


Abstract
Naloxone benzoylhydrazone (NalBzoH) is a potent muantagonist in vivo. In a cell line stably transfected with MOR-1 (CHO/MOR-1), NalBzoH also was an antagonist when examined in adenylyl cyclase studies. In binding studies, it displayed high affinity for the mu receptor, confirming its earlier characterization in brain membranes. In competition studies under equilibrium conditions, NalBzoH and diprenorphine both retained their potency in the presence of the stable GTP analog 5′-guanylylimidophosphate, consistent with their muantagonist properties, whereas the agonist DAMGO showed more than a 3-fold loss of affinity. The dissociation of3H-diprenorphine was monophasic. However, kinetic studies revealed biphasic dissociations for both 3H-NalBzoH and3H-DAMGO. The slow component of 3H-NalBzoH dissociation, corresponding to the higher affinity state, was dependent on coupling to G-proteins. It is selectively abolished by guanine nucleotides, leaving only the rapid dissociation phase. Furthermore, the slow dissociation component is eliminated by treatment of the cells with pertussis toxin, but not cholera toxin. In conclusion, NalBzoH is an unusual opioid. Functionally it is an antagonist, a classification consistent with its equilibrium binding in the presence of guanine nucleotides. Yet, kinetic studies reveal that it labels a G-protein coupled state of the receptor with high affinity.
Soon after the initial demonstration of opioid binding in brain tissue (Pert and Snyder, 1973; Terenius, 1973; Simon et al., 1973), a number of studies reported binding conditions that distinguished between agonist and antagonist binding. The first involved sodium, which enhances antagonist binding while lowering agonist binding (Pertet al., 1973; Simon and Groth, 1975). In contrast, divalent cations such as magnesium enhanced agonist binding and partially reversed the effects of sodium (Pasternak et al., 1975a). Treating membranes with protein modifying agents (Wilson et al., 1975; Pasternak et al., 1975b; Simon and Groth, 1975) or enzymes (Pasternak and Snyder, 1974, 1975) depressed agonist binding far more effectively than antagonist binding, an effect that was enhanced in the presence of sodium. Even changing the temperature of the binding assay distinguished between the two (Creese et al., 1975). These studies and others exploring other G-protein coupled receptors have led to general concepts regarding the binding of agonists and antagonists to the receptor. Overall, it is believed that agonists have highest affinity for receptors coupled to G-proteins while antagonists label coupled and uncoupled receptors with similar affinities (Dohlman et al., 1991). However, inverse agonists bind with highest affinity to uncoupled receptors (Samama et al., 1994).
NalBzoH is an unusual mixed opioid agonist-antagonist (Cheng et al., 1992; Gistrak et al., 1990; Price et al., 1989; Clark et al., 1989; Paul et al., 1990). Blocking the actions of morphine and other mu analgesics for more than a day after a single administration in vivo, higher NalBzoH doses produce analgesia throughkappa3 receptors (Gistrak et al., 1990; Paul et al., 1990).3H-NalBzoH binding also is unusual (Standiferet al., 1991; Brooks et al., 1994; Cheng et al., 1992,1995; Price et al., 1989; Clarket al., 1989). In equilibrium binding studies3H-NalBzoH displays similar affinities for bothmu and kappa3 sites. Yet,3H-NalBzoH dissociates from mu receptors in brain far more slowly than the kappa3 sites. This rate of 3H-NalBzoH dissociation from mu receptors also is far slower than 3H-naloxone despite their similar affinities. Furthermore, the mucomponent of 3H-NalBzoH binding is sensitive to guanine nucleotides, a result that was totally unexpected in view of its antagonist nature. Detailed examinations of the labeling of the mu receptor by NalBzoH have proven difficult due to the heterogeneity of opioid receptors in brain and the limited selectivity of NalBzoH. Recently, we generated a CHO cell line stably expressing MOR-1 (CHO/MOR-1) and used it to characterize the3H-morphine-6β-glucuronide binding (Brownet al., 1997). Using this cell line, we have characterized the binding of 3H-NalBzoH to a single population of mu receptors, overcoming the ambiguity of using brain membranes.
Materials and Methods
3H-DAMGO was obtained from New England Nuclear (Boston, MA), 3H-Diprenorphine was purchased from Amersham Life Sciences Inc.(Arlington Heights, IL).3H-NalBzoH was synthesized as previously described (Luke et al., 1988). All opioids and opioid peptides were the generous gift of the Research Technology Branch of the National Institute on Drug Abuse (Rockville, MD) and other chemicals were purchased from Fisher Scientific (Pittsburgh, PA).
Tissue culture.
CHO.K1 cells (ATTC, Wilmington, DE) were maintained in tissue culture flasks in F-12 media supplemented with 10% heat inactivated fetal bovine serum (Atlanta Biologicals, Atlanta, GA). Cells were grown in a 6% CO2-94% air humidified atmosphere at 37°C. Plates of cells were used at 75 to 95% confluence. Cells were lifted from the substrate for assay or subculturing after a 5 min incubation at 37°C in 5 ml of phosphate- buffered saline containing trypsin.
Cells were transfected with either cDNA encoding the clonedmu opioid receptor cloned into the Hind III site of pRcCMV (a generous gift from Dr. L. Yu) or the vector without an insert as previously described (Brown et al., 1997). Plasmid DNA (20 μg) was precipitated onto CHO.K1 cells 50 to 60% confluent using DEAE-Dextran (1 mg/ml) and chloroquine (0.1 mM) in normal culture media. After a 3.5-hr incubation at 37°C the transfection media was removed, the cells were washed thrice with phosphate-buffered saline and normal culture media was added back to the cells. After 72 hr of incubation in normal culture media, the cells were trypsinized and replanted in selection media (F-12/10% fetal bovine serum/0.4 mg/ml G418; GIBCO, Gaithersberg, MD). Individual colonies were cloned and screened for their ability to bind the nonselective opioid antagonist3H-diprenorphine (0.5 nM). After selection, the concentration of G418 was reduced to .2 mg/ml in the culture medium.
Binding assays.
Membranes from transfected CHO cells were prepared by homogenizing cells in 20 volumes of treated Tris buffer, and were prepared and frozen as described above. All binding was performed in potassium phosphate buffer (50 mM; pH 7.2) at 25°C for 150 min (3H-DAMGO) or 60 min (3H-diprenorphine and3H-NalBzoH) and filtered over glass-fiber filters (Schleicher & Schuell, Keene, NH) with a Brandel cell harvester (Cambridge, MA) as previously reported (Price et al., 1989;Clark et al., 1989). Specific binding was defined as the difference between binding in the absence and presence of levallorphan (1 μM). For dissociation studies, membranes were prebound with radioligand (1 nM) as indicated above alone or with the indicated nucleotide derivative. The dissociation was then initiated by the addition of levallorphan (1 μM) and binding determined by filtration at the indicated time. For the toxin treatments, cells were grown for 24 hr with PTX (100 ng/ml) or CTX (100 ng/ml). Membranes were then harvested and assayed as described above.
Measurement of cAMP accumulation.
Inhibition of forskolin stimulated cAMP accumulation was determined in intact CHO/MOR-1 cells as previously described (Brown et al., 1997; Standiferet al., 1991; Cheng et al., 1995). After aspirating the media, cells were washed twice with phosphate- buffered saline and incubated for 10 min at 37°C with the phosphodiesterase inhibitor IBMX (0.5 mM) in Hanks’ balanced salt solution (137 mM NaCl, 5 mM KCl, 0.6 mM Na2HPO4, 0.4 mM KH2PO4, 4 mM NaHCO3, 6 mM d-glucose, .5 mM MgCl2, 0.4 mM MgSO4 and 1 mM CaCl2). Cells were then incubated an additional 10 min at 37°C after adding forskolin (10 μM) and the various opioids. The assay was stopped by aspirating the incubation medium and adding boiling Tris buffer (25 mM; pH 7.4 at 25°C). The samples were centrifuged for 10 min at 1000 × g and the supernatant was saved for cAMP analysis.
The cAMP content of the supernatant was determined by displacing3H-cAMP binding to bovine adrenal cortex extract as previously described (Brown et al., 1997; Standiferet al., 1991; Cheng et al., 1995). Samples containing Tris buffer (1 mM; pH 7.0 at 25°C), theophylline (10 mM),3H-cAMP (0.8 pmol), an aliquot of the supernatant, and bovine adrenal cortex extract (50 μg) in a total volume of 0.2 ml were incubated for 60 min at 4°C. Hydroxyapatite suspension was added to each tube [75 μl of a 50% (w/v) suspension] and incubated for an additional 6 min. At that time 4 ml of ice-cold Tris buffer (pH 7.0 at 25°C) was added to each tube, and the suspension was filtered onto a no. 34 glass-fiber filters (Schleicher & Schuell). Filters were washed with an additional 6 ml of buffer and placed in 5 ml scintillation fluor for radioactivity determination. Specific binding was determined by subtracting the binding of 3H-cAMP to bovine adrenal cortex extract in the presence of 1 μM cAMP with no supernatant added from the binding in the absence of 1 μM cAMP. The amount of cAMP present was calculated from a standard curve determined with unlabeled cAMP.
Protein determination.
Protein concentrations were determined by the method of Lowry using bovine serum albumin as the standard (Lowry et al., 1951).
Data analysis.
Statistical analysis of the experimental data was performed with Student’s t test, or ANOVA followed by Sheffé’s post hoc test (GB-STAT) and the significance established at the P < .05 level. Binding data were analyzed by regression analysis (Prism, GraphPad Software). All assays were performed in triplicate. Results are presented as means ± S.E.M. of triplicate experiments, unless otherwise indicated.
Results
3H-NalBzoH binding in CHO/MOR-1 cell membranes.
CHO cells themselves do not express muopioid receptors, as assessed in binding or functional assays (data not shown). After the stable transfection of CHO cells with MOR-1, the CHO/MOR-1 cells possess high levels of mu opioid binding and respond to mu drugs in adenylyl cyclase studies (Brownet al., 1997). As anticipated,3H-NalBzoH binds specifically to these cells. Saturation experiments are consistent with a single population of high affinity 3H-NalBzoH binding sites (Kd 0.5 ± 0.1 nM, Bmax 450 ± 107 fmol/mg protein; fig.1).
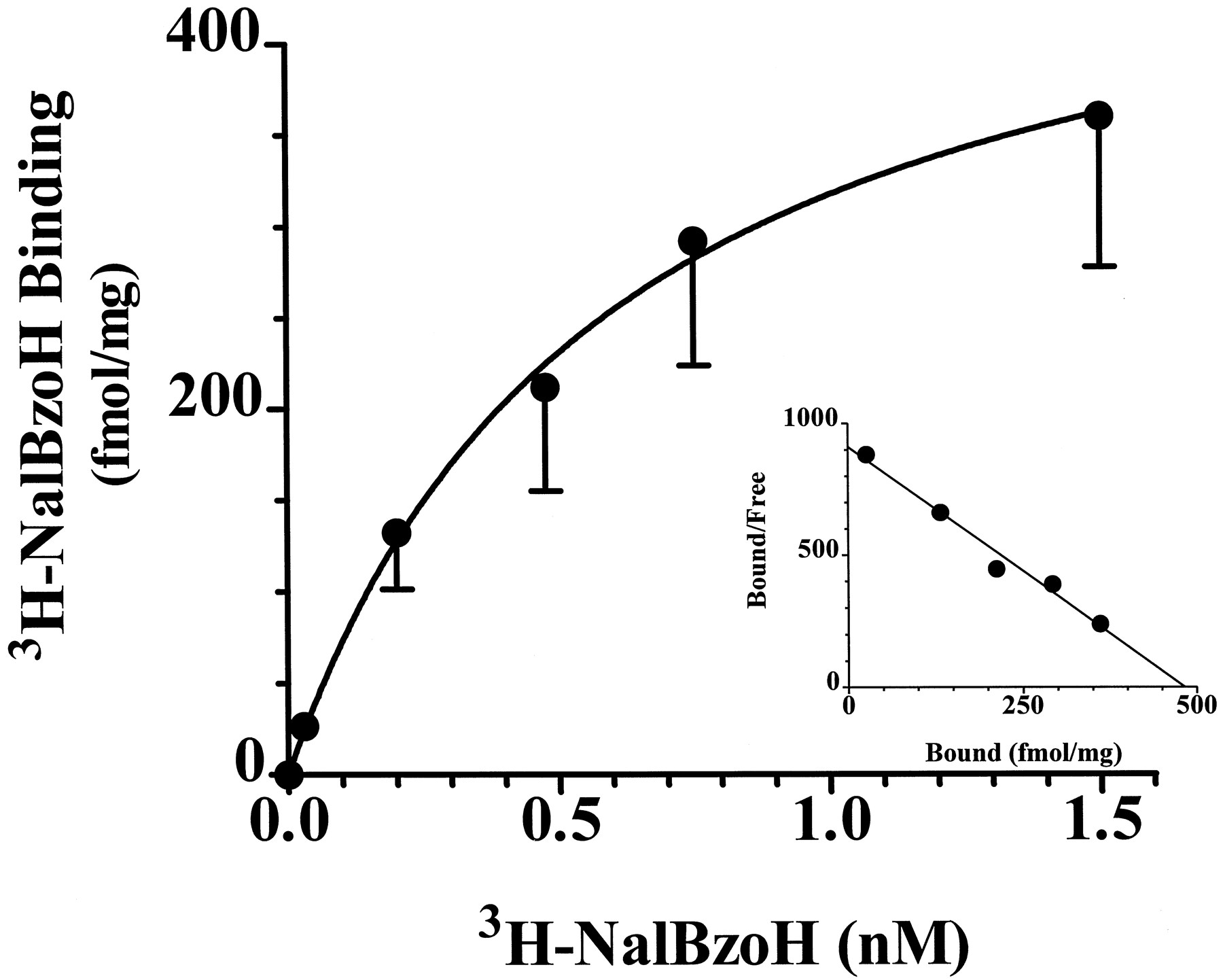
Saturation of 3H-NalBzoH binding to CHO/MOR-1 cell membranes. Increasing concentrations of3H-NalBzoH (0.05–1.5 nM) were added to CHO/MOR-1 cell membranes (0.2 mg) in a 1 ml volume in the presence and absence of levallorphan (1 μM) as described in “Materials and Methods.” Inset, Scatchard analysis of saturation binding data. The data were best fit by nonlinear regression analysis by a single site,Kd 0.5 ± 0.1 nM and Bmax 450 ± 107. This is representative experiment that as been replicated three times.
Agonist/antagonist characteristics of NalBzoH in binding and adenylyl cyclase systems.
We next performed competition studies against 3H-diprenorphine in the presence and absence of the nonhydrolyzable GTP analogue Gpp(NH)p in the CHO/MOR-1 cell membranes (table 1). The mu agonist DAMGO potently lowered 3H-diprenorphine binding in the CHO/MOR-1 cell membranes with a Hill coefficient of 0.48, presumably reflecting a mixture of G-protein coupled agonist and antagonist conformations of the receptor. Inclusion of Gpp(NH)p in the competition studies, which would be expected to convert all the receptors into an antagonist conformation, decreased the DAMGO affinity 3.5-fold and increased the Hill coefficient to 0.92. Conversely, the antagonist diprenorphine competed binding slightly more potently in the presence of Gpp(NH)p with Hill coefficients close to unity in both series of assays.
Effect of Gpp(NH)p on the competition of 3H-diprenorphine binding
The competition studies with NalBzoH corresponded closely to those of diprenorphine. The affinity of NalBzoH was slightly increased in the presence of Gpp(NH)p with a Hill coefficient close to unity in both series of assays. Additional studies in which NalBzoH competitions were performed in the presence of NaCl (100 mM) revealed aKi value of 2.6 ± 0.58 nM, which was not very different from the its Ki value in the presence of both GppNHp and NaCl (Ki 4.3 ± 0.68 nM).
We next assessed NalBzoH functionally in cyclase studies with the transfected mu receptor cell line. When tested alone, neither NalBzoH, diprenorphine nor naloxone were active in the adenylyl cyclase studies (data not shown). They neither stimulated cAMP accumulation nor did they inhibit forskolin-stimulated cAMP accumulation. However, all three drugs antagonized, in a dose-dependent manner, the inhibition of forskolin-stimulated adenylyl cyclase activity produced by DAMGO (1 μM) (fig. 2). Diprenorphine (IC50 2.28 ± 1.1 nM) was the most potent, followed by naloxone (IC50 16.3 ± 5) and NalBzoH (IC50 36 ± 14 nM). Thus, functionally NalBzoH is an antagonist with no indication of any partial agonist activity.

Reversal of DAMGO-induced inhibition of adenylyl cyclase. Increasing concentrations of each ligand were examined in CHO/MOR-1 cells treated with DAMGO (1 μM). cAMP levels were assayed as described in “Materials and Methods.” DAMGO alone inhibited forskolin-stimulated cAMP accumulation by 81.4 ± 5.3%. Results are presented as the relative percentage of inhibition of cAMP produced by DAMGO alone. IC50 values were calculated by nonlinear regression analysis: diprenorphine (2.28 ± 1.1 nM), naloxone (16.3 ± 5.0 nM) and NalBzoH (36 ± 14 nM). Results are presented as the mean ± S.E.M. of three independent determinations.
Kinetic evaluation of 3H-NalBzoH binding.
The dissociation of 3H-DAMGO in these cells was biphasic, presumably corresponding to the labeling of both G-protein-coupled agonist and uncoupled antagonist states of the receptor (fig. 3). This is consistent with the shallow competition curve and low Hill coefficient seen in the equilibrium competition studies against3H-diprenorphine. The rapid phase of dissociation had a half-life of dissociation of less than 3 min and comprised approximately 65% of the binding (table2). Its dissociation from the remaining third of the binding was slow, with a half-life of more than 30 min. In contrast, the antagonist 3H-diprenorphine dissociated from CHO/MOR-1 membranes in a single phase with a half-life of dissociation of approximately 50 min, implying that it bound both coupled and uncoupled receptors with similar affinities.

Dissociation of 3H-opioids from CHO/MOR-1 cell membranes. 3H-NalBzoH (1.0 nM),3H-DAMGO (2.0 nM) or 3H-diprenorphine (1.0 nM) were incubated with CHO/MOR-1 cell membranes to equilibrium as described in “Materials and Methods.” Dissociation was initiated by adding levallorphan (1 μM) and specific binding was determined at the indicated time afterward. 3H-NalBzoH and3H-DAMGO dissociation curves were best fit with a two-site model, and 3H-diprenorphine was best fit with a one site model using the program Prism. Results are presented as the mean ± S.E.M. of three independent determinations.
Dissociation rates of 3H-NalBzoH
3H-NalBzoH had a biphasic dissociation, suggesting that it differentially bound to the two receptor conformations. Approximately 60% of total binding dissociated quite rapidly, with a half-life of less than 5 min, while the remainder dissociated more slowly with a half-life of about 45 min. The proportions of rapidly and slowly dissociable3H-NalBzoH binding sites were similar to the two populations bound by 3H-DAMGO.
Guanine nucleotide sensitivity of3H-NalBzoH dissociation from the mu receptor.
To explore whether the two states seen in the3H-NalBzoH dissociation studies represented G-protein coupled and uncoupled states of the receptor, we examined the effects of guanine nucleotides on the dissociation of3H-NalBzoH binding from CHO/MOR-1 cell membranes. First, we determined that Gpp(NH)p did not influence the dissociation of the antagonist 3H-diprenorphine in these studies (fig. 4). Its dissociation remained monophasic with a half-life of dissociation of 30.7 min, a value similar to control studies (table 2). We then compared the dissociation of 3H-NalBzoH in the presence of Gpp(NH)p to that seen in its absence (fig. 5). Gpp(NH)p converted the dissociation of 3H-NalBzoH to a single phase with a half-life of 4.4 ± 1.0 min (table 2), corresponding to the rapid dissociation state seen in its absence.

Dissociation of 3H-diprenorphine in Gpp(NH)p-treated CHO/MOR-1 membranes. 3H-Diprenorphine (1.0 nM) was incubated with CHO/MOR-1 cell membranes for 1 hr in the presence of Gpp(NH)p (0.1 mM) as described in “Materials and Methods.” Dissociation was initiated by adding levallorphan (1 μM) and specific binding determined at the indicated time afterward. The dissociation curve was best fit to a one-site model. Results are presented as the mean ± S.E.M. of three independent determinations.

Dissociation of 3H-NalBzoH from CHO/MOR-1 membranes. 3H-NalBzoH (1.0 nM) was incubated with CHO/MOR-1 cell membranes for 1 hr in the presence or absence of either Gpp(NH)p (0.1 mM) or App(NH)p (0.1 mM) as described in “Materials and Methods.” Dissociation was initiated by adding levallorphan (1 μM) and specific binding determined at the indicated time afterwards. The dissociation curve in App(NH)p-treated membranes were best fit with a two-site model although the curve with Gpp(NH)p was monophasic. Results are presented as the mean ± S.E.M. of three independent determinations.
The effects of Gpp(NH)p were limited to guanine nucleotides. Full dissociation studies with the nonhydrolyzable ATP analogue, App(NH)p, did not influence 3H-NalBzoH dissociation (fig.5; table 2). We extended these studies by determining the effects of a variety of nucleotides on the binding remaining after 30 min of dissociation (fig. 6). In control studies, 30% of the 3H-NalBzoH binding at equilibrium remained after 30 min of dissociation. App(NH)p, ATP, UTP, CTP and TTP were inactive in this assay. In contrast, both guanine nucleotides Gpp(NH)p and GTP reduced binding in these studies from the control values of 30% of equilibrium values to approximately 5%. These findings imply that the slow phase of dissociation is dependent on a guanine nucleotide binding protein.

Effect of nucleotides on 3H-NalBzoH dissociation from CHO/MOR-1 cell membranes. Membranes were incubated with 3H-NalBzoH alone or with the specified nucleotide (0.1 mM) for 1 hr after which dissociation was initiated by adding levallorphan (1 μM) and binding determined 30 min thereafter. Binding is expressed as the percentage of specific binding remaining after the 30 min dissociation compared to the binding in control membranes which did not receive levallorphan. Results are the mean ± S.E.M. of three independent determinations. Significance was determined by ANOVA followed by Scheffe’s post hoc test.
Toxin sensitivity of 3H-NalBzoH dissociation.
The guanine nucleotides do not distinguish among the wide number of GTP-binding proteins. Therefore, we next examined the effects of CTX and PTX on the dissociation of3H-NalBzoH (fig. 7). These toxins covalently modify heterotrimeric G-proteins and alter their function. CTX constitutively activates Gsα while PTX inactivates the signaling ability of Gi and Go. CHO/MOR-1 cells were treated for 24 hr with 100 ng/ml of the respective toxin, and membranes were prepared. CTX treatment did not affect the dissociation of 3H-NalBzoH (fig. 7). The rapid phase of dissociation, representing slightly more than half of the total binding at equilibrium had a half-life of dissociation of less than 10 min while the slower phase was over 80 min (table 2). However, PTX treatment abolished the slow dissociation of3H-NalBzoH seen in the control membranes, much like Gpp(NH)p. This indicates that the slow dissociation component of3H-NalBzoH binding is dependent on either Gi- or Go-type G-proteins.

Effect of cholera and pertussis toxin treatments on3H-NalBzoH dissociation. CHO/MOR-1 cells were incubated with either PTX (100 ng/ml) or CTX (100 ng/ml) for 24 hr and membranes were then harvested for binding as described in “Materials and Methods.” Dissociation of 3H-NalBzoH was then determined. Equilibrium binding with 3H-NalBzoH (1 nM) was 72 ± 5, 66 ± 2 and 48 ± 6 fmol/mg protein in control, CTX and PTX cell membranes, respectively. Dissociation was initiated by adding levallorphan (1 μM) and specific binding determined at the indicated time thereafter. The 3H-NalBzoH dissociation from CTX-treated membranes was best fit with a two-site model although the dissociation from PTX treated membranes was best fit with a one site model. Results are the mean ± S.E.M. of three independent determinations.
Discussion
Earlier studies exploring 3H-NalBzoH binding revealed labeling of both mu andkappa3 receptors (Price et al., 1989; Clark et al., 1989; Luke et al., 1988).3H-NalBzoH labeling of mu receptors was dependent upon magnesium ions, a characteristic most commonly observed with agonists binding(Pasternak et al., 1975a). Furthermore, 3H-NalBzoH dissociated frommu receptors far more slowly than anticipated based on its affinity. This slow rate of dissociation was dramatically increased by guanine nucleotides, a sensitivity that also is typically associated with agonist binding (Childers and Snyder, 1978). Yet, all the pharmacological evidence suggested that NalBzoH was a mu antagonist (Gistrak et al., 1990; Paul et al., 1990). Detailed binding studies in brain homogenates were difficult to interpret due to the presence of a wide range of opioid receptors in the tissue other than mu receptors. The availability of a cloned mu receptor, encoded by the MOR-1 cDNA, has now opened the possibility of exploring the binding characteristics of3H-NalBzoH in a well-defined system.
3H-NalBzoH labels the mu receptors in the CHO/MOR-1 cells with high affinity, confirming the binding studies in brain. Functionally, it is an antagonist in the adenylyl cyclase system, lacking activity alone and reversing the effects of established agonists. However, the role of G-proteins in3H-NalBzoH binding is quite unusual. Under equilibrium conditions, the ability of agonists to compete the binding of radiolabeled antagonists is typically reduced by guanine nucleotides such as Gpp(NH)p while antagonists are unaffected. Thus, the reduction in the affinity of DAMGO with the addition of Gpp(NH)p in the transfected cells is not surprising. Similarly, we anticipated no decrease in affinity for either diprenorphine or NalBzoH based on their antagonist character in the functional assays and none is seen. Gpp(NH)p actually enhances the affinity of both compounds in these equilibrium competition studies. Even in the presence of both Gpp(NH)p and NaCl, NalBzoH retained its potency, consistent with an antagonist. However, kinetic approaches gave a very different picture.
The kinetic studies with 3H-DAMGO and3H-diprenorphine confirmed the traditional view of agonists and antagonists. The dissociation of3H-DAMGO is biphasic, reflecting the labeling of both G-protein-coupled and uncoupled receptors. The rapidly dissociating component, representing the lower affinity binding to uncoupled receptors, accounts for approximately 65% of total specific binding. The prominence of the uncoupled sites in this model probably reflects the overexpression of the receptor in the transfected cell line. 3H-Diprenorphine shows the anticipated monophasic pattern expected for antagonists, indicating similar affinities towards both coupled and uncoupled receptors.
3H-NalBzoH binding does not fit this pattern. As an antagonist, we expected a monophasic dissociation. However,3H-NalBzoH shows a biphasic pattern similar to that of 3H-DAMGO, implying that it differentially labels coupled and uncoupled receptors. Furthermore, the higher affinity component of 3H-NalBzoH binding is dependent on coupling of the receptor with a G-protein. The binding of neutral antagonists is not affected by G-proteins, labeling both G-protein coupled and uncoupled sites equally well (Dohlman et al., 1991). In contrast, inverse agonists label the uncoupled receptors more potently although agonists show higher affinity for the coupled receptors (Samama et al., 1994). In the3H-NalBzoH dissociation studies, the higher affinity binding component is lost after treatment with either Gpp(NH)p or pertussis toxin. This would imply that NalBzoH shows higher affinity for G-protein-coupled receptors, a conclusion that is inconsistent with these traditional hypotheses regarding antagonist binding.
In conclusion, NalBzoH binding does not correspond to a traditional agonist, antagonist or inverse agonist. Functionally, it lacks any demonstrable intrinsic activity and it shows antagonist binding characteristics in equilibrium studies. Yet, the kinetic studies reveal a very different picture. Clearly, the binding of3H-NalBzoH does not conform to the traditional receptor binding models currently used. The availability of a constitutively active mu receptor would greatly aid in the classification of NalBzoH.
Acknowledgment
The authors thank Dr. Jerome Posner for his assistance.
Footnotes
-
Send reprint requests to: Dr. Gavril W. Pasternak, Department of Neurology, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY 10021.
-
↵1 This work was supported, in part, by Grant DA06241 and Research Scientist Award K05 DA00220 from the National Institute on Drug Abuse to G.W.P. and core Grant CA08748 from the National Cancer Institute to MSKCC. G.P.B. was supported by Training Grant T32 DA07274 from the National Institute on Drug Abuse.
- Abbreviations:
- NalBzoH
- naloxone benzoylhydrazone
- PTX
- pertussis toxin
- CTX
- cholera toxin
- Gpp(NH)p
- 5′-guanylylimidophosphate
- ANOVA
- analysis of variance
- CHO
- Chinese hamster ovary
- 3H-DAMGO
- [d-Ala2, MePhe4, Gly(ol)5]enkephalin
- Received October 15, 1997.
- Accepted March 13, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}