


Abstract
The pharmacological properties of a novel selective 5-hydroxytryptamine1A (5-HT1A) receptor antagonist, NAD-299 [(R)-3-N,N-dicyclobutylamino-8-fluoro-3,4-dihydro-2H-1-benzopyran-5-carboxamide hydrogen (2R,3R)-tartrate monohydrate] were examined in vitro and in vivo and compared with the reference 5-HT1A receptor antagonist, WAY-100635 [N-(2-(1-(4-(2-methoxyphenyl)piperazin-yl))ethyl)-N-(2-pyridinyl) cyclohexanecarboxamide trihydrochloride]. The new compound had high affinity for 5-HT1A receptors in vitro with a Ki value of 0.6 nM. The only other receptors for which NAD-299 had affinity less than 1 μM werealpha-1 and beta adrenoceptors withKi values of 260 and 340 nM, respectively. Thus, the selectivity of NAD-299 for 5-HT1A receptors was more than 400 times. WAY-100635 had considerably higher affinity than NAD-299 for alpha-1 adrenoceptors (Ki = 45 nM) and dopamine D2 and D3 receptors (Ki = 79 and 67 nM, respectively). Like WAY-100635, NAD-299 competitively blocked 5-HT-induced inhibition of vasoactive intestinal peptide-stimulated cAMP production in GH4ZD10 cells and had no intrinsic activity. Both compounds were therefore 5-HT1A receptor antagonists in vitro and also behaved as such inin vivo experiments. Thus, they competitively antagonized the 8-hydroxy-2-(di-n-propylamino)tetralin-induced 5-HT behavioral effects, hypothermia, corticosterone secretion and inhibition of passive avoidance behavior without causing any actions of their own. The effective dose of NAD-299 varied between 0.03 and 0.35 μmol/kg s.c., depending on the test and the dose of 8-hydroxy-2-(di-n-propylamino)tetralin.
The selective stimulation of 5-HT1A receptors with drugs such as 8-OH-DPAT (Arvidsson et al., 1981) produces a variety of behavioral, biochemical and electrophysiological effects. These include hypothermia (Hjorth, 1985; Middlemiss et al., 1985), hyperphagia (Dourish et al., 1985; Bendotti and Samanin, 1986), antidepressant-like activity (Cervo and Samanin, 1987, 1991), effects on sexual behavior (Johansson et al., 1990), changes in the 5-HT syndrome (Middlemiss et al., 1985; Berendsenet al., 1989; Larsson et al., 1990; Rényi, 1991), inhibition of cage leaving behavior (Rényi et al., 1986), alterations in memory (Winter and Petti, 1987; Ohnoet al., 1993), disruption of a passive avoidance response (Johansson et al., 1988; Carli et al., 1992), changes in 5-HT neuronal firing rate (Blier and De Montigny, 1987), elevations in plasma corticosterone levels (Fuller, 1981; Koeniget al., 1987; Lorens and van de Kar, 1987; Kelder and Ross, 1992) as well as a variety of other biochemical effects (Cornfieldet al., 1991). Detailed characterization of these pharmacological effects became possible only with the development of selective and highly potent 5-HT1A receptor antagonists.
(S)-UH-301 was the first 5-HT1A receptor antagonist described (Hillver et al., 1990; Björk et al., 1991). However, it was also shown to have considerable potency as a DA D2 receptor agonist, a property that can complicate the interpretation of experiments. WAY-100135 and then the more selective WAY-100635 were described as highly potent and selective 5-HT1A receptor antagonists (Fletcher et al., 1993, 1994; Forster et al., 1995). They reversed the effects of 5-HT1A receptor agonists in various models and had no efficacy (“silent antagonists”). These compounds, and in particular WAY-100635, have been useful in the characterization of 5-HT1A receptor function.
We report here the basic biochemical and behavioral pharmacological characterization of a new selective 5-HT1A receptor antagonist, NAD-299 (fig. 1). The synthesis of this substance has been reported elsewhere (Evendenet al., 1995). WAY-100635 has been included in this study as a reference 5-HT1A receptor antagonist.

Chemical structure of NAD-299 in base form.
Materials and Methods
Materials.
The GH4ZD10 (rat pituitary tumor cells) cells containing rat 5-HT1A receptors and the Ltk− (mouse fibroblast) cells expressing human DA D2A (long isoform) receptors were obtained from Dr. Olivier Civelli (Vollum Institute for Advanced Biomedical Research, Oregon Health Sciences University, Portland, OR). The CHO cells expressing human D3, rat 5-HT6 and rat 5-HT7receptors were purchased from INSERM (Paris, France).
Compounds.
The standard agonist used in the studies was 8-OH-DPAT, from Research Biochemicals International Inc., Natick, MA. NAD-299 and WAY-100635 were provided from the laboratories of Astra Arcus AB. The test compounds were dissolved in saline, if not otherwise stated. Ham’s F10 medium, Earle’s balanced salt solution without Ca++ and Mg++, FCS, penicillin, streptomycin and HEPES were obtained from Gibco Ltd., Paisley, Scotland, U.K. [3H]cAMP and cAMP were obtained from Amersham International plc, Amersham, U.K. Diazepam, dithiothreitol, geneticine, NSD 1015, 5-hydroxytryptamine hydrochloride, IBMX, nicotine, oxotremorine, sodium glutamate, theophylline, tris/base and VIP were obtained from Sigma Chemical Co., St. Louis, MO. Ascorbic acid was from Merck, Darmstadt, Germany. (+)-Butaclamol hydrochloride, cimetidine, galanin, MK801 and pyrilamine were from Research Biochemicals International, Inc. (±)-Alprenolol hydrochloride was obtained from Astra Hässle, Mölndal, Sweden; methiothepine was a gift from Hoffman-LaRoche, Basel, Switzerland; methysergide was from Sandoz AG, Basel, Switzerland; paroxetine was from SmithKlineBeecham Pharmaceuticals, Betchworth, UK; and phentolamine mesylate was from Ciba-Geigy AG, Basel, Switzerland. All other compounds used were of highest purity available.
The following radioactive ligands were used (Ci/mmol in parentheses): [3H]AMPA (53), [3H]DHA (59), [3H]flunitrazepam (82.5), [125I]galanin (2200), [3H]8-OH-DPAT (130), [3H]ketanserin (64), [3H]MK-801 (20.3), [3H]nicotine (63), [3H]prazosin (78), [3H]pyrilamine (31.2), [3H]l-QNB (43), [3H]raclopride (80), [3H]SCH23390 (86), [3H]TBPS (69.2), and [3H]tiotidine (83.7), all purchased from DuPont NEN, Boston, MA. [3H]citalopram (85.7), [3H]5-hydroxytryptamine (29.7) and [3H]RX821002 (60) were obtained from Amersham International plc, UK.
Subjects.
Male Sprague-Dawley rats (B&K strain, B&K Universal, Sollentuna, Sweden), weighing 150 to 350 g, were used. The animals arrived in the laboratory at least 5 days before being used in the experiments and were housed 5 per cage under controlled conditions of temperature (21°C), relative humidity (55–65%) and light-dark cycle (12:12 h, lights on at 6 a.m.). Food (R36, Ewos, Södertälje, Sweden) and tap water were freely available in the home cage. The experiments were performed during the light phase, between 7 a.m. and 5 p.m. All injections were subcutaneous (s.c.) unless otherwise stated.
Radioligand binding studies.
The rats were decapitated and the various brain regions dissected out on ice (table1). The brain regions were frozen as tissues or homogenates in 0.32 M sucrose and stored at −20°C or −70°C until the day of the experiment. The membranes were prepared and the binding studies performed essentially as described previously (Chang et al., 1978; Speth et al., 1979;Gajtkowski et al., 1983; Hall et al., 1986;Murphy et al., 1987; Cross et al., 1989; Landet al., 1991; Rapier et al., 1990; Steeleet al., 1992; Jackson et al., 1995). The Ltk− cells expressing human DA D2A (long isoform) receptors and the CHO cells expressing human D3, rat 5-HT6 and rat 5-HT7 receptors were grown and the cell membranes prepared essentially as described by Malmberget al. (1993). Protein concentration was measured by the method of Markwell et al. (1978). The compounds were dissolved and diluted in 0.01% ascorbic acid.
The experimental conditions used in the receptor binding assays
Table 1 summarizes the incubation conditions of the binding assays for the various receptors examined. The Ki values (inhibition constants) of the test compounds were determined from inhibition curves by the iterative nonlinear curve-fitting program LIGAND (Munson and Rodbard, 1980). One- and two-site curve fitting was tested in all experiments. The one-site model gave a better fit (P > .05; F test) unless otherwise stated. TheKd values (dissociation constants) of the various radioligands used to calculate the Kivalues were determined by saturation studies and are given in table 1.
Second messenger studies.
The GH4ZD10 cells were cultured in 175-cm2 flasks in Ham’s medium with 1 mMl-glutamine supplemented with 10% FCS, 10 mM HEPES, penicillin and streptomycin at 37°C. Cells in passages 8 to 11 were used. Geneticin (G418 sulfate, 700 μg/l) was used for selection of cells transfected with receptors. The test compounds were dissolved to a 20 mM concentration in dimethyl sulfoxide and stored at −20°C until used. The stock solutions were further diluted in water containing 0.01% ascorbic acid and 0.1 mM IBMX.
The 5-HT was freshly prepared in the solution above. The cAMP assay was carried out according to Dorflinger and Schonbrunn (1983) with some minor modifications (Fowler et al., 1992). The cells were detached from the cultured flasks with Earle’s balanced salt solution supplement with 1 mM EDTA without Ca++ and Mg++. The cells were suspended in FCS-free Ham’s medium and the suspension was centrifuged at 250 × g for 6 min at room temperature. The pellets were resuspended to a density of 107 cells/ml in medium containing 0.01% ascorbic acid and 0.1 mM IBMX. Cells were preincubated in this solution for 1 h at 37°C and then diluted to a final density of 106 cells/ml. Aliquots (0.4 ml) of the cell suspension were added to Eppendorf tubes containing 0.1 ml VIP at a final concentration of 30 nM along with the test compounds and incubated for 20 min at 37°C. Each sample was carried out in duplicate. Reactions were stopped by placing the assay tubes in boiling water for 4 min after which the samples were transferred to ice water. The lysates were then centrifuged at 12,000 rpm for 4 to 5 min at 4°C, and the supernatants were frozen and stored at −20°C until analyzed. Cyclic AMP levels were determined according to the method of Brown and Ekins (1972) as modified byNordstedt and Fredholm (1990), in which free [3H]cAMP/cAMP is separated from that bound to the bovine adrenocortical protein kinase A on glass fiber filters with a semiautomatic cell harvester (Skatron AS, Tranby, Norway). Results are presented as percent of the VIP-stimulated response, set to 100%, or as relative “efficacy” which indicates the ratio of the effect of the test compound to maximum response of 5-HT in percent. The same experimental model as described above was used for Schild plots (Schild, 1949). The quantitative data (EC50 andKB) are based on at least two independent experiments.
5-HTP and DOPA accumulation.
Groups of five rats were given the test compound at the time noted before the injection of 8-OH-DPAT, 0.3 μmol/kg, or saline. NSD 1015, 100 mg/kg (in water solution with the pH brought to about 5 with sodium hydroxide), was injected 30 min later. The rats were sacrificed with a guillotine 30 min after the NSD 1015 injections. The brains were rapidly removed and the regions dissected were immediately frozen on dry ice. The samples were stored at −70°C until assayed.
Determination of 5-HTP and DOPA.
5-HTP and DOPA in striatum and hypothalamus were determined by use of HPLC with electrochemical detection according to the method of Magnusson et al.(1980). The mobile phase was 0.1 M phosphate buffer (pH 2.5)/methanol/acetonitrile (89:9:2 v/v), containing 1 mM octylsulfate. The frozen samples were weighed and homogenized in 0.1 M perchloric acid, containing 2.5 mM sodium bisulfite, 1 mM EDTA and isoprenaline as internal standard. The supernatants were injected directly onto a Supelcosil C18 (3 μm) column, connected to a detector (ESA Coulochem 5100A), set to 0.05/0.30 V.
Antagonism of 8-OH-DPAT-induced secretion of corticosterone.
The method has been described previously (Kelder and Ross, 1992). Rats were given daily injections of saline for 5 to 7 days before the start of the experiment to habituate the animals to the injections and thereby avoid acute increases in serum corticosterone. The test compounds were administered at the time noted before the injection of 8-OH-DPAT, 0.75 μmol/kg. Each experiment consisted of eight groups of five animals, and controls were always included. The rats were sacrificed 60 min after the injection of 8-OH-DPAT. The experiments were performed from 9 a.m. to 1 p.m. The trunk blood was collected in plastic tubes, and the serum obtained was stored at −70°C. Corticosterone in rat serum was assayed with a corticosterone[3H] RIA kit from ICN Biomedicals Inc., Costa Mesa, CA.
Flat body posture, forepaw treading and lower lip retraction.
The test apparatus was a clear plastic cage (Macrolon type IV cage, 34 × 56 × 19 cm), without sawdust, in which the rats were placed singly. The cages were placed in front of a mirror. The rats were placed in the experimental room, at least 1 h before the start of the experiment. In the first experiment, the effect of the test compound alone was examined. The test compound was injected 35 min before observations began as described below. In the second experiment, the test compound was given 30 min before 8-OH-DPAT (1.5 μmol/kg). Five minutes before the 8-OH-DPAT treatment the rat was placed in the test apparatus for habituation. The scoring began 5 min after the 8-OH-DPAT treatment. Five rats were tested at the same time. Each rat was studied during 1 min, once every 5 min for 20 min. The components of the 5-HT-syndrome studied were flat body posture, reciprocal forepaw treading and lower lip retraction, but other symptoms, such as tremor, head weaving, Straub tail and sedation, were noted. The components were scored by use of a ranked intensity scale where: 0 = absent; 1 = equivocal; 2 = present; 3 = intense. The experimenter was blinded to the treatment. Results are expressed as the median sum of reciprocal forepaw treading, flat body posture and lower lip retraction.
Inhibition of cage leaving.
This was performed as described by Rényi et al. (1986). The rats were housed in pairs in plastic cages (26 × 42 × 15 cm) with a sawdust-covered floor, which served as the test apparatus. The rats were first treated with the test compounds. Eighteen minutes later, the grid cover was removed from the cages and the time to climb out of the cage was measured during the next 12 min. If the rats climbed out of the cage, then, 30 min after administration of the test compound, they were injected with 0.3 μmol/kg 8-OH-DPAT. Ten minutes later the grid cover was removed, and the time taken to leave the cages was recorded in the same way.
Temperature measurements.
A YSI 4000A tele-thermometer with a flexible probe was used. The rats were divided into groups of 5. At least 3 h before the experiment, each animal was weighed and marked; the rectal temperature taken by inserting the thermometer probe 10 cm into the rat. At the start of the first experiment, either the test substance or vehicle was administered to each subject. Thirty minutes later the rectal temperature of each rat was taken (time 0) and after each group of five rats had been tested, a challenge dose of 0.9 μmol/kg 8-OH-DPAT was administered to each rat in the group. The rectal temperature was again measured 30 and 60 min after administration of 8-OH-DPAT. The rectal temperature measured 30 min after 8-OH-DPAT, the time corresponding to the maximum hypothermia, is presented here.
Passive avoidance behavior.
A shuttle box was divided equally into a light compartment and a dark compartment. Each compartment measured 21 × 22 cm floor area and the height was 22 cm. Rats were injected with the test compound or saline, 10 min later with 8-OH-DPAT (0.6 μmol/kg) or saline and 10 min later placed individually into the light compartment with no access to the dark side. After 3 min adaptation, the slide door was opened, which gave the rat free access to the dark chamber. When the animal crossed into the dark chamber (criterion, all four feet within the dark compartment), the door was shut and the animal given a scrambled shock (0.4 mA) for 5 sec through the grid floor. The animal was then removed and 24 h later the animal was tested in the absence of drug and the time (latency, seconds) for each animal to cross from the light compartment to the dark compartment determined. The cut-off time was set to 300 sec.
Statistics.
One-way ANOVA followed by Student Neuman-Keuls or Dunnett’s t-test was used for statistical analysis of the corticosterone, temperature, 5-HT, 5-HTP and DOPA results. Cage leaving behavior was compared by use of the Mann-WhitneyU-test. The data are presented as the minimal effective dose, which is the lowest dose used that significantly blocked the effect of 8-OH-DPAT. The 5-HT syndrome and passive avoidance data were analyzed by Kruskal-Wallis ANOVA; when a significant difference was indicated by the ANOVA, between-groups analysis was performed with a Mann-Whitney U-test.
Results
In Vitro Experiments
Receptor binding profile in vitro.
Table2 summarizes the binding affinities of NAD-299 for various receptors and compares them with those of WAY-100635. Both NAD-299 and WAY-100635 had high affinity for 5-HT1A receptors with Ki values less than 1 nM. A representative curve of the displacement of [3H]8-OH-DPAT from 5-HT1A receptors by NAD-299 is shown in figure 2. Apart from an affinity at alpha-1 adrenoceptors of 260 nM and atbeta adrenoceptors of 340 nM the affinity of NAD-299 was less than 1000 nM for a range of other receptors, including serotonergic (r5-HT1B, 5-HT2A, 5-HT6, 5-HT7 and 5-HT uptake site),alpha-2 adrenergic, cholinergic (muscarinic and nicotinic), dopaminergic (D1, D2 and D3), histamine (H1 and H2), GABAA, NMDA, AMPA, benzodiazepine and galanin receptors. WAY-100635 showed considerably higher affinities than NAD-299 for alpha-1 adrenoceptors and DA D2 and D3 receptors.
Comparison of the receptor binding profile of NAD-299 with that of WAY-100635

A dose-response curve of NAD-299 in inhibiting the [3H]8-OH-DPAT binding to rat hippocampal 5-HT1A receptors. Data points are the mean ± S.E. of duplicate determinations from one representative experiment performed as described under “Materials and Methods.” Where not shown, the standard error is smaller than the size of the symbol. The Hill coefficient is 0.98.
Second messenger studies.
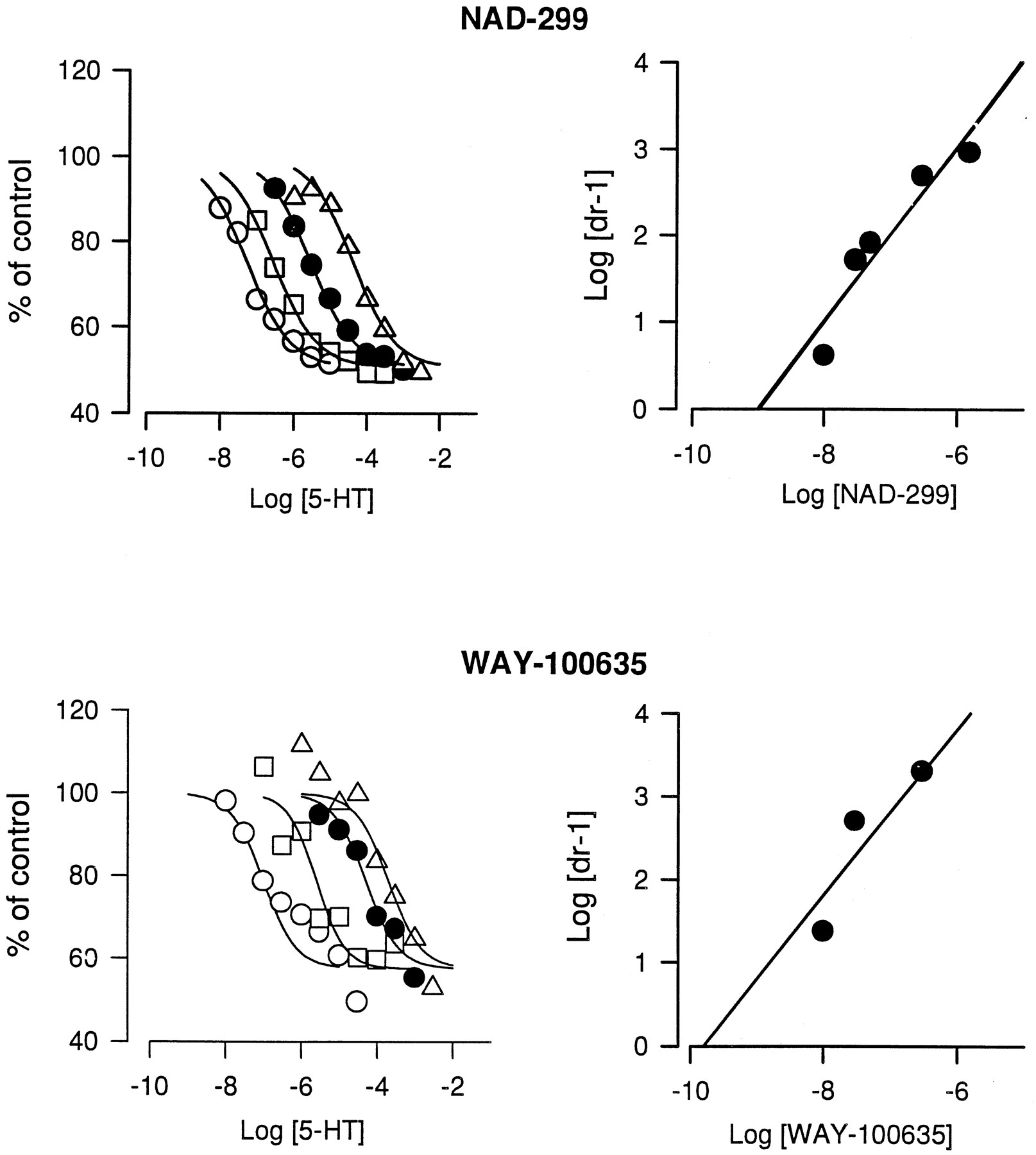
The maximal suppressive effect of 5-HT on VIP-stimulated cAMP production was about 40%. This was achieved at 1 μM 5-HT. Neither NAD-299 nor WAY-100635 exerted any intrinsic activity (efficacy) on the 5-HT1A receptor. This could be concluded from data (expressed as percent of VIP-stimulated cAMP production) obtained with the two putative antagonists at 0.1, 1 and 10 μM. NAD-299 exposure gave 101 ± 4.8, 101 ± 3.9 and 95 ± 3.7 (mean ± S.E., n = 6), respectively; and WAY-100635 exposure gave 103 ± 17, 104 ± 4 and 103 ± 8 (mean ± S.E., n = 2), respectively. The inhibitory effect of 5-HT was fully antagonized by both compounds tested (fig. 3), because the calculated apparent maximal effects of NAD-299 (87%) and WAY-100635 (86%) were not significantly different from 100%. Calculation of EC50 values of the inhibition of 5-HT-induced suppression of cAMP formation indicates that WAY-100635 was about four times more potent than NAD-299 with EC50values of 2 and 7 nM, respectively. The competitive nature of the compounds on 5-HT-inhibited cAMP production were investigated according to the description by Schild (1949). Both putative antagonists produced a parallel displacement to the right of the 5-HT concentration-response curve (fig. 4). The equiactive concentration ratios were then used for the Schild analysis. The slopes of the Schild plots yielded straight lines which did not significantly deviate from unity. The KB values were 1 nM and 0.2 nM for NAD-299 and WAY-100635, respectively.

Concentration-response curves of the antagonism of 5-HT-induced inhibition of VIP-stimulated cAMP production in GH4ZD10 cells. The concentration of VIP was 30 nM, and that of 5-HT was 0.3 μM. The amount of cAMP produced by the VIP stimulation was usually within 20 to 60 pmol/106 cells. The data shown are means of duplicate experiments. The S.E. is indicated by vertical bars.

Parallel displacement of 5-HT concentration-response curves by NAD-299 and WAY-100635 on 5-HT inhibition of cAMP production stimulated by VIP (30 μM) in GH4ZD10 cells. (left-hand figures) concentration-dependent curve shifting of 5-HT response by NAD-299 (top) and WAY-100635 (bottom). The concentrations of the antagonists are from left to right: 0, 10, 30 and 300 nM. (right-hand figures) Schild plots from the data in the left-hand figures. For NAD-299 data from an additional experiment with the concentrations 50, 300 and 1500 nM were included. The data were obtained from 20, 50 and 80% maximum responses for each antagonist concentration. The solid line is based on the experimental data.
In Vivo Experiments
NAD-299 and WAY-100635 were examined for their abilities to antagonize 8-OH-DPAT-induced effects in various in vivo test models. Because the tests required different doses of 8-OH-DPAT to produce an almost maximal effect, the dose used in each test is given in table 3, which summarizes the results of these experiments. The ED50 values were estimated by interpolation of log dose-response curves.
Summary of the 5-HT1A receptor antagonist effects of NAD-299 and WAY-100635 on various 8-OH-DPAT responses in vivo3-a
Antagonism of 8-OH-DPAT-induced 5-HTP accumulation.
8-OH-DPAT (0.3 μmol/kg s.c.) produced an almost maximal decrease in the 5-HTP accumulation under the experimental conditions used (table4). NAD-299 was 6 to 10 times less potent than WAY-100635 in antagonizing the 8-OH-DPAT-induced decrease in 5-HTP accumulation in hypothalamus and striatum (fig.5). NAD-299 by itself, at doses 1 to 30 μmol/kg, had no effect on the 5-HTP accumulation (table 4). WAY-100635 by itself produced a decrease in the 5-HTP accumulation at 5.5 μmol/kg but not at lower doses. However, this effect does not seem to result from a direct action on the somato-dendritic 5-HT1A receptors, because the same dose completely blocked the effect of 8-OH-DPAT on the 5-HTP accumulation (table 4).
The effect of the 5-HT1A receptor antagonists on the 5-HTP accumulation in hypothalamus and striatum in the rat brain

The effect of NAD-299 (circles) and WAY-100635 (squares) on the 8-OH-DPAT-induced changes in 5-HTP accumulation in hypothalamus and striatum of the rat in vivo. The test compounds were injected 15 min before 8-OH-DPAT (0.3 μmol/kg) and 45 min before NSD 1015 (100 mg/kg i.p.). The rats were sacrificed 30 min after the last injection, and the hypothalamus and striatum were rapidly dissected out and frozen on dry ice. The values are means ± S.E. (vertical bars) of 5 to 10 rats. 5-HTP accumulation (nmol/g tissue) in saline controls: 0.93 ± 0.20 (hypothalamus) and 0.37 ± 0.07 (striatum); in 8-OH-DPAT controls: 0.55 ± 0.05 (hypothalamus) and 0.22 ± 0.02 (striatum).
DOPA accumulation.
NAD-299 by itself did not significantly change the rate of DOPA synthesis in hypothalamus and striatum after DOPA decarboxylase inhibition (fig. 6). WAY-100635, on the other hand, produced a marked increase in the DOPA accumulation in striatum at the highest dose tested (5.5 μmol/kg) (fig. 6). However, the increase in DOPA accumulation induced by 8-OH-DPAT at 0.3 μmol/kg was antagonized by NAD-299 and WAY-100635 at 5-HT1A receptor-antagonizing doses.

The effects of NAD-299 (top) and WAY-100635 (bottom) alone and in combination with 8-OH-DPAT on DOPA accumulation in the hypothalamus and striatum of the rat in vivo. The test compounds or saline were injected 15 min before 8-OH-DPAT (0.3 μmol/kg) or saline and 45 min before NSD 1015 (100 mg/kg). The rats were sacrificed 30 min after the last injection, and the hypothalamus and striatum were rapidly dissected out and frozen on dry ice. The values are means ± S.E. (vertical bars) of five rats. The DOPA accumulation (nmol/g tissue) in saline-pretreated rats was: 1.64 ± 0.08 (hypothalamus) and 6.91 ± 0.34 (striatum). * denotes a significant difference, P < .05 vs. saline controls,+P < .05 vs. 8-OH-DPAT controls (Dunnett’s t-test after ANOVA).
Antagonism of 8-OH-DPAT-induced secretion of corticosterone.
8-OH-DPAT at 0.75 μmol/kg produced an almost maximal increase in the corticosterone secretion into the blood circulation (see fig. 8). WAY-100635 was about 3 times more potent than NAD-299 in antagonizing the 8-OH-DPAT-induced corticosterone secretion when injected 15 min before 8-OH-DPAT (0.75 μmol/kg) and the rats were sacrificed 60 min thereafter (fig. 7). The antagonism was competitive, because increased 8-OH-DPAT doses decreased the antagonism at fixed doses of the antagonists (fig.8). The antagonizing effects of NAD-299 (0.5 μmol/kg) and WAY-100635 (0.2 μmol/kg) had disappeared when the compounds were injected 2 h before 8-OH-DPAT, and the rats were sacrificed 1 h later (fig.9).
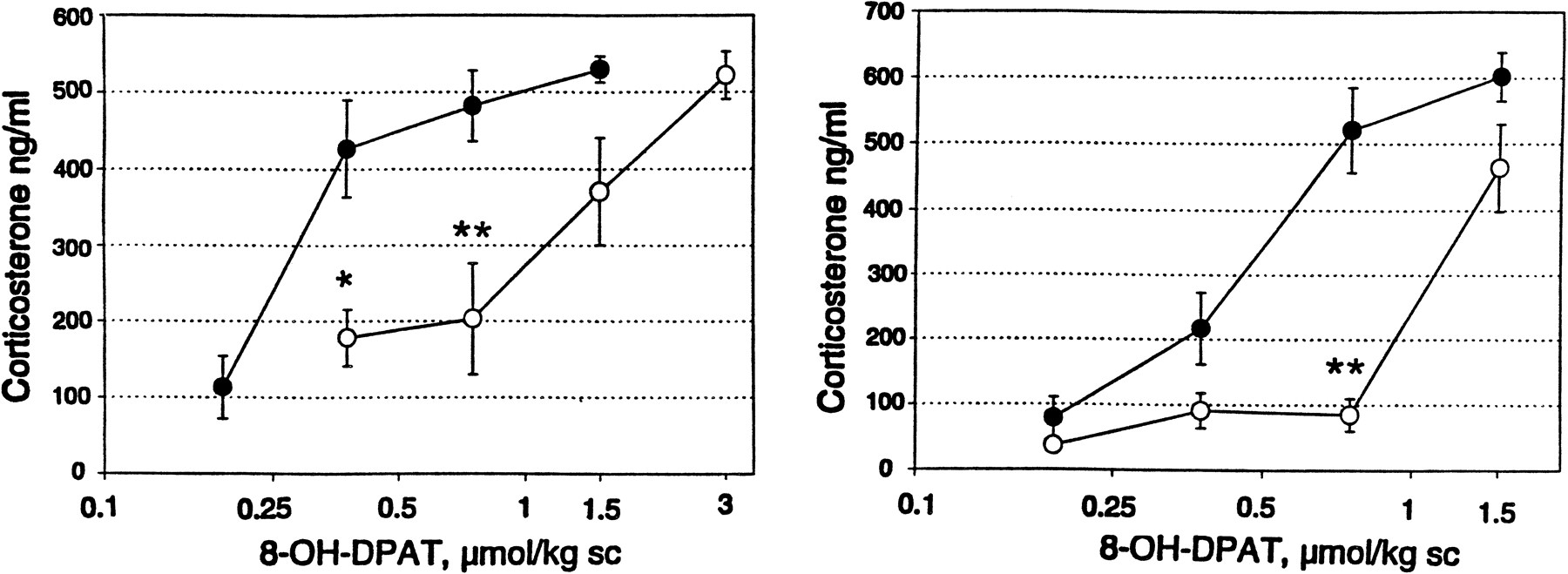
Competitive antagonism by NAD-299 (left) and WAY-100635 (right) of the 8-OH-DPAT-induced corticosterone secretion in the rat. Saline (filled circles), NAD-299 (open circles) (0.1 μmol/kg) or WAY-100635 (open circles) (0.055 μmol/kg) was injected 15 min before the various doses of 8-OH-DPAT and the rats were sacrificed 60 min after the latter injection. Corticosterone in serum was analyzed with the RIA technique. Values are means ± S.E. from five rats. * P < .05; ** P < .01 vs.corresponding saline control (Dunnett’s t-test).

Antagonism by NAD-299 (triangles) and WAY-100635 (circles) of the 8-OH-DPAT-induced secretion of corticosterone in the rat. The test compounds were injected 15 min before the injection of 8-OH-DPAT, 0.75 μmol/kg, and the rats were sacrificed by decapitation 60 min later. The trunk blood was collected, and corticosterone was analyzed with RIA technique. The antagonism is expressed as a percentage of the increase in corticosterone induced by 8-OH-DPAT (378 ± 36 ng/ml; n = 15) in saline-treated rats (control value: 30 ± 6 ng/ml; n = 15). Each value is the mean ± S.E. (vertical bars) of 10 rats and all values greater than the dose 0.01 μmol/kg are significantly different from the 8-OH-DPAT-treated controls (P < .05, Dunnett’st-test after ANOVA).

Time courses of the antagonism by NAD-299, 0.5 μmol/kg (open circles), and WAY-100635, 0.2 μmol/kg (closed circles), of the increase in corticosterone in rat serum induced by 8-OH-DPAT, 0.75 μmol/kg. The test compounds were injected at different times before 8-OH-DPAT, and the rats were sacrificed 60 min after the injection of 8-OH-DPAT. Corticosterone was analyzed with the RIA technique. The antagonism was expressed in percentage of the increase in corticosterone induced by 8-OH-DPAT. Each value is the mean ± S.E. of five rats. * denotes significant difference (P < .05) vs. the 8-OH-DPAT control (Dunnett’st-test).
Flat body posture, forepaw treading and lower lip retraction.
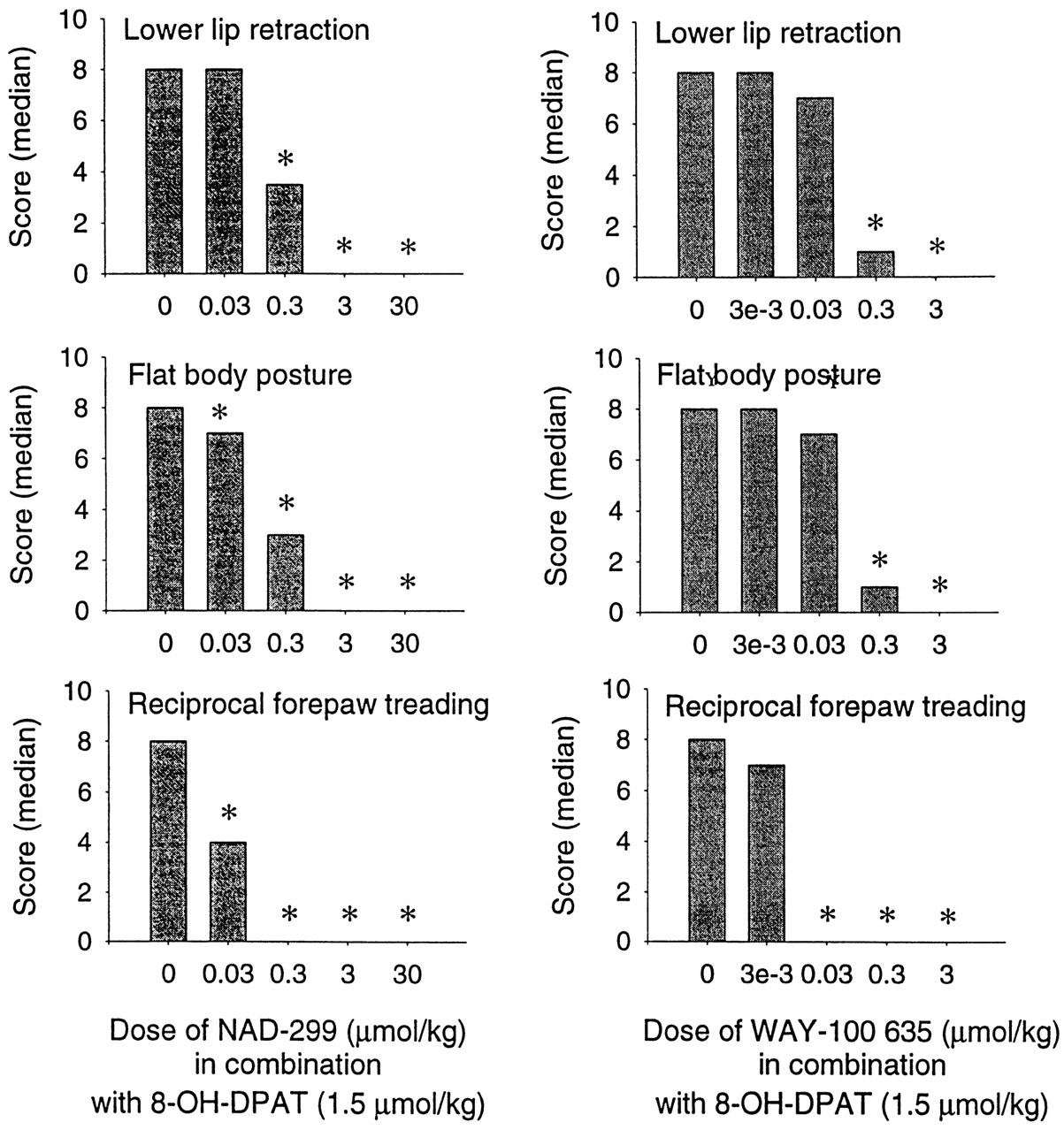
NAD-299 and WAY-100635, at the doses tested, did not exert any effects on the three behavioral measures (data not shown). In a dose finding study with 8-OH-DPAT it was found that lower lip retraction was induced already by 0.09 μmol/kg of 8-OH-DPAT, flat body posture was induced by an intermediate dose (0.4 μmol/kg), but a higher dose (1.5 μmol/kg) was needed to induce forepaw treading. Because 1.5 μmol/kg 8-OH-DPAT induced all components of the 5-HT behavioral effects, it was chosen for the study with the antagonists. NAD-299 at 0.03 μmol/kg significantly antagonized forepaw treading and flat body posture and at 0.3 μmol/kg the lower lip retraction (fig.10). Complete block of all components of the syndrome induced by 8-OH-DPAT was obtained with NAD-299 at 3 μmol/kg.

The effect of acute treatment with NAD-299 and WAY-100635 on three components of the serotonin syndrome induced by acute treatment with 8-OH-DPAT (1.5 μmol/kg). The test compound was administered 30 min before 8-OH-DPAT. The occurrence of each component was scored (from 0 to 3) 5, 10, 15 and 20 min after the last treatment. The data for groups receiving only the test compounds (no 8-OH-DPAT) are not shown (all medians were 0). n = 5 to 12 rats/group. * denotes significant difference from the saline + 8-OH-DPAT group (P < .01, Kruskal-Wallis ANOVA followed by the Mann-Whitney U-test).
Inhibition of cage leaving response.
NAD-299 and WAY-100635 by themselves did not affect the cage leaving behavior in the dose range tested. The inhibition of the cage leaving response by 8-OH-DPAT (0.3 μmol/kg) was antagonized by both compounds. The minimal effective doses (the lowest tested dose that caused significant blockade of the effect of 8-OH-DPAT) was 0.3 μmol/kg for NAD-299 and 0.05 μmol/kg for WAY-100635.
Hypothermia.
Neither of the antagonists themselves affected body temperature (data not shown). The hypothermia-inducing effects of 0.9 μmol/kg 8-OH-DPAT were blocked by the antagonists (fig.11). WAY-100635 was 7 times more potent than NAD-299 in this test (table 3).

Dose-response curves of the antagonism by NAD-299 (top) and WAY-100635 (bottom) of the hypothermia induced by 8-OH-DPAT. Both antagonists were inactive by themselves. The data are the means ± S.E. of 11 to 12 (both control groups) or 6 replicates per group. + denotes P < .01 vs. saline control, * denotes P < .01 vs. saline + 8-OH-DPAT control (Dunnett’s t-test after ANOVA).
Passive avoidance behavior.
8-OH-DPAT and other 5-HT1A receptor agonists block the acquisition of an avoidance response in the passive avoidance paradigm when administered before the training session (Johansson et al., 1989; Carliet al., 1992; Jackson et al., 1994). None of the 5-HT1A receptor antagonists studied had any effect by themselves in the passive avoidance test but antagonized the effect of 8-OH-DPAT (0.6 μmol/kg) (fig. 12). WAY-100635 was about 6 times more potent than NAD-299 when measured as the lowest dose that completely antagonized the effect of 8-OH-DPAT (table 3).

The effect of 5-HT1A receptor antagonists in counteracting 8-OH-DPAT-induced blockade of a passive avoidance learning response in rats. On day 1 the rats were treated with saline, different doses of test compound alone or in combination with 8-OH-DPAT (0.6 μmol/kg) and the learning procedure was performed as described under “Materials and Methods.” On day 2 performance was tested without giving any drugs to the rats. The figure shows the median latency time (sec) of the rats (n = 8 to 12 per group) to run into the dark chamber on day 2. The data were analyzed by Kruskal-Wallis ANOVAs and, in case of a significant effect, pairs of data were compared with use of Mann-Whitney U tests. * indicates a significant (P < .05) difference from control animals, i.e., animals that received only vehicle injections.
Discussion
The in vitro radioligand binding studies with the novel 5-HT1A receptor antagonist, NAD-299 show that it is highly selective for the 5-HT1A receptors. The only other receptors for which the compound had affinity less than 1 μM werealpha-1 and beta adrenoceptors. However, NAD-299 had a selectivity for 5-HT1A receptors vs. thealpha-1 adrenoceptors and beta adrenoceptors of about 400 times. Although the reference antagonist, WAY-100635, is 3 times more potent than NAD-299, it is less selective because of its affinities for alpha-1 adrenoceptors and DA D2Aand D3 receptors. NAD-299 also differs in selectivity from the structurally related 5-HT1A receptor antagonist, (S)-UH-301, that has considerable affinity for D2 receptors (Hillver et al., 1990; Björket al., 1991).
The 5-HT1A receptor antagonistic property of NAD-299 was demonstrated in the in vitro experiments measuring the concentration-dependent block of the inhibitory effect of 5-HT on VIP-stimulated cAMP production in GH4ZD10 cells. The results of Schild analysis of this blockade was found to be consistent with a simple reversible and competitive antagonism. TheKB values were close to theKi values calculated from the results of the binding experiments. Neither NAD-299 nor WAY-100635 had any intrinsic activity in this test, i.e., they were without any agonist effect. The GH4ZD10 cell line was chosen because of its neuronal origin and because the expressed 5-HT1A receptors are a verified model of postsynaptic receptors in rat hippocampus (Fowler et al., 1992). This cell line expresses a low amount of receptors (<50 fmol/mg protein). The low expression level, however, was not the reason for the lack of intrinsic activity since similar results were obtained (data not shown) in a CHO cell line (obtained from Dr. Philip Strange, Canterbury University, UK) containing >1 pmol human 5-HT1A receptor/mg protein. This lack of intrinsic activity was verified in the various in vivo experiments performed. Thus, no decrease in the 5-HTP accumulation in NSD 1015-treated rats, which would indicate stimulation of 5-HT1A receptors, was observed. Because of the large reserve of somato-dendritic 5-HT1A receptors this test is very sensitive to partial 5-HT1A receptor agonists and several compounds, e.g., (−)-pindolol and NAN-190 (1-(2-methoxyphenyl)-4-[4-(2-phtalimido)butyl]piperazine hydrobromide), which behave as antagonists in tests of postsynaptic 5-HT1A receptors, have been found to be partial agonists of the somato-dendritic receptors (Hjorth and Carlsson, 1986; Hjorth and Sharp, 1990). This and other functional in vivo tests,e.g., block of 8-OH-DPAT-induced behavioral effects, hypothermia and corticosterone secretion, confirm that NAD-299, like WAY-100635, is a “silent” 5-HT1A receptor antagonist.
Although these 5-HT1A receptor antagonists have no intrinsic activity, they are not always without any pharmacological effects under in vivo conditions. It has been reported that WAY-100635 can increase the firing rate of raphé 5-HT neurons in extracellular electrophysiological studies in guinea pigs (Mundeyet al., 1996) and UH-301 in rats (Arborelius and Svensson, 1992). These findings indicate that 5-HT may, under certain homeostatic conditions, exert a tonic inhibitory effect which can then be blocked by an antagonist. The observation (table 4) that a high dose (5.5 μmol/kg) of WAY-100635 decreased the 5-HTP accumulation in hypothalamus and striatum may be caused by the effects of WAY-100635 on other neuron systems, e.g., DA or noradrenaline, because WAY-100635 has some affinities to D2 receptors in vitro (table 2) and at this dose also in vivo (fig. 6) and to alpha-1 adrenoceptors in vitro (table 2). Combination of this dose of WAY-100635 with 0.3 μmol/kg 8-OH-DPAT abolished the decrease in the 5-HTP accumulation observed for both compounds alone. The origin of this interaction has not been examined in the present study.
In summary, the development and availability of the novel selective 5-HT1A antagonist, NAD-299, provides a breakthrough from a structurally new chemical class for studies of 5-HT1Areceptor pharmacology in animals and its clinical application in man.
Acknowledgments
The skillful technical assistance of Charlotte Ahlgren, Annelie Bengtsson, Ulla Haglund, Patricia Jimenez, Li-Marie Lindgren, Susanne Rosqvist, Maria Sällemark and Gun Torell-Svantesson is gratefully acknowledged. We thank Dr. Alan Cross, Astra Arcus AB, Rochester, NY, for some of the binding data.
Footnotes
-
Send reprint requests to: Dr Svante B. Ross, Behavioural and Biochemical Pharmacology, Preclinical R & D, Astra Arcus AB, S-151 85 Södertälje, Sweden.
- Abbreviations:
- DA
- dopamine
- DOPA
- l-3,4-dihydroxyphenylalanine
- 8-OH-DPAT
- 8-hydroxy-2-(di-n-propylamino)tetralin
- 5-HT
- 5-hydroxytryptamine
- IBMX
- 3-isobutyl-1-methylxanthine
- 5-HTP
- 5-hydroxytryptophan
- NAD-299
- (R)-3-N,N-dicyclobutylamino-8-fluoro-3,4-dihydro-2H-1-benzopyran-5-carboxamide hydrogen (2R,3R)-tartrate monohydrate
- NSD 1015
- 3-hydroxybenzylhydrazine dihydrochloride
- (S)-UH-301
- (S)-5-fluoro-8-hydroxy-2-(dipropylamino)tetralin
- VIP
- vasoactive intestinal peptide
- WAY-100135
- N-tert-butyl-3-(4-(2-methoxyphenyl)piperacine-1-yl)-2-phenylpropanamide dihydrochloride
- WAY-100635
- N-(2-(1-(4-(2-methoxyphenyl)piperazinyl))ethyl)-N-(2-pyridinyl)cyclohexanecarboxamide trihydrochloride
- CHO
- Chinese hamster ovary
- FCS
- fetal calf serum
- HEPES
- N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- EDTA
- ethylenediaminetetraacetic acid
- ANOVA
- analysis of variance
- AMPA
- dl-α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid
- DHA
- dihydroalprenolol
- GABA
- γ-aminobutyric acid
- NMDA
- N-methyl-d-aspartate
- TBPS
- tert-butylbicyclophosphothionate
- QNB
- l-quinuclidinyl benzilate
- Received December 31, 1996.
- Accepted June 9, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}