


Abstract
Naltrexone (NTX) exhibited approximately 3-fold higher affinity for sites labeled by [3H]U69,593 (putative κ1-selective ligand) than [3H]bremazocine (non-selective ligand) in the presence of mu anddelta receptor blockade in monkey brain membranes. This led us to test an hypothesis that NTX could display in vivo antagonist selectivity for κ1-versus non-κ1-mediated effects. Six opioid agonists were characterized by NTX apparent pA2 analysis in a 50°C water tail-withdrawal assay in rhesus monkeys. Constrained NTX pA2 values (95% confidence limits) were: alfentanil, 8.66 (8.47–8.85); ethylketocyclazocine, 7.97 (7.93–8.01); U69,593, 7.64 (7.49–7.79); U50,488, 7.55 (7.42–7.67); bremazocine, 6.92 (6.73–7.12); enadoline, 6.87 (6.69–7.05). Pretreatment with clocinnamox, an irreversible mu antagonist, confirmed that mu receptors were not involved in the antinociception produced by the kappa agonists, U69,593, U50,488, bremazocine and enadoline; however, both mu andkappa receptors mediated the antinociceptive effects of ethylketocyclazocine. The apparent NTX pA2 profile of opioid agonists correlated highly with the radioligand binding studies, which indicates that U69,593 and U50,488 produced antinociception by acting on kappa-1 receptors, whereas bremazocine and enadoline probably acted via non-kappa-1 receptors. This study provides further functional evidence ofkappa opioid receptor multiplicity in primates and suggests that NTX may be a useful tool to study this phenomenonin vivo.
Evidence for the existence of kappa opioid receptor subtypes has been reported by several laboratories based on binding experiments. Initially, it was demonstrated that the kappa opioid agonist, EKC, binds to guinea pig spinal cord membrane homogenates in a nonhomogeneous manner (Attali et al., 1982). Since that time, many in vitro studies have suggested the presence of at least two kappa opioid receptor subtypes in different preparations across species (Zukin et al., 1988; Unterwaldet al., 1991; Kim et al., 1996; Zhang et al., 1996). In general, these receptor subpopulations have been classified as kappa-1, which displays higher affinity for the arylacetamide compounds such as U50,488 and U69,593, andkappa-2, which preferentially binds some benzomorphans such as EKC and bremazocine (Zukin et al., 1988; Clark et al., 1989). Although the presence of a third subpopulation has been proposed, kappa-3, identified by [3H]naloxone benzoylhydrazone (Clark et al., 1989; Pasternak 1993), it has been suggested thatkappa-3 could be a splice variant of the orphanin receptor (Pan et al., 1995; Pasternak and Standifer, 1995; Rossiet al., 1997). In addition, subdivisions ofkappa-1 and kappa-2 receptors, based on ligand-receptor binding profiles, have been proposed (Clark et al., 1989; Rothman et al., 1990), but there is as yet a lack of supporting evidence from physiological and pharmacological studies. However, to date, only one type of kappa receptor has been cloned and apparently it is a typical kappa-1 receptor (Simonin et al., 1995; Zhu et al., 1995). Regardless of the eventual resolution of these suggested binding sites, kappa receptor heterogeneity appears to be a possibility.
In an earlier stage of the investigation of kappa opioids, a major impediment to acceptance of the notion of kappareceptor subtypes was the lack of convincing functional evidence (Traynor, 1989). However, since the beginning of the 1990s, severalin vivo studies have provided evidence for this notion. The irreversible kappa-1 opioid antagonist, (−)-UPHIT, antagonizes the antinociceptive effects of U69,593 but not bremazocine in mice (Horan et al., 1991). Further, the lack of cross-tolerance between the antinociceptive effects of U69,593 and bremazocine in mice supports the existence of kappa opioid subpopulations (Horan and Porreca, 1993). In primate studies, a selective kappa opioid antagonist, nor-binaltorphimine, antagonizes the antinociceptive effects of U50,488 and U69,593 but not other kappa agonists such as EKC, bremazocine and enadoline (Butelman et al., 1993b). Also, pretreatment with dynorphin A-(1–13) antagonizes U50,488- and U69,593-induced antinociception in the 55°C water tail-withdrawal assay, but not bremazocine- and enadoline-induced antinociception (Butelman et al., 1995a). These findings indicate that a variety of kappa agonists may produce their antinociceptive effects by acting at different subtypes of kappa receptors and support the notion of functionalkappa receptor subtypes in both primates and rodents.
In vivo apparent pA2 analysis is another tool to demonstrate the presence of different functional receptor populations (Woods et al., 1992; Bertalmio et al., 1993). This quantitative model affords a measure of thein vivo potency of an antagonist in blocking the effects of an agonist, and the finding that antagonist pA2values are different across two or more agonists is consistent with the concept that different receptor populations mediate the effects of the agonists (Takemori 1974; Tallarida et al., 1979). For example, in vitro competition binding studies show that quadazocine has higher affinity for mu receptors and lower affinity for kappa receptors in rhesus monkey brain membranes (Negus et al., 1993; Emmerson et al., 1994). Consistent with this, quadazocine is more potent in antagonizing the effects of mu agonists than kappa agonists across different behavioral preparations in this species (Bertalmio and Woods, 1987; Dykstra et al., 1987; Negus et al., 1993). Thus, quadazocine pA2 analysis can be used to differentiate between mu and kappareceptor-mediated effects in vivo.
In preliminary competition binding experiments in rhesus monkey brain membranes, NTX exhibited higher affinity for sites labeled by [3H]U69,593 than sites labeled by [3H]bremazocine in the presence ofmu and delta receptor blocking agents. This led us to test the hypothesis that NTX could differentiate κ1- from non-κ1-mediated effects in vivo. Thus, six opioid agonists were characterized by NTX apparent pA2 analysis in a thermal antinociception assay in rhesus monkeys. The functionally irreversible muantagonist C-CAM (Zernig et al., 1994) was used to ensure that the effects of some kappa agonists were not mediated bymu receptors. In addition, the binding profile of NTX was characterized further by determining Kιvalues against different radioligands and correlated with NTX in vivo antagonist selectivity. The aim of this study was to investigate whether NTX pA2 analysis is an useful tool to differentiate kappa agonists, and whether this distinction is consistent with previous findings utilizing nor-binaltorphimine and dynorphin as kappa antagonists (Butelman et al., 1993b, 1995a).
Methods
Subjects
Seven adult male and female rhesus monkeys (Macaca mulatta) with body weights ranging between 7.3 and 13.2 kg were used. They were housed individually with free access to water and were fed approximately 25 biscuits (Purina monkey chow) and fresh fruit daily. Six of the monkeys had previous experience with the tail-withdrawal procedure, and one monkey was experimentally naive. All monkeys had previously received opioids, including some of the drugs used in this study. Four of the monkeys had not undergone any experimental procedure for at least 2 months before the present study.
Warm Water Tail-Withdrawal Assay
Apparatus and procedure.
Antinociception was measured by a procedure which had been described previously (Dykstra and Woods, 1986). The subjects were seated in restraint chairs and the lower part of the shaved tail (approximately 15 cm) was immersed in warm water maintained at temperatures of 40, 50 and 55°C. Tail-withdrawal latencies were recorded manually by a computerized timer. A maximum cutoff latency (20 sec) was recorded if the subjects did not remove their tails by this time. Each experimental session began with control determinations at each temperature. The agonist then was administered by a cumulative dosing procedure with a 30-min inter-injection interval. At the start of each test cycle, graded doses of agonists were administered subcutaneously; these doses increased by 0.25 or 0.5 log units throughout the session. Subsequent tail-withdrawal latencies were determined starting 15 min after each injection. The subjects were tested one to two times at three temperatures in a varying order, with approximately 1- to 2-min intervals between tests. Experimental sessions were conducted no more than twice per week.
Experimental design.
Antagonist effects of NTX. Base-line dose-effect curves for different opioid agonists, alfentanil, EKC, U69,593, U50,488, bremazocine and enadoline, were determined twice. The antagonist effects of NTX were studied with various pretreatment doses (0.0032–0.32 mg/kg s.c.) in a random order. Typically, the antinociceptive effects of the above-mentioned opioids were redetermined 30 min after pretreatment with a single dose of NTX.
Antagonist effects of C-CAM. A single dose (0.1 mg/kg s.c.) of C-CAM was administered in combination with the above-mentioned opioids, except alfentanil. These dose-effect curves were redetermined 4 hr and 1 day after pretreatment with C-CAM. The chosen pretreatment time was in accord with former studies (Zernig et al., 1994;Butelman et al., 1995a), which had demonstrated that C-CAM had robust, insurmountable mu-selective antagonist effects between 4 hr and 3 days after administration. The time course of the antagonist effects of C-CAM against EKC was carried out 4 hr and 3, 7 and 14 days after administration in a separate experiment. In addition, C-CAM was used to deplete the functional population of mureceptors, in order to examine whether the antagonist potency of NTX against EKC and U50,488 was altered under this condition. Thus, the antinociceptive effects of EKC and U50,488 were studied after 0.1 mg/kg of NTX (30-min pretreatment) in the presence of 0.1 mg/kg of C-CAM (4-hr pretreatment). The interval between consecutive C-CAM experiments was at least 5 weeks.
The subjects were separated into two groups. Monkeys in the first group (n = 4) were used in NTX pA2studies. Monkeys in the second group (n = 3) were used to confirm the data from the first group by use of pKB analysis, which can be achieved by use of only one dose of the antagonist (Negus et al., 1993). Theoretically, under specific conditions (e.g., competitive and reversible antagonism, etc.), the antagonist pA2 and pKB values for an agonist should be identical. On the other hand, the C-CAM studies were performed in both groups (n = 7), except that the antagonism of C-CAM against U69,593, bremazocine and enadoline was performed only in the first group (n = 4).
Data analysis. Individual tail-withdrawal latencies were converted to percent of MPE by the following formula: %MPE = [(test latency − control latency)/(cutoff latency − control latency)] × 100. Mean ED50 values were obtained after log transformation of individual ED50 values, which were calculated by least-squares regression with use of the portion of the dose-effect curves spanning the 50% MPE; and 95% confidence limits (95% CL) were also determined (P < .05). In addition, dose ratios (DR) were calculated by dividing mean ED50 values in the presence of the antagonist by the base-line ED50values. The significant shifts in dose-effect curves were defined when their 95% CL of ED50 values did not overlap.
For in vivo apparent pA2 analysis, dose ratios were analyzed in a Schild plot and individual pA2 values were obtained by procedure 15 in the “Manual of Pharmacologic Calculations with Computer Programs” (Tallarida and Murray, 1987). Mean pA2 values were obtained from individual pA2 values, and the pooled pA2 values also were determined by entering all the dose ratio values in the same procedure. If the 95% CL of the slopes included −1, then apparent pA2values were redetermined by constraining the regression slope to −1 according to procedure 17 in the same computer program. Apparent pKB values were determined for individual antagonist doses by use of a modified equation (Negus et al., 1993): pKB = −log [B/(DR − 1)], where B equals the antagonist dose in moles per kilogram. Mean pKBvalues ± 95% CL also were calculated from individual pKB values for NTX. Apparent pA2 and pKB values were considered to be significantly different when their 95% CL did not overlap.
Radioligand Binding Experiments
Membrane preparation.
The brain tissue used in this study was obtained from one male adult rhesus monkey. After ketamine (10 mg/kg i.m.) administration and euthanasia by pentobarbital (100 mg/kg i.v.), the whole brain was excised rapidly and placed in ice. Cortical membranes were prepared in Tris-HCl buffer according to the methods ofEmmerson et al. (1994). Aliquots of the suspension, sufficient for experiments on one given day, were frozen at −70°C. Before use, the frozen suspension was thawed quickly, dispersed in a Dounce homogenizer and kept on ice. The protein concentration of the membrane suspensions was approximately 0.6 mg/ml, as determined by the method of Lowry et al. (1951), with bovine serum albumin as the standard.
Procedure.
Membranes (400 μg protein) were incubated in Tris-HCl buffer (50 mM, pH 7.4) with appropriate radioligand in a total volume of 1 ml for 60 min at 25°C in the presence of increasing concentrations of NTX. Mu sites were labeled with [3H]DAMGO (1 nM) and kappa sites were labeled with [3H]U69,593 (1 nM) or [3H]bremazocine (1 nM) in the presence of 1 mM DAMGO and 1 mM DPDPE to prevent binding to mu anddelta sites, respectively (Wood et al., 1989). A standard concentration of 1 nM ligand was used for each assay as the level of 3H-labeled ligand was taken into account in determining the Ki values of competing ligands. In all opioid binding experiments, nonspecific binding was defined with naloxone (10 μM). Bound and free ligands were separated by vacuum filtration and quantified by liquid scintillation counting.
Data analysis.
IC50 values were determined with use of Graphpad Prism Version 1.02 (Graphpad, San Diego, CA) and converted to Ki values according to Cheng and Prusoff (1973) or as −log values ofKi (pKi). TheKd values for [3H]U69,593 (0.95 nM) and [3H]DAMGO (0.57 nM) are from Emmerson et al. (1994); the Kd value for [3H]bremazocine, determined from saturation binding analysis, was 0.12 nM (data not shown). The relative potency of NTX in displacing each radioligand was determined by dividing theKi for NTX displacement of [3H]bremazocine by theKi for NTX displacement of the other radioligands.
Drugs.
In antinociception studies, alfentanil HCl and naltrexone HCl (National Institute on Drug Abuse, Bethesda, MD), EKC (Sterling Winthrop, Rensselaer, NY), U69,593 and U50,488 (Upjohn Co., Kalamazoo, MI), bremazocine methanesulfonate (Sandoz, Basel, Switzerland) and enadoline (Warner Lambert/Parke-Davis, Ann Arbor, MI) were dissolved in sterile water. Clocinnamox mesylate (Dr. J.W. Lewis, Bristol University, Bristol, UK) was dissolved in sterile water with the addition of a few drops of lactic acid. All compounds were administered s.c. in the back, at a volume of 0.1 ml/kg. In binding studies, the radioligands used were [3H]DAMGO (58 Ci/mmol), [3H]U69,593 (58 Ci/mmol) (Amersham Co., Arlington Heights, IL) and [3H]bremazocine (56 Ci/mmol) (NEN Life Science Products, Boston, MA). DAMGO and DPDPE were bought from Sigma (St. Louis, MO).
Results
Warm Water Tail-Withdrawal Assay
Control tail-withdrawal latencies and base-line dose-effect curves.
The monkeys used in the present study showed a consistent profile in tail-withdrawal responses. They kept their tails in 40°C water for 20 sec (cutoff latency) and removed their tails from 50 and 55°C water very rapidly (typically 1–2 sec). During the first 30 min after administration of various doses of NTX, there were no elevated tail-withdrawal latencies in 50 and 55°C water (data not shown).
All of the opioid agonists used in this study dose-dependently increased tail-withdrawal latencies in 50 and 55°C water. To avoid the convulsant behaviors that occasionally are observed with high doses of kappa agonists, agonist dosing was only continued until all of the monkeys reached 100% MPE in 50°C water. At doses that produce 100% MPE in 50°C, approximately 30 to 60% MPE were observed in 55°C water (data not shown). The base-line dose-effect curve for each agonist was measured twice and there was no significant variation between the two determinations of each base-line ED50 value. Thus, two base-line dose-effect curves in 50°C water were averaged for the data presented in table 1. The order of potency was enadoline > bremazocine > EKC > U69,593 > alfentanil > U50,488.
Antagonist effects of NTX.
The dose-effect curves for each agonist shifted dose-dependently to the right after pretreatment with various doses of NTX (0.0032–0.32 mg/kg) (fig. 1). A given dose of NTX produced different magnitudes of antagonism for different agonists, as indicated by different DRs. For example, 0.032 mg/kg of NTX caused a large rightward shift for alfentanil (DR = 49) and a moderate shift for EKC, U69,593 and U50,488 (DRs = 4–9). However, this dose of NTX only produced a slight shift (DR <2) for bremazocine and enadoline (table 1).
Figure 2 shows the Schild plots for NTX with values derived from individual DRs for each subject. The pooled pA2 values shown in each panel indicate that NTX was most potent in antagonizing the antinociceptive effects of alfentanil and much less potent against bremazocine and enadoline. The potency of NTX as an antagonist of EKC-, U69,593- and U50,488-induced antinociception was intermediate. The mean pA2values and slopes are also consistent with the profile of the pooled pA2 values (table 2). Because the 95% CL of the slopes included −1, the apparent pA2 values were redetermined by use of a slope constrained to −1, and these constrained pA2 values were also consistent with the profiles of the pooled and mean pA2 values (table 2).

Cumulative dose-effect curves in 50°C water for different opioid agonists after pretreatment with various doses of NTX. Abscissae (all panels): agonist doses in milligrams per kilogram. Ordinates (all panels): %MPE. All points represent the mean ± S.E.M. (n = 4). Dose-effect curves were determined either before NTX (base line; open circles) or 30 min after NTX pretreatment: 0.0032 (▾), 0.01 (▴), 0.032 (▪), 0.1 (•) and 0.32 (♦) mg/kg.

Schild plots obtained for NTX in the antinociception assay. Abscissae (all panels): negative log unit of the molar doses of NTX. Ordinates (all panels): log of (dose ratio − 1). Each point was converted from individual dose ratios for each subject based on the data in figure 1. The four closed symbols represent different subjects (n = 4). The pooled pA2 values and slopes are included in each panel. The 95% confidence limits are shown in parentheses; see “Data Analysis” for other details.
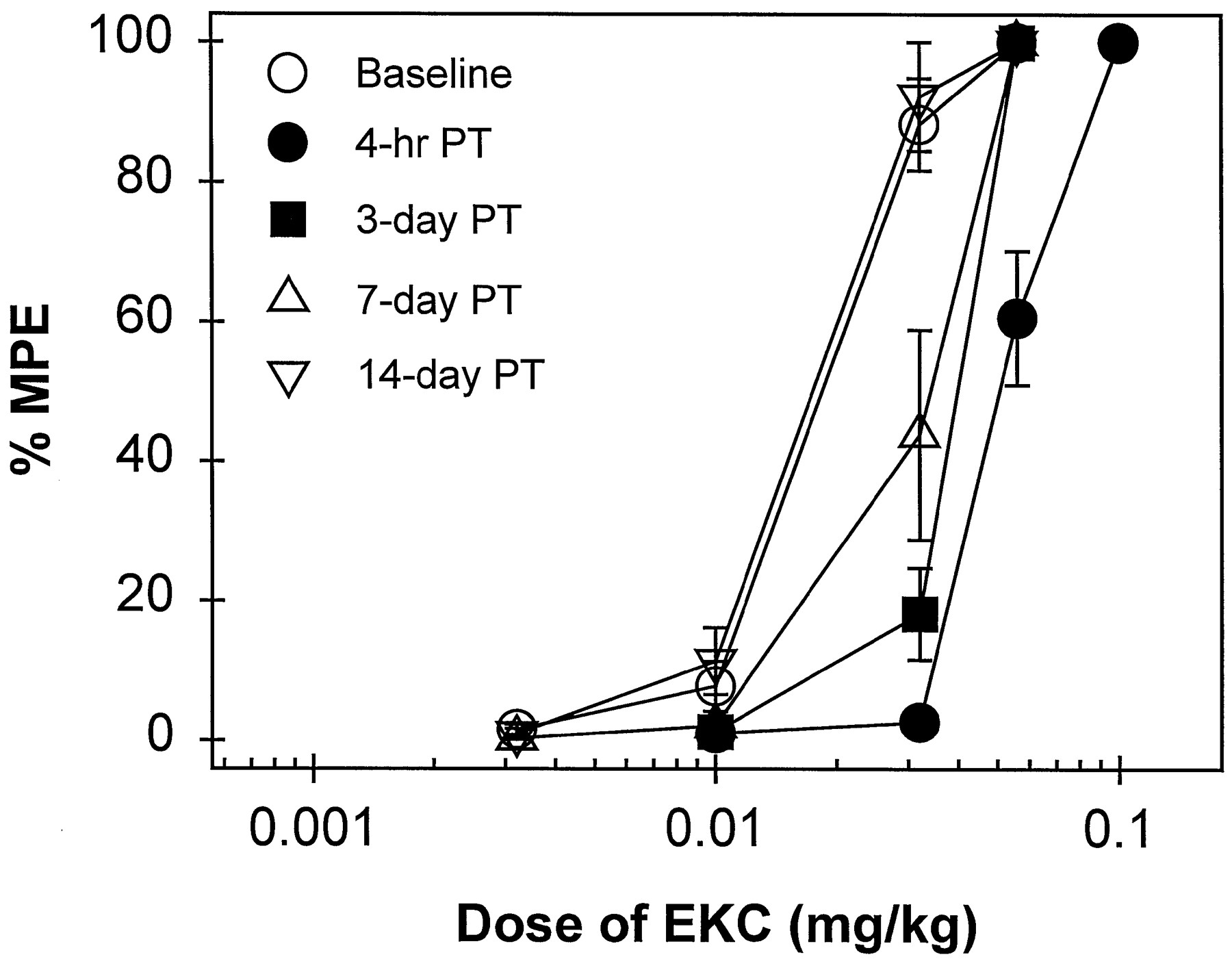
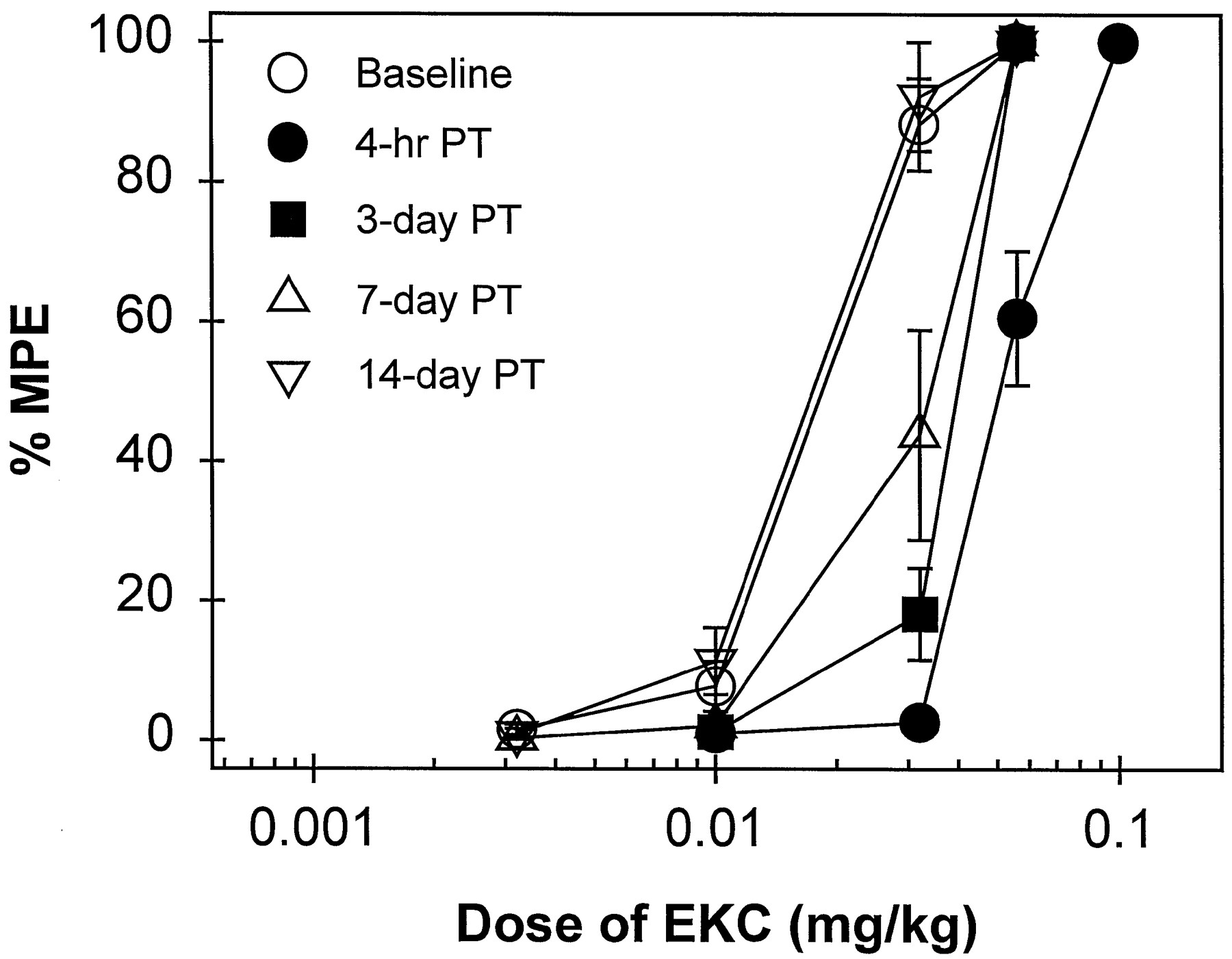
Time course of the antagonist effects of C-CAM (0.1 mg/kg) against EKC-induced antinociception in 50°C water. EKC dose-effect curves were determined either alone or 4 hr, 3, 7 and 14 days after C-CAM pretreatment (PT). All points represent the mean ± S.E.M. (n = 7). Other details are as in figure 1.

Comparison of the antagonist effects of NTX (0.1 mg/kg, 30-min pretreatment) in the absence (upper panels) or presence (lower panels) of C-CAM (0.1 mg/kg, 4-hr pretreatment) on EKC- and U50,488-induced antinociception. EKC dose-effect curves under different conditions are shown in the left panels and those of U50,488 are shown in the right panels. All points represent the mean ± S.E.M. (n = 7). Other details are as in figure 1.
Antagonist effects of C-CAM.
During the first 4 hr after administration of C-CAM (0.1 mg/kg), there were no elevated tail-withdrawal latencies in 50 and 55°C water (data not shown). When this dose of C-CAM was administered 4 hr before EKC in two groups of subjects, a rightward shift in the EKC dose-effect curve (approximately 3-fold) was observed (table 3). This rightward shift was maintained significantly for 3 days, and complete recovery was observed by 2 weeks after administration (fig. 3). However, 0.1 mg/kg of C-CAM did not shift the U50,488 dose-effect curve 4 hr after pretreatment, and it also did not antagonize U69,593-, bremazocine- or enadoline-induced antinociception 1 day after administration (table 3).
In the presence of C-CAM, NTX caused a rightward shift in the EKC dose-effect curve that differed in magnitude from that produced in the absence of C-CAM (fig. 4). NTX alone (0.1 mg/kg, 30-min pretreatment) shifted the EKC dose-effect curve by approximately 24-fold. However, in the presence of C-CAM (0.1 mg/kg, 4-hr pretreatment), the same dose of NTX only caused a 3-fold rightward shift. Consistent with this, pKB analysis indicated that the antagonist potency of NTX against EKC was reduced significantly in the presence of C-CAM, with a pKB (95% CL) of 6.9 (6.8–7.0). For comparison, 0.1 mg/kg of NTX exhibited a similar antagonist effect (10- to 11-fold shift) against U50,488 in the absence or the presence of C-CAM, with a pKB of 7.5 (7.4–7.6) (fig. 5).
To confirm the profile of the differential NTX potency in antagonizing the above-mentioned opioids, NTX pKB values were calculated in another set of subjects (n = 3). Figure 5illustrates the NTX pKB values (95% CL): alfentanil, 8.6 (8.5–8.8); EKC, 7.9 (7.8–8.0); U69,593, 7.6 (7.5–7.7); U50,488, 7.6 (7.4–7.8); bremazocine, 6.9 (6.7–7.1); enadoline, 6.7 (6.6–6.8). The results obtained with pKB analysis were similar to those of pA2 analysis. NTX showed the most potent antagonist effect for alfentanil, intermediate for EKC, U69,593 and U50,488, but much less for bremazocine and enadoline. Because the 95% CL of the pA2 and pKBvalues for these three groups of agonists did not overlap, the antagonist potency of NTX was considered to be significantly different.

Comparison of NTX pKB values (± 95% CL) for different opioid agonists. Closed symbols represent the pKB values for NTX in combination with agonists alone (n = 3). Open symbols represent the NTX pKB values against agonists in the presence of C-CAM (n = 7). Other details are as in figure 4.

Correlation of NTX antagonist potency in the pA2 study and the competition binding study. Ordinate:in vivo NTX pA2 values (moles per kilogram) for alfentanil, U69,593 and bremazocine. Abscissa: NTX pKivalues (−log of Ki values, nM) determined against [3H]DAMGO, [3H]U69,593 and [3H]bremazocine, respectively. All points represent the mean ± S.E.M. Other details are as in tables 2 and 4.
Radioligand Binding Experiments
NTX readily displaced the specific binding of [3H]DAMGO to membranes of monkey brain with an affinity (Ki) of 0.2 nM. NTX was approximately 3-fold less potent in displacing [3H]U69,593 and 10-fold less potent in displacing [3H]bremazocine (table 4). These values for [3H]DAMGO and [3H]U69,593 were similar to those in a previous report (Emmerson et al., 1994), but the NTX potency against [3H]bremazocine had not been determined previously in monkey cortex. The displacements had Hill slopes of unity confirming that NTX recognized a single binding site for each of the radioligands. The κ1 ligand U69,593 competed for itself affording a Ki value of 1.2 ± 0.2 nM with a slope of unity and also fully displaced [3H]bremazocine affording aKi value of 3.9 ± 0.3 nM, although with a Hill coefficient of 0.78 ± 0.08. The potency of NTX in antagonizing antinociception induced by different opioids was highly correlated (r2 = 0.99) with the receptor binding affinity (fig. 6).
Discussion
The kappa opioid agonists used in this study exhibited significantly different susceptibility to NTX antagonism, which suggests that their antinociceptive effects were mediated by distinct receptor subtypes. The binding studies concur in showing that NTX has differential affinity for kappa opioid receptor subpopulations. Furthermore, use of the irreversible muantagonist C-CAM confirmed that the kappa agonists, U69,593, U50,488, bremazocine and enadoline, acted selectively onkappa receptors (Butelman et al., 1993b; Franceet al., 1994), whereas both mu andkappa receptors probably mediated the antinociceptive effects of EKC (Eisenberg, 1985; Dykstra and Massie, 1988; Bodnaret al., 1991; Broadbear et al., 1994).
As expected, NTX was most potent in antagonizing the effects of themu agonist alfentanil (fig. 2). The NTX pA2 value was between 8.47 and 8.85, which is similar to the NTX pA2 value of 8.69 for alfentanil obtained in a drug discrimination procedure (France et al., 1990). This confirms that pA2 values are similar across different behavioral preparations for effects mediated by a common receptor population (Bertalmio et al., 1993; Negus et al., 1993). NTX is more potent againstmu agonists in rhesus monkeys than quadazocine or naloxone, which have pA2 values of 7.55 to 7.78 in the same preparation (France et al., 1990). In particular, the pA2 values of quadazocine for muagonists (fentanyl or alfentanil) are 7.6 to 7.9 in different procedures such as food-reinforced responding (Negus et al., 1993), antinociception (Walker, 1989) and drug discrimination (Bertalmio and Woods, 1987). However, these antagonists show a different order of affinity for sites labeled by [3H]DAMGO (quadazocine > NTX > naloxone) in rhesus monkey brain membranes (Emmerson et al., 1994). The reasons for this discrepancy are unknown, but it could be explained in part by differential pharmacokinetic profiles; for instance, NTX has high lipophilicity and is well-distributed rapidly after systemic administration (Gmerek et al., 1986; Franceet al., 1987), but quadazocine has a relatively slower onset and longer duration of action (Bertalmio and Woods, 1987).
The potency of NTX in antagonizing the antinociceptive effects of U69,593 and U50,488 was similar (pA2 values, 7.42–7.79). On the other hand, the NTX pA2values for bremazocine and enadoline were significantly lower (6.69–7.12) (fig. 2). This indicates that U69,593 and U50,488 produced antinociception by acting on kappa-1 receptors, for which NTX has higher affinity, whereas bremazocine and enadoline probably act on non-kappa-1 receptors. However, it is unknown whether the effects of bremazocine and enadoline were mediated bykappa-2 receptors, or by different kappa receptor subtypes for which NTX has similar lower affinity. Intriguingly, this distinctive profile among kappa agonists afforded by NTX pA2 analysis is consistent with previous primate studies. In the same preparation, pretreatment with nor-binaltorphimine (3.2 mg/kg) only antagonizes U69,593 and U50,488 but not bremazocine and enadoline (Butelman et al., 1993b). Recently, it had been reported that nor-binaltorphimine, even at a dose of 10 mg/kg, still preferentially antagonizes U50,488 but not enadoline (Butelmanet al., in press). This may indicate that nor-binaltorphimine is a selective kappa-1 antagonistin vivo. Although the opioid peptide dynorphin A-(1–13) acts as a low-efficacy agonist in the 55°C water preparation, it also selectively antagonizes U69,593 and U50,488 but not bremazocine and enadoline (Butelman et al., 1995a). Although it is unclear why the arylacetamide congener, enadoline, does not producekappa-1-mediated effects, a rodent study also provides evidence that this compound acts differently from U69,593 and U50,488, based on qualitative and quantitative differences in nociceptive and cardiovascular parameters (Herrero and Headley, 1993). These data together with the present study strongly support the notion of functional kappa opioid receptor subtypes.
NTX had different affinities for the kappa receptor population labeled by [3H]U69,593 and that labeled by [3H]bremazocine (table 4). This agrees with the differential binding profile of NTX seen in rats (Nocket al., 1990). The affinity of NTX determined against the various radioligands, given as pKi values, correlates very well with the NTX pA2 values determined by antagonism of antinociception mediated by the mu agonist alfentanil and the kappa agonists U69,593 and bremazocinein vivo (fig. 6). Although the correlation is good, the slope is not 1 because the absolute potencies are very disparate. This is to be expected because the administered dose of NTX, which may not directly relate to the concentration in the vicinity of the receptor, is used in calculating the in vivopA2.
To obtain more information on the kappa population labeled by [3H]U69,593 and [3H]bremazocine in monkey brain, the ability of the unlabeled U69,593 to displace both radioligands was studied. There was a 3-fold difference in the determined Ki value, but the fact that U69,593 displaced all of the specifically bound [3H]bremazocine, even with a shallow slope, suggests that the difference between the kappa sites, at least when measured by binding assay, is small. [3H]Bremazocine is expected to label allkappa sites (Wood et al., 1989), so the obtainedKi value of 3.9 nM represents a combination of the affinity of U69,593 for its own site (presumablykappa-1) and an additional site labeled by [3H]bremazocine in the presence ofmu and delta receptor blockade. However, this site is unlikely to be the classical kappa-2 site as defined by binding in other species, because the benzeneacetamide class of κ-ligands have an affinity of >5000 nM for the kappa-2 site (Wood and Traynor, 1989; Kim et al., 1996). On the other hand, NTX does displace [3H]bremazocine binding with slope of unity, whereas the slope of displacement by U69,593 is shallow. This may suggest that additional kappasubsites are recognized by [3H]bremazocine for which U69,593, but not NTX, has differential affinity. To date, because of lack of kappa subtype selective antagonists and lack of identification of separate clones, there have been difficulties in resolving the controversial issue of the exact nature of pharmacologically and biochemically defined kappa subtypes. Such subtypes may be the result of a specific gene, but may arise from tissue-specific alterations from the same gene. Nevertheless the behavioral and binding studies concur in showing that NTX has differential affinity for kappa receptor subpopulations.
In vivo apparent pA2 analysis has some specific assumptions. In particular, this quantitative model (Arunlakshana and Schild, 1959) can be applied only under the circumstances in which an agonist and antagonist compete for the same receptor site in a reversible manner (Takemori 1974; Tallarida et al., 1979). It is also assumed that measurement of the antagonist effect should be related to the peak tissue concentration, although the proportionality between administered dose and tissue concentration may not hold over a large dose range. In the present study, NTX (0.0032–0.32 mg/kg) produced parallel rightward shifts in all agonist dose-effect curves, and the slopes of a regression line in the Schild plots were not significantly different from −1. These observations indicate that the NTX antagonism was competitive and reversible at opioid receptors and could be used to distinguish selective opioid agonists. Moreover, in vivo pKBanalysis was performed in a separate group of monkeys (fig. 5). The results were similar to the data of the first group based on in vivo pA2 analysis. Thus, in vivopKB analysis also can provide an appropriate estimate of the relative antagonist potency under these conditions.
The pharmacological profile of EKC, however, could not be described adequately by NTX pA2 analysis alone, because its profile of activity was changed in monkeys treated with C-CAM. C-CAM has been characterized previously as a functionally irreversiblemu antagonist without any initial agonist effect in different preparations across species (Comer et al., 1992;Burke et al., 1994; Zernig et al., 1994; Butelmanet al., 1996). Pretreatment with C-CAM (0.1 mg/kg) did not antagonize the antinociceptive effects of U69,593, U50,488, bremazocine and enadoline, confirming that these compounds induced antinociception through non-mu receptors. However, the EKC dose-effect curve was shifted to the right approximately 3-fold after 4 hr pretreatment with C-CAM. This antagonism was maintained for 1 week, then returned to control potency by 2 weeks (fig. 3), which is similar to the time course of C-CAM against mu agonists in this assay (Zerniget al., 1994). Initially, EKC was characterized as akappa agonist in antinociception, based on the finding that 24 hr pretreatment of β-FNA, a selective irreversible muantagonist, did not antagonize EKC (Dykstra et al., 1987). However, β-FNA has kappa agonist effects particularly at the beginning of the postinjection period (Ward et al., 1982; Dykstra et al., 1987). For example, even after 24 hr pretreatment, β-FNA augments the diuretic effects of EKC (Dykstraet al., 1987). It is possible that the residual κ-effects of β-FNA interfere with its antagonism of EKC. Nevertheless, several rodent studies have found that β-FNA can antagonize EKC in different antinociceptive assays and suggested that EKC can produce antinociception via mu receptors (Hayes et al., 1986; Clark et al., 1988; Horan et al., 1993; Broadbear et al., 1994). In the presence of C-CAM, the antagonist potency of NTX was reduced against EKC, but not against U50,488 (fig. 4). In monkeys, 4 hr pretreatment with C-CAM (0.1 mg/kg) substantially depletes the functional population of mureceptors (Zernig et al., 1994). Under this condition, the reduced NTX pKB value of 6.9 suggested that non-kappa-1 receptors mediated the EKC-induced antinociception (fig. 5).
Although the present study indicated that both mu andkappa receptors mediated the antinociceptive effects of EKC, the slope of the Schild plot for NTX antagonism against EKC was not less than unity. It is possible that differences in pA2 values for NTX versus themu component of EKC and the kappa component of EKC are not sufficiently different to alter the Schild plot slope to less than unity when measured in vivo. However, the slope of −1 may indicate that the contribution to antinociception from activations of mu and kappa receptors is not equal and that mu receptors contribute the major component of the antinociceptive profile in the absence of C-CAM. Consistent with the present findings, another primate study showed similar results (Dykstra and Massie, 1988). In a shock-titration antinociceptive procedure, the pA2 value for quadazocine in combination with EKC was 7.04 (slope = −0.96), which was intermediate to those obtained with mu and kappaagonists, which indicates that EKC has both mu andkappa agonist properties. Furthermore, studies have demonstrated that EKC has mu-mediated effects such as respiratory depression and the occurrence of cross-tolerance with morphine in antinociception (Porreca et al., 1982; Eisenberg 1985; Butelman et al., 1993a). Taken together, the evidence supports the notion that EKC has mu agonist properties in addition to its kappa agonist properties (Gmerek et al., 1987; Dykstra and Massie, 1988; Craft and Dykstra, 1992;Broadbear et al., 1994). On the other hand, it is worth noting that unique properties of mixed mu/kappaagonists are difficult to characterize without selective irreversible receptor antagonists and that an imbalance between the efficacy ofmu/kappa agonists can contribute to a differential pharmacological profile. For example, butorphanol has been evaluated as a mu partial agonist in rhesus monkeys without detectable kappa-mediated effects (Butelman et al., 1995b); however, after pretreatment with C-CAM, butorphanol exhibits kappa-mediated diuretic effects (Vivian et al., 1997). The discriminative stimulus properties of EKC arekappa-mediated (France et al., 1994). If selective irreversible kappa antagonists were available, it would be very interesting to investigate whether EKC exhibitsmu-like discriminative stimulus properties when the functional population of kappa receptors were depleted.
In supporting the present findings, France et al. (1994)have reported that a single dose of the opioid antagonist quadazocine caused differential shifts among kappa agonists. In particular, bremazocine and enadoline dose-effect curves were shifted to the right by 2- to 3-fold, whereas quadazocine caused 10- to 30-fold rightward shifts for U69,593 and U50,488. This distinction is not consistent with other studies in which quadazocine pA2 analysis did not differentiatekappa agonists (Dykstra et al., 1987; Pitts and Dykstra, 1994). The reason behind these inconsistent reports with quadazocine is unknown. However, if quadazocine differentiates opioid agonists across a lesser pA2 range (e.g., 6.1–7.9) than NTX (6.6–8.9), it would be more difficult to use quadazocine to distinguish kappa receptor subgroups when few subjects are used. Until the binding affinity of quadazocine for sites labeled by [3H]U69,593 and [3H]bremazocine is available, it is difficult to clarify this discrepancy.
In summary, the present study demonstrates that NTX apparent pA2 analysis is useful for differentiating selective mu, kappa-1 and non-kappa-1 agonists in rhesus monkeys. Correlated with its radioligand binding profile, NTX pA2 analysis indicates that U69,593 and U50,488 produce antinociception by acting on kappa-1 receptors, but bremazocine and enadoline on non-kappa-1 receptors. This provides further functional evidence forkappa opioid receptor multiplicity in primates in mediating thermal antinociception. Rodent studies have reported that the κ-ligands, divided into two groups by this study, also have distinctive cardiovascular and neurophysiological profiles (Herrero and Headley, 1993; Schoffelmeer et al., 1997). Further studies may explore these differences in designing more effectivekappa opioids.
Acknowledgments
The authors express their gratitude to Dr. Albert J. Bertalmio for helpful advice for this manuscript, Dr. Carol A. Paronis for pilot studies and Ms. Min Zhang and Eunice Hong for excellent technical assistance.
Footnotes
-
Send reprint requests to: Dr. James H. Woods, Department of Pharmacology, Medical School, University of Michigan, 1301 MSRB III, Ann Arbor, MI 48109-0632.
-
↵1 Animals used in these studies were maintained in accordance with the University Committee on the Use and Care of Animals, University of Michigan, and Guidelines of the Committee on the Care and Use of Laboratory Animals of the institute of Laboratory Animal Resources, National Health Council (Department of Health, Education and Welfare, Publication ISBN 0–309-05377–3, revised 1996).
-
↵2 This research was supported by USPHS Grant 00254. Preliminary results were presented at the 59th annual meeting of the College on Problems of Drug Dependence, Nashville, TN, 1997.
-
↵3 Present address: Rockefeller University, New York, NY
- Abbreviations:
- C-CAM
- clocinnamox
- Enadoline
- CI-977
- DR
- dose ratio
- EKC
- ethylketocyclazocine
- NTX
- naltrexone
- %MPE
- %maximum possible effect
- DAMGO
- [d-Ala2,(Me)Phe4,Gly(ol)5]enkephalin
- DPDPE
- [d-Pen2,d-Pen5]enkephalin
- β-FNA
- β-funaltrexamine
- Received August 29, 1997.
- Accepted January 9, 1998.
- The American Society for Pharmacology and Experimental Therapeutics
References
ED50 values (mg/kg) for different opioid agonists either alone or after pretreatment with various doses of NTX in the 50°C water antinociceptive assay
Apparent pA2 values for NTX in antinociception
ED50 values (mg/kg) for kappa opioid agonists after either 4-hr or 1-day pretreatment with C-CAM (0.1 mg/kg)
Comparison of the affinity of NTX measured by displacement of the binding of different radioligands in monkey membranes




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}