climbing elicited by apomorphine (0.75 mg/kg s.c.). E) Inhibition of conditioned avoidance responses F) Inhibition of head-twitches evoked by DOI (2.5 mg/kg i.p.). Data are means ± S.E.M.; N = 4 to 18 per value. Results of one-way ANOVA were as follows. Amphetamine-induced locomotion: S 18126, F(7,47) = 3.6, P < .01; L 745,870, F(6,43) = 2.0, P > .05; raclopride,F(3,61) = 12.0, P < .01. Cocaine-induced locomotion: S 18126, F(3,24) = 5.1, P < .05; L 745,870,F(3,21) = 1.4, P > .05; raclopride, F(4,30) = 8.12, P < .01. Dizocilpine-induced locomotion: S 18126,F(3,32) = 1.4, P > .05; L 745,870, F(3,21) = 1.2, P > .05; raclopride, F(5,37) = 8.1, P < .01. DOI-induced head-twitches: S 18126, F(4,26) = 4.7, P < .05; L 745,870, F(2,12) = 0.5, P > .05; raclopride, F(4,38) = 6.1, P < .01. VEH = vehicle. Asterisks indicate significance of differences from vehicle values in Dunnett’s test, the Fisher test (apomorphine-climbing) or the paired Wilcoxon test (conditioned avoidance responses). * P < .05.
Influence on extracellular levels of DA, NAD and 5-HT in the FCX, accumbens and striatum.
Injection of vehicle did not markedly modify basal levels of monoamines, although there was a slight and transient increase in levels of NAD (fig.9B). Raclopride potently and dose-dependently increased FCX dialysate levels of DA (fig. 9), and a further increase in its dose to 2.5 mg/kg s.c. did not yield any additional effect (not shown). At a dose of 0.16, raclopride also significantly increased levels of NAD, although those of 5-HT were not significantly modified. At a maximally effective dose (0.16) for increasing FCX levels of DA, raclopride also, and more markedly, increased dialysate levels of DA in both the nucleus accumbens and the striatum. At a dose of 0.16 mg/kg s.c., S 18126 failed to modify levels of DA, NAD or 5-HT in FCX (fig. 10). Over a higher dose range (2.5–40.0 mg/kg s.c.), S 18126 dose-dependently and markedly increased levels of both DA and NAD in FCX, whereas those of 5-HT were not significantly modified. In contrast to the FCX, levels of DA were not significantly modified by a maximally effective dose of S 18126 (40.0) in either nucleus accumbens or striatum. L 745,870 (fig.11) also failed to modify FCX levels of DA, NAD and 5-HT at a dose of 0.16 mg/kg s.c. Although it tended to increase levels of DA and NAD at a higher dose (2.5), this action was not significant.
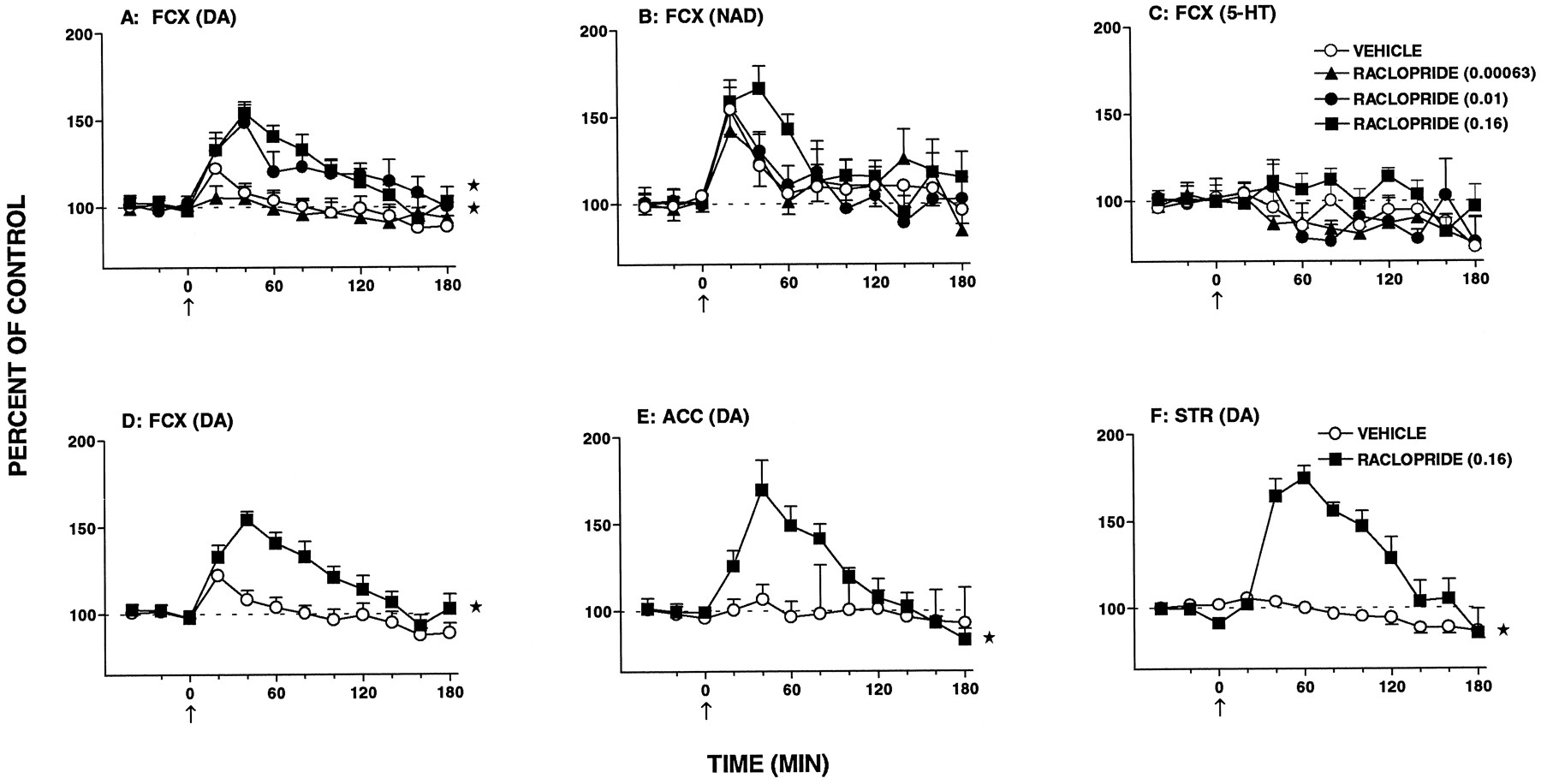
Influence of raclopride on extracellular levels of DA, NAD and 5-HT in single samples of FCX, nucleus accumbens (ACC) and striatum (STR) dialysates of freely moving rats. Data presented in panel D are the same as those in panel A (0.16 mg/kg s.c.) and permit easier comparison with panels E and F. Data are means ± S.E.M.;N = 5 to 10 per value of DA; NAD and 5-HT levels are expressed as a percentage of basal preinjection values (= 100%). Absolute levels (in picograms per 20 microliters) were as follows. FCX, 1.25 ± 0.08, 1.09 ± 0.11 and 0.71 ± 0.06 for DA, NAD and 5-HT, respectively; nucleus accumbens, 5.48 ± 0.77 for DA and striatum, 16.94 ± 2.99 for DA. For comparison of individual values with vehicle-treated group, a factorial ANOVA with drugs as the between-subjects factor was performed over 20 to 180 min. FCX: raclopride (0.00063), DA, F(1,16) = 0.2, P > .05; NAD,F(1,14) = 0.1, P > .05 and 5-HT, F(1,10) = 1.6, P > .05; raclopride (0.01), DA, F(1,17) = 9.8, P < .01; NAD, F(1,15) = 0.1, P > .05 and 5-HT,F(1,9) = 0.1, P > .05; raclopride (0.16), DA,F(1,19) = 14.5, P < 0.1; NAD, F(1,19) = 6.5, P < .05 and 5-HT, F(1,12) = 1.5, P < .05. Nucleus accumbens: raclopride (0.16), DA, F(1,11) = 9.4, P < .01. Striatum: raclopride (0.16), DA, F(1,14) = 22.3, P < .01. The asterisks indicate significance of the difference between the drug-treated group and the vehicle-treated group. * P < .05.
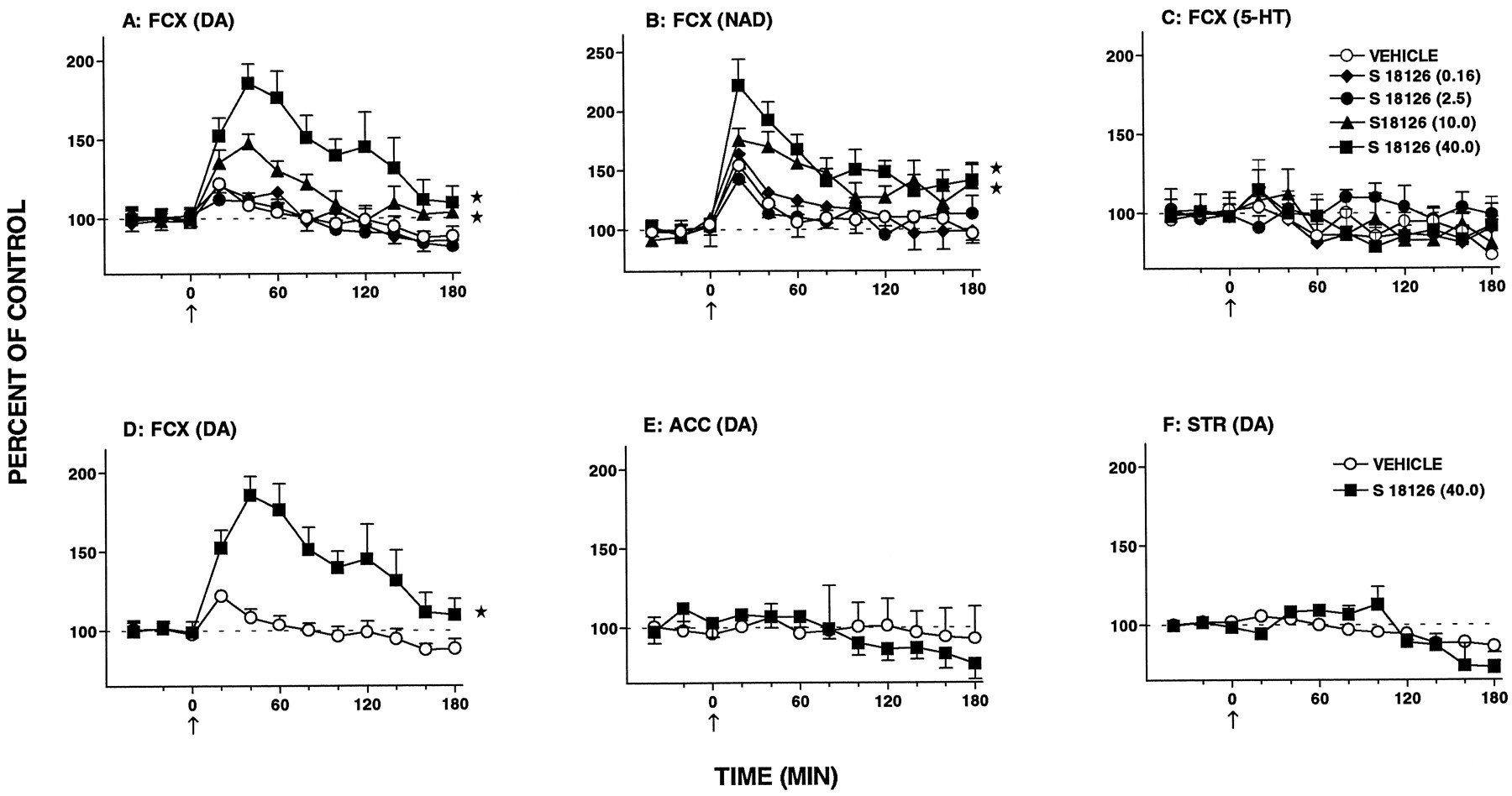
Influence of S 18126 on extracellular levels of DA, NAD and 5-HT in single samples of FCX, nucleus accumbens (ACC) and striatum (STR) dialysates of freely moving rats. Data in panel D are the same as those in panel A (40.0 mg/kg s.c.) and permit easier comparison with panels E and F. Data are means ± S.E.M.;N = 5 to 8 per value of DA; NAD and 5-HT levels are expressed as a percentage of basal preinjection values (= 100%). For comparison of individual values with vehicle-treated group, a factorial ANOVA with drugs as the between-subjects factor was performed over 20 to 180 min. FCX: S 18126 (0.16), DA, F(1,18) = 0.1, P > .05; NAD, F(1,16) = 0.1, P > .05 and 5-HT,F(1,11) = 0.1, P > .05; S 18126 (2.5), DA,F(1,17) = 0.4, P > .05; NAD, F(1,15) = 0.1, P > .05 and 5-HT, F(1,9) = 4.4, P > .05; S 18126 (10.0), DA, F(1,18) = 8.5, P < .01; NAD,F(1,15) = 6.7, P < .05 and 5-HT, F(1,10) = 0.1, P > .05; S 18126 (40.0), DA, F(1,19) = 11.3, P < .01; NAD, F(1,17) = 11.8, P < .01 and 5-HT,F(1,10) = 0.1, P > .05. Nucleus accumbens: S 18126 (40.0), DA, F(1,12) = 0.49, P > .05. Striatum: S 18126 (40.0), DA, F(1,10) = 0.02, P > .05. The asterisks indicate significance of the difference between the drug-treated group and the vehicle-treated group. * P < .05.

Influence of L 745,870 on extracellular levels of DA, NAD and 5-HT in single samples of FCX dialysates of freely moving rats. Data are means ± S.E.M.; N = 6 to 8 per value. DA, NAD and 5-HT levels are expressed as a percentage of basal preinjection values (= 100%). For comparison of individual values with vehicle-treated group, a factorial ANOVA with drugs as the between-subjects factor was performed over 20 to 180 min. L 745,870 (0.16), DA, F(1,18) = 0.1, P > .05; NAD,F(1,16) = 0.2, P > .05 and 5-HT, F(1,10) = 5.0, P > .05; L 745,870 (2.5), DA, F(1,19) = 1.8, P > .05; NAD, F(1,17) = 3.4, P > .05 and 5-HT,F(1,11) = 0.9, P > .05.
Activity in models predictive of extrapyramidal actions.
Raclopride elicited catalepsy and PRL secretion and inhibited induction of gnawing by the DA releaser methylphenidate (table5; fig.12). It also induced ataxia in the rotarod procedure in mice and reduced spontaneous locomotor activity (table 5; fig. 12). In contrast, S 18126 and L 745,870 displayed only weak activity, even at high doses (table 5; fig. 12). S 18126 did not modify the cataleptic actions of haloperidol (2.5 mg/kg s.c.): vehicle/haloperidol (n = 4), 29.4 ± 0.6 secvs. S 18126 (0.01 mg/kg s.c.)/haloperidol (n= 4), 26.7 ± 1.4 sec, S 18126 (0.16 mg/kg s.c.)/haloperidol (n = 4), 29.3 ± 0.7 sec, S 18126 (2.5 mg/kg s.c.)/haloperidol (n = 4), 29.0 ± 1.0 sec and S 18126 (40.0 mg/kg s.c.)/haloperidol (n = 4), 24.7 ± 1.9 sec, P > .05.
Actions of drugs in models predictive of extrapyramidal activity

Influence of S 18126, compared with L 745,870 and raclopride, on several parameters predictive of the induction of an extrapyramidal syndrome or disruption of motor behavior. A) Induction of catalepsy. B) Inhibition of the gnawing elicited by methylphenidate (40.0 mg/kg i.p.). C) Inhibition of latency to fall in the rotarod test. D) Inhibition of spontaneous locomotion in mice. E) Inhibition of spontaneous locomotion in rats. F) Increase in PRL levels. Data are means ± S.E.M.; N = 4 to 10 per value. Results of ANOVA were as follows. Catalepsy: S 18126, F(5.41) = 0.5, P > .05; L 745,870, F(2,12) = 1.2, P > .05 and raclopride, F(5,38) = 14.5, P < .01. Gnawing: S 18126,F(2,11) = 2.0, P > .05; L 745,870, F(2,17) = 2.3, P > .05 and raclopride, F(4,22) = 47.2, P < .01. Ataxia: S 18126, F(3,21) = 15.4, P < .01; L 745,870, F(3,17) = 41.2, P < .01 and raclopride,F(5,38) = 9.9, P < .01. Locomotion, mice: S 18126,F(3,20) = 7.7, P < .05; L 745,870, F(3,22) = 3.0, P < .05 and raclopride, F(5,37) = 9.3, P < .01. Locomotion, rat: S 18126, F(6,53) = 7.8, P < .01; L 745,870, F(4,28) = 3.8, P < .05 and raclopride,F(4,17) = 6.5, P < .01 and PRL: S 18126,F(2,15) = 1.4, P > .05; L 745,870, F(2,10) = 0.9, P > .05 and raclopride, F(6,14) = 12.9, P < .01. Asterisks indicate significance of differences from vehicle values in Dunnett’s test. * P < .05.
Correlation analyses with benzamides.
In analogy to raclopride, several further benzamides displayed activity in the functional models employed herein (table6), although with markedly different potencies. These models included a test of activity at postsynaptic dopaminergic receptors (inhibition of 7-OH-DPAT-induced hypothermia), two models of antipsychotic activity (inhibition of apomorphine-induced climbing and inhibition of conditioned avoidance responses) and a model predictive of extrapyramidal properties (induction of catalepsy). A comparison of their potencies, together with those of S 18126 and L 745,870, with their affinities at hD4 receptorsfailed to reveal a significant degree of correlation (table7). In contrast, there was a pronounced correlation between drug potency in each procedure and affinity at hD2 receptors (table 7; fig.13). (Correlation coefficients were also significant for affinities at hD3 receptors. However, because the drugs used herein all have similar affinities at hD2 and hD3 sites, the respective, putative roles of D2 and D3 receptors could not be differentiated by this analysis (Millan et al., 1995a). The present data suggest that D2 (or D3) rather than D4 receptors play a role in the functional models employed. Further, they provide an indication of the doses at which S 18126, L 745,870 and raclopride—all of which were situated close to the regression curves of the correlation analyses—exert their actions at D2 receptor in vivo (fig. 13). Bearing in mind the relative affinities of S 18126 and L 745,870 at hD4vs. hD2 receptors, doses about 100-fold lower may thus be predicted to be D4-selective (see “Discussion”).
Influence of S 18126, compared with L 745,870, raclopride and other benzamides, on several dopaminergic parameters
Correlation matrix of in vitro affinities with functionalin vivo actions for S 18126, L 745,870, raclopride and several other benzamides (based on data presented in table 6)
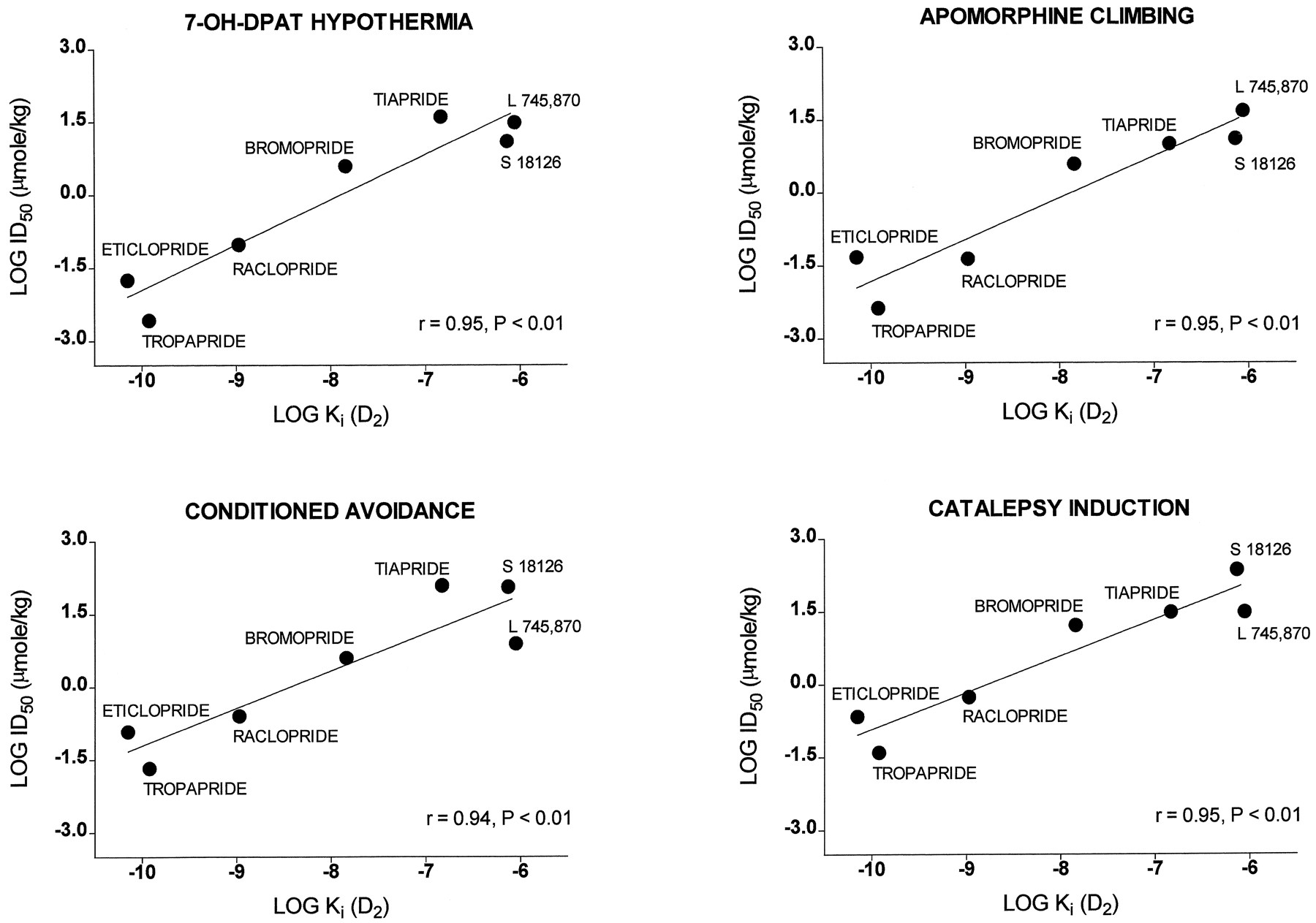
Correlation analysis of drug affinity at hD2 receptors vs. activity in various functional models. Note that S 18126 and L 745,870 are both close to regression lines. For r values, see table 7.
Activity of S 18126 upon p.o. administration.
Upon p.o. administration, S 18126 displayed activity in several models of potential antipsychotic activity, with a maximal effect comparable to that seen by the s.c. route (table 8). It was also active in the rotarod test upon p.o. injection, although only at very high doses. The median ratio for activity by the p.o. as compared with the s.c. route was 1.6.
Discussion
Interaction of S 18126 with recombinant hD2 and hD4 receptors.
Both equilibrium competition binding and functional G protein activation studies demonstrated the marked (> 100-fold) selectivity of S 18126 for hD4 receptorsvs. other dopaminergic receptor types. In line with previous observations (Asghari et al., 1995; Bristow et al., 1997; Kugalowski et al., 1996; Millan et al., 1995a and b; Patel et al., 1996b), this marked selectivity of S 18126 for hD4 receptors was shared by L 745,870 and opposite to the receptor profile of raclopride, which exhibited high activity at hD2 and hD3receptors yet low affinity for D4 receptors. S 18126 likewise displayed pronounced selectivity for hD4 receptors compared with a broad range of about 50 other binding sites (table 2and “Results”) with the exception of ς1 sites, the potential significance of which is discussed below.
S 18126 did not stimulate [35S]-GTPγS binding at hD4 receptors, which indicates an absence of agonist activity (Newman-Tancredi et al., 1997a). However, it potently inhibited DA-stimulated [35S]-GTPγS binding with a Kb value (2.2 nM; fig. 3) resembling itsKi value (2.4 nM) derived from competition binding studies. When examined by Schild analysis, the potency of S 18126 (pA2 = 8.9) was similar to theseKb and Ki values. Taken together, these data demonstrate that S 18126 behaves as a potent and competitive (neutral) antagonist at hD4 receptors. The present data corroborate the previously reported activation of hD4 receptors by NAD (Lanau et al., 1997;Newman-Tancredi et al., 1997a) and show that NAD-stimulated [35S]-GTPγS binding is blocked by S 18126; this result offers further evidence that the action of NAD is indeed mediated by D4 receptors. There are indications of a perturbation of adrenergic transmission in psychotic disorders. Thus, the functional properties of antipsychotic agents may involve a blockade of the actions of NAD not only at adrenergic but also, perhaps, at hD4 receptors (Van Kammen et al., 1990; seeNewman-Tancredi et al., 1997b). S 18126 and L 745,870 only weakly inhibited DA-stimulated [35S]-GTPγS binding at hD2 receptors, which demonstrates that their in vitro selectivity for hD4vs.hD2 receptors extends to a functional model. In contrast, raclopride potently inhibited [35S]-GTPγS binding at hD2 receptors (Kb = 1.4 nM, similar to Ki = 1.1 nM), an effect consistent with its high affinity and potent in vivo antagonist properties at these sites (Asghari et al., 1995; Millan et al., 1995a and b).
Like other studies of novel, D4-selective antagonists (Hartmann et al., 1996; Holland et al., 1996;Merchant et al., 1996; Patel et al., 1996a andb), and within the framework of a possible clinical development, the aforementioned studies were performed on heterologously expressed, recombinant hD4 receptors. Because hD4receptors differ from their rat counterparts in the third, intracellular loop region, which is responsible for G protein coupling, the possibility that S 18126, L 745,870 or other antagonists at hD4 receptors possess differential intrinsic activity at rat vs. human D4 receptors cannot be formally excluded (Asghari et al., 1995). Nevertheless, comparative studies of rat and human D4 receptors, as well as analyses of polymorphisms in the third intracellular loop of the hD4receptor, suggest that this is unlikely (Hadley, 1996). Furthermore, the ligand recognition site of the human and rat D4receptors shows a high degree of homology (Hadley, 1996; Makoff, 1992;O’Malley et al., 1992).
Actions of S 18126 at presynaptic dopaminergic receptors in vivo: modulation of dopaminergic transmission.
There is substantial anatomical (Baik et al., 1995, Bouthenetet al., 1991; Diaz et al., 1995), electrophysiological (Bowery et al., 1996; Lejeune and Millan, 1995; Mercuri et al., 1997), behavioral (Sangeret al., 1996) and biochemical (Bowery et al., 1996; Cooper et al., 1996; Gainetdinov et al., 1996; Gobert et al., 1995 and 1996; O’Hara et al., 1996; Tang et al., 1994; Tepper et al., 1997) evidence that terminal- and dendritically localized D2 and possibly D3, autoreceptors control the activity of mesocortical, mesolimbic and nigrostriatal dopaminergic neurons. Correspondingly, raclopride increased cerebral DA synthesis and the spontaneous electrical activity of ventrotegmental area-localized dopaminergic neurons and antagonized the inhibitory influence of apomorphine on their firing rate. The low affinity of raclopride for D4 sites largely excludes their involvement in its actions, and although circular arguments should be avoided, the weak influence of S 18126 and L 745,870 on dopaminergic transmission probably reflects their residual affinity for D2autoreceptors. Thus, consistent with neuroanatomical studies that have failed to detect D4 receptors on dopaminergic neurons (Ariano et al., 1997; Defagot et al., 1997;Primus et al., 1997), selective blockade of D4receptors neither directly nor indirectly (via a postsynaptic feedback loop, for example) modifies the activity of ascending dopaminergic neurons. Although the selective D4antagonist U 101,387 was reported to interfere with the development of sensitization to amphetamine-induced DA release in the nucleus accumbens, this phenomenon does not necessarily reflect a role of D4 receptors in the control of the activity of dopaminergic neurons per se and U 101,387 does not modify the activity of mesolimbic or hypothalamic dopaminergic neurons (Feldpauschet al., 1996; Merchant and Yamamoto, 1996; Merchant et al., 1996; Piercey et al., 1996; Stone et al., 1996). Thus the observation that knockout mice lacking D4 receptors show an increase in basal DA synthesis in the striatum is unlikely to reflect the loss of a putative, D4autoreceptor-mediated control of nigrostriatal dopaminergic pathways (Pugsley et al., 1996).
In conclusion, comparing two selective D4 antagonists, S 18126 and L 745,870, with the D2/D3 antagonist raclopride, and employing measures of the electrical, releasing and synthetic activity of mesocortical, mesolimbic and nigrostriatal dopaminergic pathways, the present studies suggest that selective blockade of D4 receptors does not modify the activity of cerebral dopaminergic networks.
Actions at postsynaptic dopaminergic receptors in vivo.
Activation of postsynaptic D3 and/or D2 receptors contributes to the decrease in CT elicited by 7-OH-DPAT and PD 128,907 (Audinot V. et al., unpublished observations; Millan et al., 1995a), and correspondingly, raclopride potently inhibited their hypothermic actions. By contrast, S 18126 and L 745,870 were weakly active. Employing a series of benzamides that possess similar affinities at hD2 and hD3 receptors but contrasting affinities at hD4receptors (Baldessarini et al., 1996; Giulani and Ferrari, 1997; Nasello et al., 1991; Rumigny et al., 1984;Steele et al., 1993), we found these modest actions of S 18126 and L 745,870 to correlate with their low affinities at D3 and D2 receptors. These data suggest that D4 receptors are not involved in the modulation of CT, a finding pertinent to the clinical problem of malignant hyperthermia that is provoked by neuroleptic agents (Heiman-Patterson, 1993). In a “hemiparkinsonian” model of activity at postsynaptic D2receptors, induction of contralateral rotation by quinpirole in unilateral substantia nigra-lesioned rats (Creese and Fraser, 1987), S 18126 and L 745,870 were virtually inactive compared with raclopride. These observations are in line with the conclusions of an in vitro study suggesting that D4 receptors are unlikely to be involved in the therapeutic actions of antiparkinsonian agents (Newman-Tancredi et al., 1997a).
In vivo actions of S 18126 and L 745,870 at D4 receptors: estimation of active dose ranges.
An important question concerns the dose ranges over which S 18126 and L 745,870 express their putative actions at D4 receptorsin vivo. In a previous study, employing an ex vivo binding approach, Patel et al., (1997) established the dose range over which L 745,870 occupies ς1 sites. On the basis of this “surrogate” action of L 745,870, they calculated the dose range over which it should act as a selective D4receptor antagonist in vivo. Herein, we adopted a complementary strategy whereby we determined the doses of S 18126 and L 745,870 required for the expression of their actions at D2receptors in vivo. On the basis of their relative affinities at hD4vs. hD2 sites, it was possible to calculate the theoretical dose ranges over which they act at D4 receptor antagonists in vivo. For example, in the apomorphine-induced climbing model (see the correlation analysis of figs. 12 and 13), S 18126 and L 745,870 were active at doses of 4.5 and 15.8 mg/kg s.c., respectively. Dividing these doses by the ratio of their affinities at hD2vs. hD4sites (308 and 362 for S 18126 and L 745,870, respectively) yields theoretical, “D4-active” doses of 0.01 and 0.04 mg/kg s.c. for S 18126 and L 745,870, respectively. This dose of L 745,870 corresponds well to the predictions of Patel et al. (1997)mentioned above. These calculations serve, then, as anestimate of the approximate dose ranges over which S 18126 and L 745,870 are likely to express actions selectivelyvia D4 receptors in vivo and provide a framework for a discussion of their in vivo properties. Nevertheless, they should not be regarded as definitive values in the absence of direct measurement of their occupation of cerebral populations of D4 receptors in vivo, which is not, unfortunately, currently feasible. Such information, as well as the identification of specific functional responses unambiguously attributable to D4 receptors, will be necessary for a further understanding of their pathophysiological role.
Actions in models of potential antipsychotic activity.
In contrast to the potent activity of raclopride in several paradigms predictive of antipsychotic properties, S 18126 and L 745,870 either were inactive or were active only at high doses corresponding to those effective in the D2/D3 models we have described. In addition, further analysis—incorporating the effects of several benzamides—of the apomorphine-induced climbing and conditioned avoidance response models revealed that the actions of S 18126 and L 745,870 correlated with their affinities at D2(D3) rather than D4 receptors. In line with these observations, the selective D4 receptor antagonists U 101,837 and CP 293,019 are also little active in “traditional” models of potential antipsychotic activity based on blockade of the motor actions of apomorphine and amphetamine in rodents (Merchant et al., 1996; Rubinstein et al., 1997;Zorn et al., 1996). Collectively, then, these data suggest that selective blockade of D4 receptors is unlikely to control the productive symptoms of schizophrenia.
One general problem with such experimental models is that they are designed to predict the therapeutic activity of a particular drug class—in this case, D2 antagonists. Consequently, drug activity may be correlated with, rather than causal of, clinical efficacy. Thus even the diversity of models utilized herein might not, arguably, be appropriate to the prediction of antipsychotic activity for agents lacking D2 receptor affinity—specifically, D4 receptor antagonists. In fact, in a recent clinical study of acute, hospitalized psychotic patients, L 745,870 proved to be devoid of antipsychotic (antiproductive) properties (Bristow et al., 1997; Krameret al., 1996). These clinical findings are in line with the present data in suggesting that selective D4 receptor blockade is unlikely to control productive symptoms in neuroleptic-responsive patients. Nevertheless, additional clinical data for other selective D4 antagonists are required before definitive conclusions can be reached. Furthermore, it remains possible that a subpopulation of neuroleptic-resistant patients who are not responsive to haloperidol and other drugs that block mesolimbic D2 receptors might respond to selective D4receptor antagonists.
It has also been suggested that the distinctive, “atypical” profile of clozapine in treating neuroleptic-resistant patients and deficit-cognitive symptoms might be attributable to its proportionately greater occupation of D4 than of D2 receptors as compared with neuroleptics (Seeman, 1992; Seeman et al., 1997). The limited clinical trials performed to date with L 745,870 (Kramer et al., 1996) are not sufficient for a rigorous assessment of the potential utility of D4 receptor blockade in controlling deficit-cognitive symptoms. These probably reflect a functional perturbation of the frontal/prefrontal cortex (“hypofrontality”), perhaps involving a hypoactivity of mesocortical dopaminergic pathways. Correspondingly, the marked D2 antagonist properties of neuroleptics may even enhance or induce (“secondary”) deficit symptoms (Andreasen, 1996; Chuaet al., 1997; Kahn and Davis, 1995). Clozapine, but not haloperidol, selectively reinforces dopaminergic transmission in the FCX as compared with subcortical structures (Moghaddam and Bunney, 1990; Rivet et al., 1996) and it might be hypothesized that blockade of D4 receptors contributes to this action. However, at a D4 receptor-selective dose, neither S 18126 nor L 748,870 modified the release of DA in the FCX. These data suggest that D4 receptor blockade is not involved in the influence of clozapine on FCX levels of DA and NAD. Nevertheless, recent studies of the selective D4 receptor antagonists RO 61-6270, CP 293,019 and U 101,387 have shown that, by analogy to clozapine and in contrast to neuroleptics, they preferentially induce expression of the immediate-early gene c-fos in the FCX as compared with the striatum (Hartman et al., 1996; Holland et al., 1996; Merchant et al., 1996). Although no unambiguous evidence that these changes reflect selective blockade of D4 receptor is available, U 101,387 was active at low doses (≈ 0.2 mg/kg s.c.). Thus, because D4 receptors are present in a high concentration in FCX (Meador-Woodruff et al., 1996; Tallman et al., 1997; Wang et al., 1996), their relationship to the cognitive-deficit symptoms of schizophrenia warrants further exploration.
In a further evaluation of a possible role of D4 receptor blockade in the amelioration of deficit symptoms, we examined other potential similarities between the actions of S 18126 and L 745,870 and clozapine. It has been suggested that preferential blockade of the hyperlocomotion elicited by PCP, which provokes both productive and deficit symptoms of schizophrenia in the human, as compared with amphetamine, which elicits only productive symptoms, may be predictive of a clozapine-like profile of antideficit properties (Maurel-Remyet al., 1995). However, in contrast to clozapine, both S 18126 and L 745,870 failed to block the hyperactivity provoked by PCP. Further, in a drug discrimination paradigm employing animals trained to recognize clozapine, we recently found that S 18126 and L 745,870 do not generalize to clozapine, which indicates that D4 receptor antagonism does not make a major contribution to the global, “psychological” influence of clozapine (Millan et al., 1997b). Thus there is currently little concrete evidence that selective D4 receptor blockade should improve the deficit symptoms of schizophrenia (see also Rothet al., 1995). However, the clinical evaluation of selective D4 antagonists in patient populations displaying predominantly deficit symptoms appears imperative before concrete conclusions can be reached.
Finally, there is neuroanatomical and behavioral evidence that cortical (temporal, frontal and entorhinal), striatal, thalamic and hippocampal populations of D4 receptors play a role in the modulation of cognition and attention, specifically in the control of information access to, and processing in, the cortex (Ariano et al., 1997; Joyce and Meador-Woodruff, 1997; Mrzljak et al., 1996). Correspondingly, an evaluation of whether D4receptor blockade may improve cognitive-attentional symptoms in schizophrenic patients would seem to be of interest.
Influence on FCX monoaminergic transmission.
Although a low, D4-selective dose of S 18126 failed to modify FCX levels of DA, it dose-dependently increased levels of DA and NAD, but not 5-HT, in the FCX over a higher dose range, and this action was specific inasmuch as levels of DA and NAD were not modified in accumbens and striatum. By analogy to the influence of D4 antagonists on c-fos expression in the FCX (vide supra), this pattern of effects of S 18126 resembles that of clozapine (Moghaddam and Bunney, 1990; Rivet et al., 1996). Further, it may be distinguished from the effects of both raclopride and haloperidol, which preferentially increase dialysate levels of DA in the striatum and accumbens as compared with the FCX (Gobert et al., 1998;Moghaddam and Bunney, 1990; Nomikos et al., 1994; Rivetet al., 1996) (fig. 10). The actions of raclopride may be attributed to its blockade of D2/D3autoreceptors. However, the weak actions of S 18126 at these sites cannot provide a full explanation for its effects because 1) DA levels should also have been increased in limbic and striatal regions, which was not the case, 2) the increase in FCX levels of DA was more pronounced for S 18126 than for raclopride and 3) S 18126 did not increase the firing rate of ventrotegmental dopaminergic neurons. A blockade of D2/D3 autoreceptors would also not provide an explanation for the S 18126-induced increase in FCX levels of NAD, although these were also increased by raclopride and may reflect a general state of drug-induced arousal (Cenci et al., 1992; Gobert et al., 1998; see Millan et al., 1997). Thus the processes underlying the selective induction in FCX release of DA (and NAD) by S 18126 remain to be clarified.
A further interesting observation was that changes in DA synthesis and release were not invariably modified in parallel. For example, S 18126 elicited a mild increase in DOPAC/DA ratios in the accumbens and striatum without affecting dialysate levels of DA. Although these differences were not marked, they are of interest because as they support findings of previous studies suggesting that the intracellular transduction mechanisms that underlie modulation of DA synthesis arenot identical to those that control its release. For example, there is evidence that a decrease in intracellular Ca++ concentrations suppresses DA synthesis by reducing the activation (phosphorylation) of tyrosine hydroxylase, the rate-limiting enzyme for DA formation (El Mestikawy et al., 1986; Haycock and Haycock, 1991). On the other hand, a modulation of current flow via K+ channels appears to play a major role in the control of DA release (Tang et al., 1994). It would be interesting to examine this issue further.
Extrapyramidal syndrome and other side effects.
Blockade of postsynaptic D2 receptors in the basal ganglia (striatum) underlies the induction of extrapyramidal motor side effects by antipsychotics, whereas antagonism of D2 receptors on hypophyseal lactotrophs enhances circulating levels of PRL (Ben-Jonathan et al., 1989; McDonald et al., 1984; Millan et al., 1995a; Nilsson et al., 1996). The present data show that, unlike raclopride, S 18126 and L 745,870 do not elicit either PRL secretion or catalepsy, a motor response predictive of extrapyramidal side effects in the human. Furthermore, as compared with raclopride, even high doses of S 18126 and L 745,870 only marginally influenced stereotyped gnawing, a dopaminergic motor response mediated in the striatum (Creese and Fraser, 1987), and they had little effect on general motor behavior. The correlation analyses undertaken herein with several benzamides further support the argument that the induction of catalepsy reflects blockade of D2 rather than D4 receptors. In addition, L 745,870 fails to provoke motor effects in primates (Bristowet al., 1997), whereas the selective D4antagonists U 101,387 and CP 293,019 were also reported to modify little motor behavior and PRL secretion in rodents (Merchant et al., 1996; Zorn et al., 1996). Moreover, whereas mice lacking D2 receptors display a Parkinson-like disruption of motor coordination, mice with a null mutation for D4receptors show little modification of motor function (Ariano et al., 1997; Paulus et al., 1996; Pugsley et al., 1996). Similar findings have indicated that PRL secretion and catalepsy are not elicited by a selective loss of activity of D3 receptors (Accili et al., 1996; Millanet al., 1995a and 1997a). However, blockade of D3 receptors may actively oppose D2receptor-mediated catalepsy and facilitate motor activity (Acciliet al., 1996; Hall and Strange, 1997; Hanner et al., 1996; Millan et al., 1995a; Sautel et al., 1995; Waters et al., 1993). In contrast, there is no evidence for such effects of D4 receptor antagonism, and S 18126 did not modify the induction of catalepsy by haloperidol (see “Results”).
Collectively, then, these observations indicate that the selective blockade of D4 receptors does not provoke the extrapyramidal, motor and endocrine side effects associated with treatment by neuroleptics. Rather, antagonism of D2receptors is responsible for these symptoms.
S 18126 possesses negligible affinity at histaminic, muscarinic and adrenergic receptors, which indicates that it should lack the autonomic and cardiovascular side effects, such as sedation, weight gain (both H1-histaminic), disturbed vision, constipation, tachycardia (all M1-muscarinic) and orthostatic hypotension (alpha-1 adrenergic) associated with the use of clozapine and other antipsychotics possessing activity at these receptors (Cunningham-Owens, 1996; Gerlach, 1991).
The putative significance of actions at ς1receptors.
S 18126 possesses marked affinity at ς1binding sites, a blockade of which has often been implicated in the actions of antipsychotic drugs (see Wyrick and Booth, 1995). In fact, more recent studies suggest that ς1 sites are not involved in the motor actions of haloperidol and other neuroleptics (Matsumoto R. et al., 1996; Meltzer et al., 1992;Yamamoto et al., 1995; Walker et al., 1993) whereas clozapine, which does not elicit an extrapyramidal syndrome, isdevoid of ς1 affinity (Cunningham-Owenet al., 1996; Gerlach, 1991). This observation indicates that an improved, “atypical” antipsychotic profile doesnot require marked activity at ς1 receptors. The inactivity of S 18126 in models predictive of antiproductive properties and its lack of an extrapyramidal syndrome, together with the potent activity of raclopride (which is devoid of affinity at ς1 binding sites), constitute additional evidence that ς1 sites are not of major importance in this regard. Raclopride also displays antipsychotic (and extrapyramidal) properties in the human (British Isles Raclopride Study Group, 1992). Furthermore, studies with very-high-affinity ς1 ligands have found only modest antipsychotic activity, even at high doses (Akunne et al., 1997; Poncelet et al., 1993). The lack of intracellular transduction mechanisms, or of in vivo functional responses unambiguously attributable to activation of ς1 sites, continues to call into question their significance. Indeed, ς1 sites have recently been cloned, and their primary structure displays marked similarities to a sterol C8-C7 isomerase isolated from fungi and involved in the synthesis of ergosterol, the fungal equivalent of cholesterol (Hanner et al., 1996;Kekuda et al., 1996; Moebius et al., 1997). Correspondingly, ς1 sites may be affiliated with postsqualene cholesterol synthesis in mammals (Moebius et al., 1997). The tissue distribution of ς1 sites is consistent with this proposed role, inasmuch as they are far more concentrated in peripheral tissues, such as liver and pancreas, than in the brain (Hanner et al., 1996; Kekuda et al., 1996). Notwithstanding these observations, there is increasing interest in the potential modulation of neuronal activity by steroids (Paul and Purdy, 1992; Yamamoto et al., 1995). Thus we cannot exclude the possibility that a pronounced action at ς1 sites might indirectly modify the activity of dopaminergic receptors and other mechanisms involved in the antideficit actions of antipsychotic agents. This remains to be evaluated. Currently, it would be imprudent to contend that an action of S 18126 at ς1 sites imparts therapeutic benefits in the treatment of schizophrenia, and the significance, if any, of its actions at ς1 sites remains to be demonstrated.
General discussion.
First, we exploited the residual affinities of S 18126 and L 745,870 at D2 (D3) receptors in order to predict the dose ranges over which they exert actions via the selective occupation of D4receptors in vivo. This strategy is important in the evaluation of drug actions at novel sites for which functional andex vivo/in vivo binding models are lacking. Second, the conclusions of the present study are based on the actions of two chemically distinct D4 antagonists, S 18126 and L 745,870, compared with those of the benzamide D2/D3antagonist raclopride. This allows for rigorous conclusions about the effects, or lack of effects, of selective blockade of D4receptors. However, certain phasically mediated effects at D4 receptors on mood or other parameters might, putatively, be revealed only by selective D4agonists, and such studies remain to be undertaken. In addition, any putative effects of “inverse” agonists at D4 receptors remain to be evaluated (Bristow et al., 1997; Gainetdinov et al., 1996). Third, the present data suggest that selective D4 receptor blockade does not elicit an extrapyramidal syndrome or disrupt motor behavior. Fourth, the present findings provide little support for the concept that blockade of D4receptors is associated with antiproductive properties and suggest that D4 antagonist properties are unlikely to underlie the atypical antipsychotic profile of clozapine. Nevertheless, an evaluation of a putative influence of D4 receptor antagonist properties on cognitive-attentional function in schizophrenia would be of interest, particularly because the procognitive actions of clozapine are limited by its antagonist properties at muscarinic receptors (Cunningham-Owen et al., 1996; Goldberg et al., 1993). Fifth, over a high, non-D4-selective dose range, S 18126 did, in fact, mimic certain of the actions of clozapine, inasmuch as it reinforced mesocortical—but not mesolimbic or nigrostriatal—release of DA and exerted mild antiproductive actions at doses that did not elicit catalepsy or PRL secretion. It is possible that these actions of S 18126 at high doses reflect an extremely high degree of D4vs. modest D2 receptor occupation (Seeman, 1992;Seeman et al., 1997). In any case, it might be of interest to explore the potential clinical utility of S 18126—and L 745,870—over such high dose ranges. Sixth, S 18126 expressed its actions on p.o. administration at doses close to those active by the s.c. route, a result that suggests significant bioavailability in rodents. The pharmacokinetics of lower, orally administered, D4 receptor-selective doses of S 18126 in the human remain to be established.
Conclusions.
In conclusion, S 18126 is a novel, orally active competitive, selective and potent antagonist at hD4receptors, and ς1 receptors represent its only other significant activity. Employing S 18126, in parallel with the selective D4 antagonist L 748,870 (which has far lower ς1 affinity), the D2/D3antagonist raclopride and several other benzamides, the present data, which suggest that blockade of D4 receptors is unlikely to result in antipsychotic and extrapyramidal actions. However, a possible influence of selective D4 receptor blockade on the cognitive-attentional symptoms of schizophrenia justifies further evaluation, and clinical studies with selective D4antagonists are required before definitive conclusions can be reached. The availability of S 18126 and other D4 antagonists should improve our understanding of the potential physiological and therapeutic significance of D4 receptors.
Acknowledgments
We thank C. Chaput, L. Cistarelli, L. Defaye, B. Denorme, V. Dubreuil, S. Girardon, H. Gressier, C. Melon, S. Monneyron, V. Pasteau, N. Richard and S. Veiga for technical support and C. Le Roy for secretarial assistance.
Footnotes
-
Send reprint requests to: Dr. Mark J. Millan, Institut de Recherches Servier, Psychopharmacology Department, 125, Chemin de Ronde, 78290—Croissy-sur-Seine (Paris, France).
- Abbreviations:
- AD
- active dose
- CHO
- Chinese hamster ovary
- CP 293
- 019, (7R, 9a5)-7-(4-fluorophenoxy)methyl 2-(5-fluoropyrimidin-2-yl) 2,3,4,6,7,8,9,9a-ortahydro-1H-pyrido[1,2-a]pyrazine
- CT
- core temperature
- DA
- dopamine
- DOI
- (1-[2,5-dimethoxy-4-iodophenyl]-2-aminopropane)
- DOPAC
- dihydroxyphenyl acetic acid
- ED
- effective dose
- FCX
- frontal cortex
- 5-HT
- serotonin
- IC
- inhibitory concentration
- ID
- inhibitory dose
- L 745
- 870, 3-(4-[4-chlorophenyl]piperazin-1-yl)methyl-1H-pyrrolo[2,3b]pyridine
- NAD
- noradrenaline
- 8-OH-DPAT
- (±)-8-dihydroxy-2-(di-n-propylamino) tetralin
- PCP
- phencyclidine
- (+)-PD 128
- 907, (+)-(4aR,10bR)-3,4,4a,10b-tetrahydro-4-propyl-2H,5H-[1]benzopyrano-[4,3-b]-1,4-oxazin-9-ol)
- PRL
- prolactin
- RO 61-6270
- 2-aminobenzoic acid 1-benzylpiperidin-4-yl ester
- S 18126
- {2-[4-(2,3-dihydro benzo[1,4]dioxin-6-yl)piperazin-1-yl methyl]indan-2-yl}
- U 101
- 387, 5(−)-4-{4-[2-(isochroman-1-yl)ethyl]piperazin-1-yl} benzenesulfonamide
- Received October 9, 1997.
- Accepted May 12, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}