


Abstract
Activity-dependent neurotrophic factor (ADNF) is a glia-derived protein that is neuroprotective at femtomolar concentrations. A 14-amino acid peptide of ADNF (ADNF-14) has been reported that protects cultured neurons from multiple neurotoxins. Structure-activity relationships of peptides related to ADNF-14 now have been determined. A 9-amino acid core peptide (ADNF-9) has been identified that has greater potency and a broader effective concentration range (10−16 to 10−13 M) than ADNF or ADNF-14 in preventing cell death associated with tetrodotoxin treatment of cerebral cortical cultures. Deletions or conservative amino acid substitutions to ADNF-9 resulted in reduced potency, narrower effective concentration range and/or decreased efficacy. Removal of the N-terminal serine or the COOH-terminal isoleucine-proline-alanine from ADNF-9 produced a significant reduction in survival-promoting activity. Comparative studies of ADNF-9 action in mixed (glia plus neurons) vs.glia-depleted neuronal cultures indicated that ADNF-9 can act directly on neurons, although the potency of the peptide was 10,000-fold greater in mixed cultures. Kinetic studies showed that exposure to ADNF-9 for only 2 hr was sufficient to produce a 4-day protection against the cell-killing action of tetrodotoxin. Treatment with bafilomycin A1 (an inhibitor of receptor-mediated endocytosis) for 2 hr prevented the ADNF- and ADNF-9-mediated neuroprotection. ADNF-9, like ADNF-14, was neuroprotective against N-methyl-d-aspartate and the β-amyloid peptide (amino acids 25–35), and had a much broader range of effective concentrations than ADNF-14. These studies identify ADNF-9 as an attractive lead compound for the development of therapeutic agents against neurodegenerative diseases.
VIP has neurotrophic action on neurons in the central nervous system and growth-promoting action in postimplantation embryos (Brenneman and Eiden, 1986; Brenneman et al., 1985a; Gressens et al., 1993). In addition to these developmentally important functions, VIP also has been shown to be neuroprotective (Brennemanet al., 1988; Said et al., 1996; Gozes et al., 1996). Previous studies have indicated that the neuroprotective actions of VIP are contingent on the presence of glial cells (Brenneman et al., 1987; Brenneman et al., 1990a) expressing high-affinity VIP binding sites (Gozes et al., 1991). VIP has been shown to be a secretagogue for several astroglia-derived substances that can increase the survival of developing CNS neurons; these substances include interleukin-1 (Brenneman et al., 1995; Brenneman et al., 1992), protease nexin-1 (Festoff et al., 1996) and ADNF (Brenneman and Gozes, 1996). The structure-activity relationships of neuroactive peptides derived from ADNF are the focus of this work.
ADNF is a neuroprotective, glia-derived neurotrophic protein (14,000 Da and pI 8.3 ± 0.25) isolated by sequential chromatographic methods (Brenneman and Gozes, 1996). The protein was named activity-dependent neurotrophic factor because it protects neurons from death associated with electrical blockade. The purification of ADNF was achieved by biochemical fractionation of conditioned medium from VIP-stimulated astrocyte cultures. The isolation assay measured neuronal survival in developing spinal cord cultures treated with TTX, an agent that blocks synaptic activity. TTX produces cell death in neurons that are dependent on ongoing electrical activity for their survival (Brennemanet al., 1983). VIP synthesis and release from cultured spinal cord neurons have been shown to be prevented by treatment with TTX (Brenneman et al., 1985a; Agoston et al., 1991). The hypothesized effect is that TTX blocks the release of VIP and therefore the release of the ADNF necessary for neuronal survival. Previous studies indicated that the cell death produced by TTX in dissociated spinal cord cultures was apoptotic (Gozes et al., 1997). ADNF was shown to prevent TTX-mediated increases in apoptosis as determined by an in situ terminal deoxynucleotidyl transferase assay. Antiserum to ADNF was found to increase the number of apoptotic neurons, which indicates the importance of endogenous ADNF for the survival of a subpopulation of neurons. The anti-ADNF effect on apoptosis was prevented by cotreatment with purified ADNF. Although the identity of the ADNF-dependent neurons has not been characterized fully, previous studies indicated that cholinergic neurons are among those affected (Gozes et al., 1997).
During the course of studies to characterize the structure of ADNF, an active peptide fragment was discovered: ADNF-14 (Brenneman and Gozes, 1996). This peptide had strong homology, but not identity, to the intracellular stress protein hsp60 (Gozes and Brenneman, 1996). Although survival-promoting activity was not detected for recombinant hsp60, a peptide derived from hsp60 (VLGGGCALLRCIPA) was shown to have neuroprotective action, though at reduced efficacy and 10,000-fold less potency than ADNF-14 (VLGGGSALLRSIPA). Furthermore, neutralizing antisera raised against ADNF recognized the ADNF-14 peptide (Gozeset al., 1997). In this report, we have identified ADNF-derived, neuroprotective peptides and have examined in particular the stability, mechanism of action and potential utility of ADNF-9, a short peptide with activity that surpasses that of the parent protein and ADNF-14 with regard to potency and, more important, has a broader range of effective concentrations.
Materials and Methods
ADNF purification.
ADNF purification from the conditioned medium of VIP-treated astrocytes was performed as before, utilizing DEAE sephacel chromatography followed by size exclusion and hydrophobic interaction (Brenneman and Gozes, 1996) on fast-performance liquid chromatography.
Peptide syntheses.
Peptides synthesis was conducted as previously described (Gozes et al., 1995; Gozes et al., 1996). Products were purified on Sephadex G-25 and reverse-phase HPLC on a semipreparative C8 column (Lichrosorb RP-8; Merck Darmstat, FRG). Elution of peptides was produced by linear gradients established between 0.1% trifluoroacetic acid in water and 0.1% trifluoroacetic acid in 75% (v/v) acetonitrile in water. Peptides showed the desired molar ratios of the constituent amino acids. Molecular weights were ascertained by mass spectroscopy (VG Tofspec, Laser Desorption mass spectrometer, Fison Instruments, Loughborough, England). Sequences were determined with a gas phase Applied Biosystems model 470A protein microsequencer coupled to an Applied Biosystems model 120A PTH analyzer. Each peptide was dissolved in PBS as a 1 mM solution, diluted appropriately with PBS and tested. With the exception of the freeze-thaw stability studies, all peptides were tested without freezing the stock solutions.
Cell cultures.
Cerebral cortical cultures derived from newborn rats (Hill et al., 1993) were used for the neuron survival assays. Two preparations of cerebral cortical cultures were used: 1) mixed neuronal and glial cultures and 2) glia-depleted, neuronal cultures on poly-l-lysine. For mixed cultures, dissociated cerebral cortical tissue was seeded on a confluent layer of astroglial cultures derived from rat cerebral cortex (McCarthy and de Vellis, 1980). We placed 250,000 cells into a 35-mm dish in a volume of 1.5 ml. The mixed cultures were maintained in medium (Brennemanet al., 1987) consisting of 5% horse serum in minimal essential medium supplemented with defined medium components (Romijnet al., 1984) and 5′-fluoro-2-deoxyurdine (15 μg/ml) plus uridine (3 μg/ml). For the glia-depleted, neuronal cultures, 35-mm culture dishes were coated with 10 μg/ml of poly-l-lysine hydrobromide (30–70 kDa, Sigma Chemical Co., St. Louis, MO) dissolved in 0.15 M sodium borate (pH 8.4). The poly-l-lysine solution was removed after 1 hr, and the dishes were rinsed three times with PBS. After removal of the last saline rinse, dissociated cerebral cortical cells (300,000 cells) were added to the culture dish. After 30 min, the medium was removed, along with unattached cells and debris, and replaced with the nutrient medium, including the mitotic inhibitors. The cultures grown on poly-l-lysine were not totally free of astrocytes as determined by immunocytochemical analysis with antiserum to glial fibrillary acidic protein; rather, astrocytes constituted less than 10% of the total cells in the cultures. This is in contrast to the mixed cultures, which were more than 95% astrocytes. Four days after adding the cerebral cortical suspension either to astrocyte feeder cultures or to the poly-l-lysine-coated dishes, the culture preparations were given a complete change of medium (before peptide treatment). Cultures were given their respective treatments once and were assayed for neuronal survival after a 4-day incubation period. Neuronal cell counts were conducted after fixation with glutaraldehyde (Brenneman et al., 1987). Neuronal identity was established with sister cultures immunocytochemically stained with antiserum against neuron specific enolase (Schmechel et al., 1978). Neurons were counted in 40 fields in each culture dish without knowledge of the treatment group. Previous studies demonstrated that 1 μM TTX blocked synaptic activity in CNS cultures (Ransom and Holz, 1977) and produced decrements as measured with many neuronal parameters, including choline acetyltransferase, tetanus toxin fixation, saxitoxin binding (Brenneman, 1986) and neuronal survival (Brenneman et al., 1983). All statistical comparisons were made with an analysis of variance followed by the Student-Newman-Kuels multiple comparison of means test.
Results
Structure-activity studies.
Peptide sequence analysis of ADNF revealed an area of sequence homology to hsp60 (Brenneman and Gozes, 1996), and peptides spanning this area of homology were synthesized. The structure-activity relationships of these ADNF-related peptides were assessed in mixed neuronal-glial cell cultures cotreated with TTX.
The first objective was to find shorter peptides that retained the full biological activity of ADNF or ADNF-14. As summarized in figure 1A, a 9-amino acid peptide (SALLRSIPA: ADNF-9), the C-terminal portion of the parent peptide ADNF-14, captured the full survival-promoting efficacy of the parent molecule, exhibiting an even greater potency than that of purified ADNF. In addition, ADNF-9 was effective over a much broader range of concentrations than was ADNF or ADNF-14. The biological activity of ADNF-9 was very sensitive to amino acid deletions, additions or substitutions. Removal of the N-terminal serine (ALLRSIPA) produced a complete loss of neurotrophic activity (fig. 1A). Addition of a single glycine to the N-terminus of ADNF-9 produced a 100-fold loss in potency with no apparent loss in efficacy (fig. 1B). Cultures treated with SALLRS showed a 6-order-of-magnitude reduction in potency and a 25% decrease in efficacy compared with ADNF-9. Deletions of the COOH-terminus of ADNF-14 were also performed. Removal of the last two amino acids (PA) resulted in 1000-fold reduction in potency and a 50% loss in efficacy (fig. 1C). Deletion of five amino acids (RSIPA) resulted in complete loss of neuroprotective activity (fig. 1C). Removal of the last four amino acids (SIPA) produced a 50% loss in efficacy with no apparent change in potency (fig. 1D). Similarly, removal of six amino acids (LRSIPA) resulted in no detectable activity (fig. 1D). Treatment with RSIPA alone had little effect on neuronal survival (fig. 1D). These data indicated the critical importance of the N-terminal serine and the carboxy-terminal isoleucine-proline-alanine of ADNF-9. Peptides lacking these residues did not possess full biological activity. Additional experiments supported this conclusion. A conservative substitution of threonines for the serines in the 9-amino acid core peptide resulted in a greater than 50% loss of efficacy and a 10-fold decrease in potency (fig. 1E). On the basis of the previous observation (Brenneman and Gozes, 1996) that a homologous peptide sequence of hsp60 also had survival-promoting activity, a hybrid molecule consisting of ADNF-14 and an hsp60 sequence was synthesized. Addition of an N-terminal hsp60 sequence to ADNF-14 resulted in VEEGIVLGGGSALLRSIPA, which exhibited the same efficacy as ADNF-9; however, this longer peptide had a narrower range of active concentrations and reduced potency compared with ADNF-9. The dose-response of the elongated peptide was similar to that of intact ADNF (fig. 1A). Taken together, the data indicated that ADNF-9 had exceptional neuroprotective properties, so it was chosen for further studies.
Kinetics and mechanism of action for ADNF-9.
The time course of ADNF-9-mediated neuroprotection from TTX is shown in figure 2. Cerebral cortical cultures were treated with ADNF-9 for different time periods, and then the culture medium was removed and replenished with fresh growth medium plus TTX to ensure electrical blockade. The data shown are the number of surviving neurons after the 4-day test period (•). The dotted line represents neuron counts from cultures treated with 10−15 M ADNF-9 and TTX during the entire 4-day test period. Results indicated that the full biological response to ADNF-9 was produced within 2 hr of incubation. In the same experiment, media from ADNF-9-treated cultures were tested on sister cultures (○). Results indicated that after 15 min of exposure to cells, ADNF-9 activity was no longer detectable in the culture media. Together, these data implied a rapid utilization and disappearance of peptide from the incubation medium, which resulted in a long-term protection from neuronal cell death produced by TTX.
During the isolation of ADNF, remarkable stability after freeze-thaw cycles was observed. As shown in figure 3, purified ADNF exhibited virtually no loss in survival-promoting activity after 5 freeze-thaw cycles with dry ice. In contrast, ADNF-9 dissolved in PBS lost more than 90% of its activity after a single freezing. However, there was no visible loss of peptide solubility after freezing. In addition, an electrophoretic analysis (SDS gel under reducing conditions) revealed no apparent loss of solubilized peptide after a freeze-thaw cycle, compared with freshly solubilized, nonfrozen control peptide. Unless specifically stated otherwise, all experiments were conducted with ADNF-9 that was dissolved in PBS on the day of the experiment.
To begin testing whether ADNF-9 might act through a mechanism of receptor-mediated endocytosis, cultures were treated with BFA, a potent and specific inhibitor of the vacuolar-type H+ ATPase, the enzyme responsible for forming the pH gradient in acidic compartments (Bowman et al., 1988). Cultures were pretreated with BFA for 30 min, and then ADNF-9 was added for 2 hr. At the end of this incubation, the treated medium was removed and replaced with fresh medium. Neurons were counted after a 4-day incubation period. As shown in figure 4, when neurotoxicity was produced by TTX, acute BFA treatment completely prevented the neuroprotective action elicited by the 2-hr exposure to ADNF-9. Acute BFA had no effect on neuronal survival, and it produced no further reduction in cell counts when coadministered with TTX. Separate experiments with purified ADNF (1 fM) showed that cell counts increased from 193 ± 3 in TTX-treated cultures to 292 ± 13 in ADNF/TTX-treated cultures after a 2-hr incubation (P < .001). Control cultures had cell counts of 309 ± 8. As described for the experiments in figure 4 with ADNF-9, the cultures were rinsed in fresh medium after a 2-hr incubation and then treated again for 4 days with TTX, after which the cultures were fixed and assessed for neuronal cell counts. As in the case of ADNF-9, the increase in neuronal survival after ADNF treatment was completely prevented by BFA cotreatment (200 ± 6). These data indicate that the neurotrophic action of ADNF-9 and ADNF is mediated through a BFA-sensitive process.
A second series of experiments examined the effect of chronic BFA treatment for 4 days. Treatment with ADNF-9 and/or TTX for 4 days produced effects (data not shown) that were not significantly different from those observed in the 2-hr experiment (see fig. 4). However, a 4-day treatment with BFA produced a reduction in neuronal cell counts (234 ± 7) that was not significantly different from that observed after TTX treatment (228 ± 4). Furthermore, the cell counts after chronic cotreatment with TTX and BFA (235 ± 15) were not different from that observed with either agent tested separately. Control cultures had cell counts of 358 ± 12.
The cellular basis of ADNF-9 action was addressed by comparing the responses of glia-depleted, neuronal cultures to mixed neuronal-glial cultures derived from the cerebral cortex. As shown in figure 5, ADNF-9 prevented neuronal cell death produced by TTX in both culture preparations, but with greatly differing potencies. As evident in the previous experiments with test cultures comprising neurons and glia, ADNF-9 had a subfemtomolar EC50, with an attenuation of the biological response at picomolar concentrations or greater. In contrast, ADNF-9 was 10,000 times less potent in protecting glia-depleted, neuronal cultures than neurons grown on a confluent layer of astroglia. Furthermore, there was no evidence of attenuation of the neuroprotective effect in the glia-depleted, neuronal cultures, even with micromolar amounts of peptide.
ADNF-9 neuroprotection from clinically relevant neurotoxins.
The majority of experiments with ADNF and ADNF-9 have examined the protective properties of these agents against TTX, a substance previously used to study the role of electrical activity on neurodevelopment in CNS cultures (Brenneman et al., 1983;Brenneman et al., 1984; Brenneman et al., 1990b). To extend these studies, ADNF-9 has been tested for neuroprotective actions against clinically relevant neurotoxic substances including β-amyloid peptide (Alzheimer’s disease neurotoxin) and NMDA (excitotoxicity). Neuroprotection was measured in a tissue culture system comprising both neurons and abundant glial cells. As shown in figure 6, ADNF-9 provided potent protection from neuronal cell death produced by β-amyloid peptide (amino acids 25–35) after a 4-day incubation with cerebral cortical cultures. As observed with TTX-treated cultures, the EC50of the response against β-amyloid was 0.1 fM, with attenuation evident at picomolar or greater concentrations. Furthermore, neuronal cell death produced by a 4-day treatment with 10−5 M NMDA was prevented by cotreatment with subfemtomolar concentrations of ADNF-9 (fig. 7). Together, these data indicate a potent protection from several neurotoxic agents that are believed to play roles in the etiology of neurodegeneration.
Discussion
A peptide of nine amino acids (ADNF-9: SALLRSIPA) has been discovered that prevents neuronal death at femtomolar concentrations while maintaining full efficacy over a 1000-fold concentration range. The basis for this discovery is ADNF, an astroglia-derived protein released by VIP. These studies have revealed amino acids that are important for this extraordinary activity: the N-terminal serine and the COOH-terminal amino acids isoleucine-proline-alanine. The VLGGG portion of ADNF-14 (VLGGGSALLRSIPA) is not essential to the survival-promoting activity of ADNF. SALLRS did show biological activity (fig. 1B) but at a much higher EC50 compared with ADNF-9 and ADNF-14. Modulations of the N-terminal site of ADNF-9 (elongation) also resulted in a decreased EC50 as well as a narrower effective concentration range, a result that indicates the importance of the N-terminal portion of the molecule for the biological action. These studies reinforce the sensitivity of the biological activity of neuroactive peptides to even single-amino acid, conservative substitutions.
Recent studies indicate that peptides derived from larger molecules can mimic the biological activity of the parent molecule. Thus the N-terminal domain of growth-inhibitory factor is sufficient to produce growth inhibition (Uchida and Ihara, 1995), and synthetic peptides (cyclized dimers) derived from nerve growth factor (NGF) prevent neuronal cell death but do not promote neurite extension (Longoet al., 1997). ADNF-9 is the first example of a short peptide fragment of a neurotrophic factor whose biological activity surpassed that of the parent protein. The potency, efficacy, activity range, size and hydrophobic nature of ADNF-9 support its candidacy for future drug development against neurodegeneration. However, the mimicking effects of ADNF-9 for ADNF have been investigated only for survival-promoting activity in CNS cultures. It is unlikely that ADNF-9 captures all biological activities of intact ADNF or VIP.
The mechanism through which ADNF-9 mediates femtomolar neuroprotection is not known. However, experiments performed in the present study have provided clues to how the peptide may function and have indicated characteristics of the peptide that are important to the design of future studies. The time course of ADNF-9 activity revealed that a very short exposure (2 hr) to cerebral cortical cultures could elicit full efficacy that persisted over a 4-day test period. As shown in figure 2, the survival-promoting activity present in the medium of ADNF-9-treated cultures was not detectable after 15 min of exposure to cells. This rapid disappearance of detectable biological activity associated with the peptide is probably mediated by peptide degradation and/or uptake into the cells. Paradoxically, the cultures treated for 15 min with ADNF-9 followed by the addition of fresh medium did not show full biological activity compared with cultures treated for 2 hr or 4 days. A simple interaction of the peptide with neurons cannot explain these kinetic data. We hypothesize that other substance(s) in the conditioned medium of the ADNF-9-treated cultures played a role in mediating the ADNF-9 neurotrophism. The fact that the conditioned medium from the test cultures treated for 15 min had no detectable activity suggested a complex response combining immediate intracellular changes produced by the peptide in the surviving neurons and glia with the secretion of additional factors into the conditioned medium. It is possible that the 15-min exposure was not long enough to collect sufficient secreted modulating factors for a full biological response.
ADNF-9 elicited marked differences in the pharmacological response between glia-depleted, neuronal cultures and the mixed neuronal-glial cultures. ADNF-9, like ADNF and ADNF-14, exhibited attenuated activity at picomolar concentrations or greater when tested in mixed cultures. The mechanism of this response has not been determined, but several explanations should be considered: 1) receptor down-regulation, 2) stimulation of another receptor with an opposite pharmacological response, 3) partial agonist-antagonist activity and 4) self-association of ligand at higher concentrations. The observation that neuroprotection by ADNF-9 of glia-depleted, neuronal cultures did not exhibit attenuation, even at μM concentrations, suggested yet another explanation involving possible cellular interactions with other effector molecules. The fact that the cellular composition of the test culture influenced the attenuation response implied that this biological response probably cannot be accounted for by receptor down-regulation, partial agonists or self-association. Rather, we suggest that the ligand interacts with both neurons and nonneuronal cells to produce the effect. We hypothesize that ADNF-9 interacts with non-neuronal cells to produce modulatory substance(s) that shift the dose-response to the left and attenuate the biological response at higher concentrations. Thus the attenuation of the biological response of this peptide may be due to the generation of modulatory effects of ADNF-9 on non-neuronal cells. The modulation may involve proteases, protease inhibitors, allosteric receptor effectors or components of macromolecular complexes with the ligand. Regardless of mechanism(s), the explanation resides in multicellular interactions elicited by the peptide that regulate both the potency of the biological response and the inactivation of the ligand. It is our further speculation that many of the inverted-U-shape dose-response characteristics of various neuropeptides, growth factors and cytokines may be attributable to such modulatory, cell interaction-based responses.
ADNF-9 exhibited an almost complete loss of biological activity after freezing in PBS. This loss of activity with such a small molecule was unexpected and suggested a higher order of structure or a decrease in solubility. However, no apparent loss of solubilized peptide was evident after a freeze-thaw cycle. We hypothesize that ADNF-9 forms macromolecular structures in physiological salt solution and that this complex is important both for the biological activity of the peptide and for its vulnerability after freezing. ADNF-9 may be useful to study interactions of solvents, peptide aggregation and biological activity. Indeed, in the case of peptide mimics of NGF, neuroprotective activity was not detected in the monomeric form of the peptide fragment; rather, a dimer of cyclized peptides was required (Longo et al., 1997).
The concept that growth, differentiation, survival and maintenance of neurons are regulated via the uptake at the nerve terminal and retrograde transport of the trophic molecule (the neurotrophic hypothesis) is a central idea in neurobiology, especially in developmental neurobiology (Matsumoto et al., 1994). Previous studies have shown that growth factors such as insulin-like growth factor-1 (Prager et al., 1994), epidermal growth factor (Vieira et al., 1996; Galcheva-Gargova et al., 1995), platelet-derived growth factor (Mori et al., 1994) and interleukin-1 (Korherr et al., 1997) all function through receptor-mediated endocytosis (RME). RME is a general mechanism for the uptake of macromolecules (Brown and Goldstein, 1979;Goldstein et al., 1979) that mediate different regulatory processes through diverse signal transduction pathways [tyrosine kinase activation, for example, in the case of epidermal growth factor (Multhaup, 1997)]. After activation, receptors mediate ligand-induced internalization to endosomes, which become acidified by the action of vacuolar H+-ATPase (Yocum et al., 1995). Although it is not clear whether ADNF receptors are present on nerve terminals or the peptide is retrogradely transported, the present studies with BFA suggest that RME may be involved in the mechanism of action of ADNF and ADNF-9.
BFA is a specific inhibitor of vacuolar H+-ATPase, and it has been used to study the role of RME (Bowman et al., 1988). Studies of BFA action in other systems have shown a number of relevant effects, including an increase in apoptosis in PC12 cells (Kinoshita et al., 1996). The cell death effect required protein synthesis and was preceded by growth arrest and chromatin condensation. The chronic BFA effect in the present studies supports these previous observations and suggests that activity-dependent neurons in the cerebral cortex are among the cells vulnerable to this agent. The demonstrated protective effects of ADNF and the apoptosis-producing effects of the ADNF antiserum in CNS cultures (Gozes et al., 1997) may be related to the BFA-associated mechanisms on apoptosis. We would speculate that the deleterious effect of chronic BFA may be due to interference with a neurotrophic pathway of endogenous ADNF or other trophic substances. Other studies have shown that BFA can inhibit mitogen-induced DNA synthesis during the G1 phase of the cell cycle. In this regard, ADNF has been shown to be a potent mitogen that can regulate growth in whole-embryo cultures (Dibbern et al., 1997).
The possible involvement of RME was investigated as a mechanism of ADNF-9 entry into cells. Acute BFA was shown to prevent ADNF-9 from protecting neurons from cell death produced by TTX. Chronic BFA also reduced the number of surviving neurons to the same degree as TTX. Furthermore, treatment of cultures with both reagents resulted in no additional reduction in neuronal survival, which supports the hypothesis that these agents are working on the same population of neurons through a common pathway of apoptotic regulation (Kinoshitaet al., 1996; Gozes et al., 1997). These data did not prove, but were consistent with, the hypothesis that ADNF-9 neuroprotection was mediated through RME and that the vulnerable population of neurons was both BFA-sensitive and dependent on electrical activity for survival. The fate of the ADNF-9 after entry into the cell is not known. However, the observation that a 4-day protection occurred after a 2-hr incubation period strongly implies a long-lasting response of the cells, perhaps involving transcriptional control of proteins that regulate apoptosis.
ADNF-9 was shown to protect neurons against death associated with β-amyloid peptide (amino acids 25–35). Although the precise role of β-amyloid in Alzheimer’s disease is the subject of ongoing debate, there is growing evidence that this peptide can be neurotoxic. Previous studies have suggested that multiple mechanisms may account the β-amyloid toxicity. For example, the toxic effects vary with brain regions. In the cortex, superoxide dismutase activity and peroxide production increased after β-amyloid treatment (Cafe et al., 1996), whereas in the hippocampus, β-amyloid treatment did not induce the production of superoxide anions (Prehn et al., 1996). In human neurons, β-amyloid down-regulated the expression of the antiapoptotic protein Bcl-2 and increased the levels of Bax, a protein known to promote cell death (Paradis et al., 1996). The β-amyloid treatment has been shown to result in the generation of free radicals (Markesbery, 1997), leading to lipid peroxidation and impaired glucose transport (Mark et al., 1996), uncoupling G-protein linked receptors and producing neuronal cell death. The generation of free radicals may also involve the production of mutations and mitochondrial dysfunction (Schapira, 1996). The accumulation of peroxide can be induced by fragments of the β-amyloid peptide in rat cortical neurons, such as the fragment used in the current study [amino acids 25–35; (Cafe et al., 1996)]. Our results imply that ADNF-9 provides neuroprotection against oxidative stress associated with the recognized actions of β-amyloid peptide. Furthermore, the neurotoxicity exhibited by the β-amyloid peptide (25–35) may also involve the NMDA-responsive glutamate receptor (Brorson et al., 1995). Neuronal cell death produced by chronic NMDA was prevented by ADNF-9, a result that suggests another application for neuroprotection. The effects of ADNF-9 on necrosis are unknown. However, the ADNF-9-mediated protection from TTX, β-amyloid peptide and NMDA toxicity suggests that systems treated with other oxidative and apoptotic agents should be examined to establish the breadth of effectiveness of ADNF-related peptides.
This study has extended the observation that a peptide can mimic the action of a neurotrophic protein. The following pharmacologic properties of an ADNF peptide have been described: 1) ADNF has a core 9-amino acid sequence that exhibits extraordinary potency and breadth of neuroprotective properties, 2) the mechanism of action of ADNF-9 involves a BFA-sensitive pathway, 3) freezing in PBS renders the biological activity of ADNF-9 inactive and 4) the dose-dependent attenuation of the biological response of ADNF-9 is contingent on cellular interactions. The potential of this peptide as lead compound for the treatment of neurodegeneration is enhanced by its potency, small size, broad range of effective concentrations and demonstrated protective properties in vitro.
Acknowledgments
Professor Illana Gozes is the incumbent of the Lily and Avraham Gildor Chair for the Investigation of Growth Factors. We thank Drs. Joanna Hill, Catherine Spong, Raquel Castellon, Lura Williamson and Grethen Gibney for their helpful suggestions with this work. The assistance of Rachel Zamostiano and Ayelet Reshef is also acknowledged.
Footnotes
-
Send reprint requests to: Dr. Douglas E. Brenneman, Chief, Section on Developmental and Molecular Pharmacology, Laboratory of Developmental Neurobiology, National Institute for Child Health and Human Development, Building 49, Room 5A38, National Institutes of Health, Bethesda, MD 20892.
-
↵1 Supported in part by the U.S.-Israel-Binational Science Foundation (BSF).
- Abbreviations:
- ADNF
- activity-dependent neurotrophic factor
- TTX
- tetrodotoxin
- PBS
- phosphate-buffered saline
- NMDA
- N-methyl-d-aspartate
- VIP
- vasoactive intestinal peptide
- SDS
- sodium dodecyl sulfate
- hsp60
- heat shock protein 60
- BFA
- bafilomycin A1
- RME
- receptor-mediated endocytosis
- Received August 14, 1997.
- Accepted January 22, 1998.
- The American Society for Pharmacology and Experimental Therapeutics
References

Structure-activity relationship of ADNF peptides. A) Comparison of purified ADNF (•) with ADNF-9 (○) and ADNF-9 without the N-terminal serine (▴). The peptides are identified by their primary amino acid sequence. Cerebral cortical cultures comprising both neurons and glia were treated for 4 days with various concentrations of peptide. Cultures were all cotreated with 1 μM TTX. Additions were made only once. The molarity of the purified ADNF was based on a total amino acid analysis of purified ADNF with an apparent molecular weight of 14,000 Da. Each point is the mean of two determinations. All duplicates agreed within 5% of the mean. The dotted line is the mean number of surviving neurons in control cultures at the end of the treatment period. Neuronal cell counts for ADNF- and ADNF-9-treated cultures were greater than the control levels because these substances also prevent naturally occurring neuronal cell death in these cultures (Brenneman et al., 1985b). B) ADNF-related peptides are compared to ADNF-9 (•) in the test described in figure 1A. The primary sequences of the tested peptides are given. Each point is the mean of duplicate determinations. The values agree within 5% of the mean. The test cultures are sister cultures of those shown in figure 1A. C and D) ADNF-related peptides are compared to purified ADNF (▴). The primary sequence of each of the tested peptides is given next to its symbol. Each point is the mean of duplicate determinations, which agree within 5% of the mean. The peptides shown in figures 1C and 1D were tested in the same experiment. The explanation of the control cell counts is as described for figure 1A. E) Effect of a N-terminal elongation and conservative substitution on the survival-promoting activity of ADNF-9 (○). The primary sequence of the tested peptides is given. The experimental design is as described in figure 1A. Test cultures are sister cultures to those shown in figures 1A and 1B. Each value is the mean of duplicate determinations, which agree within 5% of the mean.
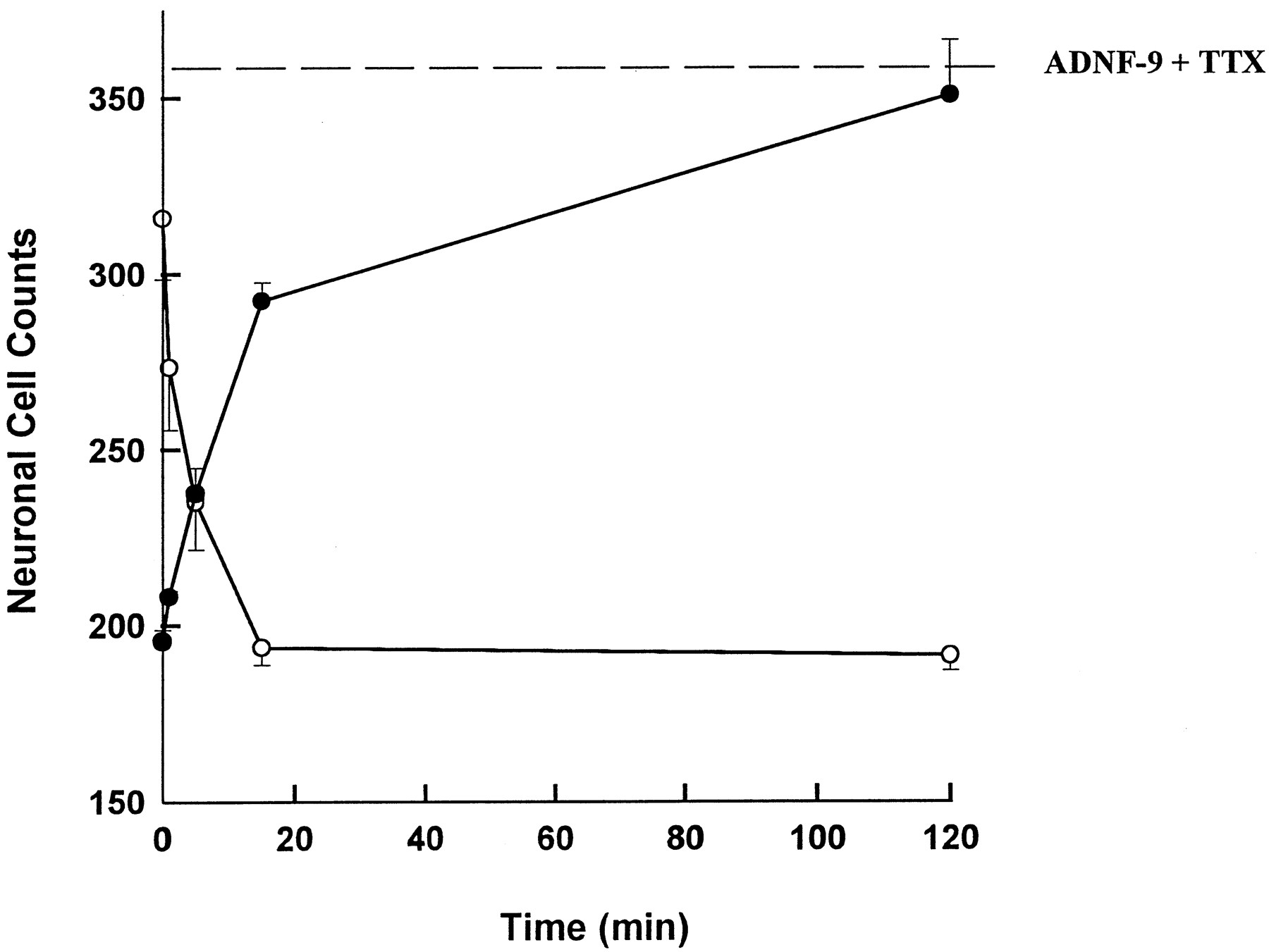
Time course of ADNF-9 neuroprotection in TTX-treated cerebral cortical cultures. ADNF-9 (10−15 M) was added to cultures containing 1 μM TTX for 1, 5, 15 or 120 min. The medium of treated cultures was removed and replaced with fresh medium containing 1 μM TTX (•). These cultures were allowed to incubate for 4 days and then fixed with glutaraldehyde before neuronal cell counts. The dotted line represents the mean of neuronal cell counts for cultures treated with 10−15 M ADNF-9 and 1 μM TTX for 4 days without any change of medium; this represents maximal response of the survival assay. The media removed from the time course cultures were added to another series of cultures (○) to test for any remaining ADNF-9 neuroprotective activity in the medium. Each value is the mean ± the S.E. of four determinations. Neuronal cell counts from control cultures were 315 ± 17. Statistical analysis of counts from cultures treated with TTX and ADNF-9 (•) indicated significant increases in survival compared with TTX alone after incubation times ≥ 5 min (P < .05). Treatment with ADNF-9 for 2 hr resulted in neuronal cell counts that were not significantly different from cell counts after treatment with ADNF-9 and TTX for 4 days (dotted line). Analysis of the medium from ADNF-9-treated cultures (○) indicated significant decreases in residual neuroprotective activity from control values at all time-points tested (P < .05).

Comparison of the bioactivity of purified ADNF (○) and ADNF-9 (•) after repeated freeze-thaw cycles. Purified ADNF [10−13 M in PBS (pH 7.4)] was frozen on dry ice for 5 cycles. Between freeze-thaw cycles, the ADNF was brought to room temperature and aliquots were taken for testing in TTX-treated cerebral cortical cultures comprising both neurons and glia. ADNF-9 was dissolved from fresh powder in PBS. The peptide freeze-thaw cycles were performed in tandem with the purified ADNF. No significant changes in the survival-promoting activity of ADNF were detected after any of the 5 cycles compared with unfrozen ADNF. In contrast, significant decreases in neuronal cell counts were observed in cortical cultures cotreated with ADNF-9 and TTX after one freeze-thaw cycle (P < .001). Four additional freeze-thaw cycles produced no significant additional losses in survival-promoting activity. Neuronal counts for control cultures were 241 ± 5. Treatment duration was 4 days.

Effect of bafilomycin A1 on ADNF-9-mediated increases in neuronal survival of electrically blocked cerebral cortical cultures. C = control, T = TTX, P = ADNF-9 peptide, B = BFA, BT = TTX and BFA and BPT = cotreatment with TTX, ADNF-9 and BFA. After a complete change of medium, BFA was added to the cultures 30 min before the addition of peptide or TTX for a further 2 hr of incubation. At the conclusion of the 2-hr incubation, the cultures were rinsed with PBS three times, and then fresh medium was added to the cultures. The replenished medium also contained 1 μM TTX for the following treatment groups: T, BT, PT and BPT. The BFA was prepared as a 320 μM stock in dimethyl sulfoxide and stored frozen at −20°C. The final concentration of BFA in the dish was 80 nM, after serial dilutions in PBS. ADNF-9 was dissolved in filtered (0.2 μm) PBS. ADNF-9 was serially diluted with PBS to give a final concentration of 10−15 M in the dish. The concentration of TTX was 1 μM. Statistical comparisons indicated that BFA (B) produced neuronal cell counts that were not significantly different from controls. The increase in neuronal cell counts associated with ADNF-9 plus TTX (PT) were significantly greater than all other treatment groups (P < .01). The decrease in counts after TTX (T) treatment were significantly less than that of control (P < .001). BPT treatment produced a significant decrease in counts compared with PT (P < .001). Each value is the mean ± the S.E. of triplicate determinations.

The effects of ADNF-9 on neuronal survival in cerebral cortical cultures: comparison of neuronal cultures with mixed neuronal plus glial cultures. Mixed cultures (○) were cotreated with various concentrations of ADNF-9 and 1 μM TTX. Glia-depleted, neuronal cultures (•) were treated with the same peptide preparation. For the mixed cultures, significant increases in neuronal cell counts were observed at ADNF-9 concentrations ≥10−17 to 10−9 M (P < .01) compared with cultures treated with TTX alone. A significant attenuation of the survival-promoting activity of ADNF-9 occurred at 10−12 M compared with 10−13 M (P < .05). In the glia-depleted, neuronal cultures, significant increases from TTX-treated cultures were observed at all concentrations greater than 10−13 M (P < .001). Each value represents the mean ± the S.E. of at least three determinations. Neuronal cell counts for controls were 538 ± 8 for the PLL cultures and 539 ± 9 for the mixed cultures.

ADNF-9 prevents neuronal cell death produced by β-amyloid peptide (25–35). Mixed neuronal-glial cultures cotreated with 25 μM β-amyloid peptide and varying amounts of ADNF-9. The peptides were added on day-4 cultures and were assessed 4 days later. The dotted line represents the cell counts of control cultures at the conclusion of the treatment period. Significant increases in neuronal cell counts were observed at concentrations from 10−16 M to 10−11 M (P < .01). Each point is the mean ± the S.E. of at least three determinations.

ADNF-9 prevents neuronal cell death produced by 10 μm NMDA. This experiment was performed as described for figure 6 except that NMDA rather than β-amyloid peptide was tested. Significant increases in neuronal cell counts were observed at ADNF-9 concentrations from 10−17 M to 10−8 M (P < .01). Each point is the mean ± the S.E. of three determinations.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}