


Abstract
Recent studies propose that ς site ligands antagonize N-methyl-d-aspartate (NMDA) receptors by either direct, or indirect mechanisms of inhibition. To investigate this question further we used electrical recordings to assay actions of seventeen structurally diverse ς site ligands on three diheteromeric subunit combinations of cloned rat NMDA receptors expressed inXenopus oocytes: NR1a coexpressed with either NR2A, 2B or 2C. The ς site ligands had a wide range of potency for antagonizing NMDA receptor currents. Steady-state IC50 values ranged between ∼0.1 to >100 μM. In all cases inhibition was non-competitive with respect to glycine and glutamate. Five structurally related ς ligands [eliprodil, haloperidol, ifenprodil, 4-phenyl-1-(4-phenylbutyl)-piperidine and trifluperidol] were strongly selective for NR1a/2B receptors. The other drugs were weakly selective or nonselective inhibitors. There was no correlation between ς site affinity and potency of NMDA receptor antagonism for any subunit combination. Inhibition of NR1a/2B receptors by the selective antagonists was independent of voltage whereas inhibition by the weakly selective antagonists was voltage dependent. Potency of 10 ς ligands was cross-checked on NMDA currents in cultured rat cortical neurons. There was close correspondence between the two assay systems. Our results argue that antagonism of NMDA receptor currents by the ς ligands tested is due to direct effects on the receptor channel complex as opposed to indirect effects mediated by ς receptors. Inhibition occurs via sites in the NMDA receptor channel pore, or via allosteric modulatory sites associated with the NR2B subunit.
The ς receptors are membrane-associated binding sites of unknown physiological function that have been implicated in regulating psychotomimetic behaviors in animal models of schizophrenia (Martin et al., 1976; Su, 1982; Tam and Cook, 1984; Taylor and Dekleva, 1987; Snyder and Largent, 1989; Walker et al., 1990). Radioligand binding studies indicate that there are at least three, and possibly more, types of ς site; denoted ς1, ς2, ς3 etc. (Hellewell and Bowen, 1990; Quirion et al., 1992; Booth et al., 1993). The ς1 site is modulated by guanosine 5′-triphosphate (GTP) and, in rat brain synaptoneurosomes, has effects on phosphatidylinositol turnover stimulated by muscarinic acetylcholine receptor activation (Bowenet al., 1988). A protein with the radioligand binding profile of the ς1 site has recently been cloned (Hanneret al., 1996). The protein is unrelated to any known mammalian receptor. Indeed, the only detected sequence homology is with fungal proteins involved in synthesis of sterols. The ς2site may mediate the motor effects of many ς ligands (Matsumotoet al., 1990), and the regulatory effects of ς ligands on K+ channels (Wu et al., 1991). The ς3 site is less well characterized, but appears to influence dopamine metabolism in rat and guinea pig forebrain (Boothet al., 1993).
A structurally diverse range of compounds bind to ς sites (e.g., Largent et al., 1988; Walker et al., 1990; Rothman et al., 1991; de Costa and He, 1994). Confusion has arisen in the field because many of the early ς ligands interact with other types of receptor (Walker et al., 1990). This has made it difficult to positively ascribe biochemical, physiological and behavioral effects of ς ligands to actions specifically at ς sites. One example of poor specificity is the direct interaction between ς site ligands and NMDA receptors (Zukin et al., 1984; Tam and Cook, 1984; Lockhart et al., 1995).
Previous studies have characterized inhibition of NMDA receptor responses by ς ligands such as pentazocine and DTG (Church and Lodge, 1990; Fletcher et al., 1993). For these drugs inhibition appeared to be consistent with blockade of the channel pore, probably via the PCP site. Sigma ligands such as ifenprodil, belonging to the N-substituted 4-benzyl-piperidine structural class, similarly antagonize NMDA receptors (Karbon et al., 1990;Legendre and Westbrook, 1991; Carter et al., 1991; Churchet al., 1994). For these compounds inhibition is mechanistically distinct from the PCP site and is characterized by pronounced selectivity for receptor subtypes comprised of NR1/2B subunit combinations (Moriyoshi et al., 1991; Monyeret al., 1992; Williams, 1993). The ς-site ligand haloperidol, belonging to the butyrophenone structural class, is also an NMDA receptor antagonist (Fletcher and MacDonald, 1993; Cougenour and Cordon, 1997; Ilyin et al., 1996). In the initial studies (Fletcher and MacDonald, 1993), inhibition of NMDA responses by haloperidol showed dependence on glycine concentration, leading to the proposal that haloperidol is a partial agonist for the glycine coagonist site. In subsequent studies (Ilyin et al., 1996), inhibition of NMDA responses by haloperidol was found to be insurmountable with respect to glycine and glutamate, and, as with ifenprodil, to have selectivity for NR1/2B receptors.
To complicate matters further, low, systemically administered doses of ς site ligands such as DTG and (+)-pentazocine increase the sensitivity of CA3 hippocampal neurons to locally applied NMDA (Monnetet al., 1990, 1992). Other ς site ligands, such as haloperidol and R(+)-3-PPP, inhibit this facillitation, suggesting that the drugs are behaving as ς receptor agonists and antagonists. Similar types of effect are seen in vitro, for example, in the regulation of NMDA-induced release of [3H]-norepinephrine from rat hippocampal slices (Gonzales-Alvear and Werling, 1995; Monnet, et al., 1996), and [3H]-dopamine release from rat striatal slices (Gonzales-Alvear and Werling, 1994). Collectively, these results suggest that ς receptors modulate NMDA receptor function, probably via a second messenger system (Debonnel, 1993; Monnet et al., 1996).
Most recently, indirect mechanisms have also been proposed to explain the inhibitory effects of many ς ligands on NMDA receptors. For example, Yamamoto et al. (1995) report that there is a close correlation between affinity for ς1 sites and potency for inhibition of TCP binding in cultured rat forebrain neurons (Yamamotoet al., 1995). Similarly, Hayashi et al. (1995)report that in cultured rat cortical neurons inhibition of NMDA-induced Ca++ influx by ς site ligands is independent of PCP site potency but correlates well with affinity for ς sites (Hayashiet al., 1995). In each case, the authors suggest that inhibition of NMDA receptors is due to indirect modulation mediated by ς receptors. If correct, this result is important because it means that NMDA receptor inhibition can be used as a simple functional assay of ς receptor activation and inhibition (Walker et al., 1990). However, contrary to these findings, Fletcher et al.(1995) assayed some of the same ς ligands for inhibition of NMDA-activated membrane currents responses in cultured rat hippocampal neurons and concluded that inhibition is due to direct antagonism of NMDA receptors.
To investigate the issue further we used whole cell electrical recordings to assay 17 ς site ligands for inhibition of three cloned NMDA receptor subunit combinations expressed in Xenopusoocytes. To confirm that data from the cloned NMDA receptors is relevant to neuronal receptors we tested 10 ς ligands for inhibition of NMDA responses in cultured rat cortical neurons. We then compared potency, subtype-selectivity and mechanism of NMDA receptor inhibition with previously reported affinities for ς sites and PCP sites, as determined by binding assays. A diverse group of ς ligands was chosen to lessen the chances of generating false correlations due to a limited sample of compounds. A preliminary report of this work has appeared previously (Ilyin et al., 1995).
Methods
Expression of cloned NMDA receptors in Xenopusoocytes.
cDNA clones encoding NR1a, NR2A, NR2B and NR2C rat NMDA receptor subunits were generously provided by Dr. P. H. Seeburg (Heidelberg University, Heidelberg, Germany) (Moriyoshi et al., 1991; Monyer et al., 1992). For the NR1 splice variants we adopt the terminology used in Sugihara et al.(1992), denoting the isoform with the lower case letter. Clones were prepared by standard techniques and cRNA was synthesized with T3 RNA polymerase. Xenopus oocytes were prepared using previously described procedures (Woodward et al., 1995), and were stored in Barth’s medium with composition (in mM): NaCl, 88; KCl, 1; CaCl2, 0.41; Ca(NO3)2, 0.33; MgSO4, 0.82; NaHCO3, 2.4; 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid (HEPES) 5; pH 7.4, with 0.1 mg/ml gentamycin sulfate. Oocytes were injected with between 4 to 40 ng of NMDA receptor-encoding cRNAs. The NR1a + NR2A combination was injected at 1:4 or 1:1 ratios, depending on expressional potency of cRNA preparations. Other subunit combinations were injected 1:1. Drugs were dissolved in Ringer solution with composition (in mM): NaCl, 115; KCl, 2; CaCl2, 1.8; HEPES, 5; pH 7.4. Solutions were applied to oocytes by bath perfusion or using a linear array system of capillary tubes (Hawkinson et al., 1996). Membrane current responses were recorded with a conventional two electrode voltage-clamp.
Neuronal cultures and electrical recordings.
Primary cultures of mixed cortical neurons were prepared using a modification of procedures described previously (Whittemore et al., 1995;Ilyin et al., 1996). Briefly, cortices were obtained from Sprague-Dawley (Charles River, Hollister, CA) rat embryos [gestation day (E) 16–17]. Cells were dissociated by incubation in trypsin followed by light triturization and were plated into poly-d-lysine-coated 35-mm culture dishes at a density of 3 to 5 × 104/cm2 in Neurobasal medium (GIBCO, Gaithersburg, MD) supplemented with 5% fetal calf serum, 0.5 mM l-glutamine and 0.25 μM l-glutamate. Cultures were maintained at 37°C in a humidified incubator (5% CO2/95% air). The first feeding was after 4 days with the same medium minus the glutamate. Subsequent feedings were twice weekly thereafter. Neuronal recordings were made with cultures maintained 5 to 12 days in vitro using methods described previously (Ilyinet al., 1996). The internal pipette solution was either (in mM): 145, CsCl; 10, Cs-HEPES (pH = 7.4); 0.5, CaCl2; 10, EGTA, with 2, adenosine 5′-triphosphate (ATP) and 1, GTP (∼290 mOsmol), or the same solution with the CsCl replaced by 134 KF and the ATP and GTP omitted. Whole-cell recordings were made with an Axopatch 200A amplifier (Axon Instruments, Inc., Foster City, CA). Currents were filtered at 2 kHz and analyzed using software provided by the laboratory of Dr. Ricardo Miledi (University of California, Irvine, CA).
Data analysis.
Data were analyzed as reported in Woodwardet al. (1995) and Ilyin et al. (1996). Briefly, if antagonists gave, or were presumed to give, full inhibition of NMDA responses the concentration-inhibition data were fit with equation 1: where I is the measured current, Icontrol is the current in the absence of antagonist, IC50 is the concentration of drug that causes 50% inhibition of the control response and n is the slope factor of the inhibition curve. If antagonists did not inhibit the NMDA responses fully then concentration-inhibition data were fit with equation 2:
where I is the measured current, Icontrol is the current in the absence of antagonist, IC50 is the concentration of drug that causes 50% inhibition of the control response and n is the slope factor of the inhibition curve. If antagonists did not inhibit the NMDA responses fully then concentration-inhibition data were fit with equation 2:
Drugs.
(+)-SKF 10,047, (−)-SKF 10,047, carbetapentane, DTG, haloperidol, R(+)-3-PPP, S(−)-3-(3-hydroxyphenyl)-N-propylpiperidine (S(−)-3-PPP)ifenprodil, NMDA, (+)-pentazocine, (−)-pentazocine, rimcazole and trifluperidol were from Research Biochemicals Inc. (Natick, MA). BD 1008, 4-IBP, IPAG and 4-PPBP were from Tocris/Cookson (St. Louis, MO). Eliprodil was synthesized and generously provided by Dr. C. F. Bigge (Parke-Davis, Pharmaceutical Research, Ann Arbor, MI). Other drugs and reagents were from Sigma Chemical Co. (St. Louis, MO). Drugs was initially made as dimethylsulphoxide (DMSO) or water stocks over the range 0.01 to 100 mM depending on potency. Ringer solutions were made by 300- to 1000-fold dilution of stocks. At these concentrations DMSO caused <5% inhibition of NMDA responses in oocytes. Structures of ς ligands are given in figure1.

Structures of ς ligands tested.
Results
Inhibition of cloned NMDA receptor responses in Xenopusoocytes.
Inhibition of cloned NMDA receptors by ς site ligands was measured on responses elicited by saturating, or near saturating, concentrations of agonists. The standard holding potential was −70 mV. As described previously (Williams, 1993; Woodward et al., 1995), the NMDA response followed a multiphasic time course consisting of a transient spike of current, due to secondary activation of Ca++-gated Cl− channels, and a slowly developing second phase that corresponds more closely to current passing directly through NMDA receptor channels (fig.2). Oocytes were pretreated for 30 to 60 sec with ς site ligands and potency of inhibition was assessed from reductions in amplitude of the second phase of the response. Potency was expressed in terms of the concentration of antagonist that induced 50% inhibition of the control response (IC50 value). A sample experiment assaying(−)pentazocine on NR1a/2B receptors is shown in figure 2. The 17 ς site ligands assayed in this study inhibited NMDA responses with a wide range of potencies and varying degrees of subunit selectivity. Sample concentration-inhibition curves for four ligands are given in figure 3. The complete set of IC50 values is given in table 1.

Sample records illustrating inhibition of NMDA responses by the ς site ligand (−)-pentazocine. The oocyte was expressing NR1a/2B subunits. Drugs were applied as indicated by bars. In the first five records and in the final record the initial spike of current, due to transient activation of Ca++-gated Cl− channels, has been cropped during preparation of the figure. Current amplitudes were measured at the second, more slowly developing phase (arrow). Small, transient increases in current are apparent upon wash of 3 and 10 μM pentazocine. Responses were separated by 3- to 6-min wash to minimize response rundown. Inward current is denoted by downward deflection; holding potential was −70 mV.

Concentration-inhibition curves showing subunit-selectivity profiles of four ς site ligands: (−)-pentazocine and DTG, two weakly selective antagonists, and 4-PPBP and trifluperidol, two NR2B-selective antagonists. In these and following graphs data points are the mean ± S.E.M. Response amplitudes for NR1a/2A are expressed as a fraction of currents elicited by 10 μM glycine and 100 μM glutamate. Amplitudes for NR1a/2B and NR1a/2C are expressed as fractions of currents elicited by 1 μM glycine and 100 μM glutamate. FR, fractional response. Smooth curves are best fits of equation 1 to the data, except for 4-PPBP and trifluperidol inhibition of NR1a/2B that were fit with equation 2. IC50 values (μM) and slopes for the fits to data for NR1a/2A, NR1a/2B and NR1a/2C receptors are, respectively: (−)-pentazocine, 0.6, −1.1; 0.82, −0.83; 0.28, −0.88; 4-PPBP, 110, −1.3; 2.2, −0.92; 240, −1.4; DTG, 4.7, −1.1; 4.7, −0.80; 6.0, −0.93; trifluperidol, 69, −0.81; 1.2, −1.0; 390, −1.0 (see table1). The minimum values (equation 2) for 4-PPBP and trifluperidol on NR1a/2B receptors were 0.05 and 0.06, respectively.
Inhibition of NMDA-activated membrane current responses by ς site ligands
Five compounds had >50-fold subunit-selectivity for inhibition of NMDA receptor currents. In each case selectivity was directed towards NR1a/2B subunit combinations. The selective inhibitors were eliprodil, haloperidol, ifenprodil (see also, Williams, 1993; Ilyin et al., 1996; Kew et al., 1996), and the haloperidol analogues 4-PPBP and trifluperidol. Antagonism of NR1a/2B receptors by these selective antagonists was characterized by incomplete or biphasic inhibition curves. For ifenprodil, between 10 to 15% of the current was not blocked upon saturation of the high affinity component (Williams, 1993; Kew et al., 1996). For the other drugs, demonstrating a second component of inhibition was prevented by limited solubility (Ilyin et al., 1996); these curves appeared incomplete, with 5 to 15% of the current remaining unblocked. In oocytes from some frogs, 30 to 100 μM haloperidol selectively induced 10 to 100% potentiation of NR1a/2A receptor responses (Ilyin et al., 1996). The effect was particular to haloperidol and was not investigated further. Two compounds had weak subunit selectivity;i.e., >3-fold but <10-fold. These were carbetapentane, which was slightly more potent against NR1a/2A compared to NR1a/2C, and BD 1008, which had some selectivity for NR1a/2B. DTG, IPAG, rimcazole, the two enantiomers of 3-PPP, SKF 10,047, and pentazocine, were essentially nonselective; i.e., potencies varied by <3-fold between different subunit combinations. Inhibition of NMDA responses by the more potent nonselective inhibitors was complete. Overall, potency of inhibition ranged between ∼100 nM for (+)-SKF 10,047 to >100 μM for a number of antagonists. One compound, 4-IBP, was essentially inactive against all subunit combinations up to its solubility limit in saline of ∼30 μM.
The issue of ς receptor agonism vs. antagonism was addressed by assaying for interactions between ς site ligands. Specifically, we tested whether inhibition of NR1a/2C responses by (+)-SKF 10,047 was affected by haloperidol, and whether inhibition of NR1a/2C responses by DGT was affected by 4-IBP. Expressed in terms of fractional currents, NR1a/2C responses were reduced to 0.75 ± 0.01 by 0.1 μM (+)-SKF 10,047. The fractional response upon coapplication of 100 μM haloperidol was 0.74 ± 0.02 (n = 4). Similarly, NR1a/2C responses were reduced to 0.85 ± 0.02 by 1 μM DTG, and were 0.81 ± 0.02 upon coapplication of 30 μM 4-IBP (n = 4). Coapplication of the second ς ligand had no significant effect on levels of inhibition in either case (P > .7 and > .2, respectively). Also, we tested whether 1- to 2-min incubations in 1 or 10 nM DTG increased steady-state NMDA currents (Monnet et al., 1990,1992). The fractional current for NR1a/2A, NR1a/2B and NR1a/2C receptors was 0.98 ± 0.02, 0.98 ± 0.01, and 0.97 ± 0.01, respectively, with 1 nM DTG, and 0.99 ± 0.01, 0.97 ± 0.02, and 0.98 ± 0.01, respectively, with 10 nM DTG (n = 3 and 4). In no case was there an increase in current.
Mechanisms of inhibition on cloned NMDA receptors in oocytes.
Mechanism of antagonism was investigated in three ways: 1) by testing whether inhibition was dependent on agonist concentration, 2) by testing if potency of inhibition was dependent on membrane voltage and 3) for selected compounds, testing if washout of inhibition was dependent on channel activation.
1) As a quick check for dependence on agonist concentration we selected a concentration of antagonist that caused 40–80% inhibition of control currents and then tested whether simultaneously increasing glycine and glutamate concentrations by 10-fold, or in some cases 100-fold, changed this level of inhibition. For the non-selective and weakly selective compounds these assays were done using NR1a/2C receptors. Here, agonist concentrations were changed from 1 μM glycine and 100 μM glutamate, to 100 μM glycine and 1 mM glutamate. For compounds with high levels of subunit-selectivity assays were done using NR1a/2B receptors. Here, agonist concentrations were changed from 10 μM glycine and 100 μM glutamate, to 100 μM glycine and 1 mM glutamate. Results are summarized in figure 4. Only two ς site ligands showed significant changes in apparent potency upon raising agonist concentrations. These were rimcazole, which had an 8% reduction in potency for NR1a/2C (P = .02), and ifenprodil, which had an 11% reduction in potency for NR1a/2B (P = .05). The other compounds all showed <10% changes in apparent potency upon raising agonist concentrations (P > .22).

Effect of increasing agonist concentration on inhibition of NMDA responses by ς site ligands. Histograms indicate levels of inhibition induced ς site ligands (abbreviated) at the fixed concentrations indicated. Inhibition expressed as a fractional response. Solid bars are with control concentrations of glycine and glutamate, open bars are with 10- or 100-fold increases in agonist concentrations (see text). Nonselective antagonists were tested using NR1a/2C receptors (upper and middle panels), selective antagonists were tested using NR1a/2B receptors (lower panel). *indicates P < .05;n = 3 to 5 for each antagonist.
Voltage dependence was assessed by selecting a concentration of antagonist that caused a 40 to 75% reduction in current at the standard holding potential of −70 mV, and then measuring levels of inhibition caused by the same concentration of antagonist at −20 and −110 mV. Experiments were done using NR1a/2B or NR1a/2C receptors. Sample records comparing voltage dependence of inhibition for (−)-SKF 10,047 and 4-PPBP are given in figure 5. Results are summarized in figure 6. All the inhibitors that had no subunit-selectivity, or were only weakly selective, inhibited the NMDA responses in a voltage-dependent manner (P = 7 × 10−6 to .01). In contrast, the five compounds that showed strong selectivity inhibited NR1a/2B receptors by a mechanism that was independent of voltage (P = .49–.81). When three of these were tested on NR1a/2C receptors, however, the low potency inhibition was found to be voltage-dependent (P = 5 × 10−5 to .024).

Sample records illustrating assays to test voltage-dependence of inhibition for (−)-SKF 10,047 and 4-PPBP. Records were taken from a single oocyte expressing NR1a/2B receptor subunits. (upper records) Levels of inhibition induced at a holding potential of −20 mV. Currents were measured on the second phase of the response (arrow). (lower records) Inhibition induced by the same concentration of antagonist at a holding potential of −110 mV.

Current-voltage relationships for inhibition of NMDA receptors by ς site ligands. (upper panels) Voltage-dependence of inhibition for weakly selective and nonselective antagonists. Experiments were done in oocytes expressing NR1a/2C receptor subunits. (lower panels) Voltage-dependence of inhibition for strongly selective antagonists. (left panel) Low potency inhibition was assayed in oocytes expressing NR1a/2C receptor subunits. (right panel) High potency inhibition was assayed in oocytes expressing NR1a/2B receptor subunits. Inhibition was induced by fixed concentrations of ς ligands as indicated; n = 3 to 5 for each data point.
Studying use-dependence for the washout of NMDA response antagonism in oocytes was restricted by the slow speed of drug application and wash. Only in the case of potent inhibitors, with slow dissociation kinetics, was it possible to distinguish inhibitor unbinding from the washout rate of the perfusion system. For the strongly selective inhibitors, previous studies had showed that inhibition by ifenprodil and haloperidol was not associated with use-dependence; at least, not in the sense of open channel or channel-trap blockade (Williams, 1993;Ilyin et al., 1996). In our study we assayed use-dependence of washout for the four most potent nonselective inhibitors; the two isomers of pentazocine and SKF 10,047. For each drug washout of inhibition required activation of the NMDA receptor-channel complex. Washing the oocyte for 4 to 6 min in the absence of receptor activation was almost wholly ineffective at reducing levels of inhibition, whereas the same interval of wash coupled with receptor activation was sufficient to completely remove blockade. These experiments were done on NR1a/2C receptors that have the most stable steady-state phase of response. Sample records from an experiment with (+)-SKF 10,047 are given in figure 7.

Sample records illustrating use-dependence of washout for inhibition induced by (+)-SKF 10,047 on NR1a/2C receptors (upper panel). Inhibition with 5 μM (+)-SKF-10,047 washed out in 5 min with the continuous presence of agonist (lower panel). Washing the oocyte for 5 min in the absence of agonist failed to reverse inhibition. Washout of inhibition occurred only upon reactivation of the channel.
Inhibition of NMDA responses in cultured rat cortical neurons.
Neuronal NMDA responses had similar amplitudes and time courses to those described previously (e.g., Fletcher et al., 1993, 1995; Ilyin et al., 1996). IC50values of ς site ligands were measured at a holding potential of −70 mV on the plateau phase of the response. Assays of inhibition were made using cultures that had been maintained for <12 days in vitro, when the neurons are expressing predominantly NR1/NR2B subunits (Zhong et al., 1994; Ilyin et al., 1996). Ten ς ligands, covering a wide range of potencies on the cloned NMDA receptors, were assayed for antagonism of neuronal NMDA responses. Results are given in table 1. IC50 values from the neuronal recordings corresponded closely to the values for inhibition of NR1a/2B receptors. We also tested whether low concentrations of DTG had any positive modulatory effects on the neuronal NMDA responses. Neurons were treated with DTG for 0.5 min during established steady-state responses. Under these conditions the current was unaffected by 3 and 10 nM DTG (n = 3) (not illustrated). Thresholds for the inhibitory effects of DTG were approximately 0.3 μM.
Discussion
Inhibition of NMDA receptors by ς site ligands.
Recent reports offer conflicting explanations as to the mechanism by which ς site ligands inhibit NMDA receptors. On the one hand, binding and Ca++ imaging studies suggest that inhibition is mediated indirectly via ς sites (Hayashi et al., 1995; Yamamotoet al., 1995). On the other, electrophysiological studies indicate that inhibition is due to direct effects on NMDA receptors (Fletcher et al., 1993; Fletcher et al., 1995). We reasoned that NMDA receptor subtype-selectivity might reconcile some of the discrepancies in data and differences in interpretation that have arisen between previous studies. Instead of reconciling differences, however, our results strongly support the position that, for the seventeen ς ligands tested, inhibition of NMDA responses results from direct actions on the receptor-channel complex. The reasons are as follows: 1) We find no correspondence between patterns of NMDA receptor subtype-selectivity and differences in ς subtype-selectivity. 2) We find no correlation between potency of NMDA receptor inhibition and affinity for ς sites. 3) Antagonism by the non-selective, or weakly selective, NMDA receptor inhibitors is voltage-dependent and, for the more potent antagonists, is use-dependent. Both effects imply involvement of binding sites located in a transmembrane channel pore. 4) Antagonism by the NR2B-selective inhibitors (ifenprodil, trifluperidol, eliprodil, haloperidol and 4-PPBP) correlates with potency for displacement of radiolabeled ifenprodil, where the ifenprodil binding site is associated with the NMDA receptor.
Patterns of NMDA receptor subtype-selectivity.
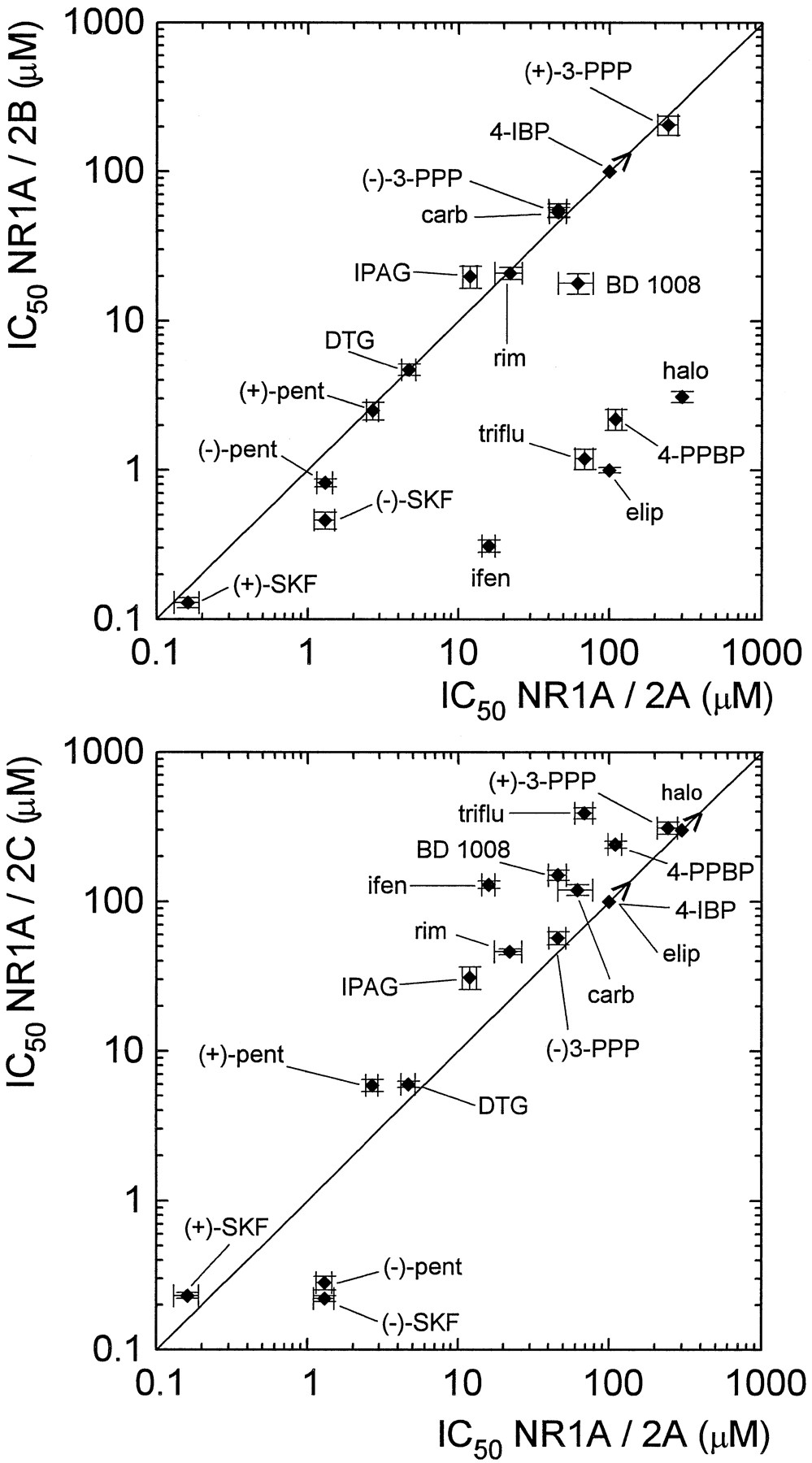
Plotting the correlation of potency for inhibition of NR1a/2A or NR1a/2C responsesvs. potency for NR1a/2B responses illustrates the point that five of the ς site ligands are selective for NR1a/2B, whereas the remaining compounds are essentially non-selective (fig. 8, upper panel). The correlation coefficient (r) for the latter group is 0.98 (n = 11, with calculations restricted to pairs of definite values). The slope shows highly significant deviation from zero (P < .0001). Plotting the correlation of potency for NR1a/2A vs. NR1a/2C illustrates that none of the compounds tested has strong selectivity for these receptor subtypes (r = 0.98, P < .0001,n = 14) (fig. 8, lower panel).

Upper panel, correlation between potency of inhibition for NR1a/2A and NR1a/2B receptors. Lower panel, correlation between potency of inhibition for NR1a/2A and NR1a/2C receptors. In this and next graphs, > denotes an indefinite minimum value. The direction of the symbol (vertical, horizontal or diagonal) indicates which value, or whether both values, are indefinite.
Plotting potency of NR1a/2B antagonism vs. potency of inhibition of rat neuronal NMDA responses also gives a strong correlation; values measured herein for cortical neurons and reported previously for rat hippocampal neurons (Fletcher et al., 1995) (table 2), (r = 0.97, P < .0001, n = 10 for the cortical neurons alone:r = 0.93, P < .0001, n = 18 including data from hippocampal neurons) (fig. 10, upper panel). Thus, if NMDA receptor inhibition in neurons occurs via ς sites, then the same mechanism of action would appear to be implicated in the oocyte recordings and it is difficult to argue that results from the cloned receptors are irrelevant when it comes to considering effects on neuronal NMDA responses.
Binding affinity of ς ligands to ς1, ς2 and PCP sites; comparison with potencies in assays of whole cell [3H]-TCP displacement, inhibition of NMDA-induced Ca++ signals and inhibition of NMDA-activated membrane currents

Upper panel, Correlation between potency of inhibition for NR1a/2B receptors and neuronal NMDA receptors. Solid symbols, inhibition of NMDA responses in rat cortical neurons cultured for <12 days. Open symbols, inhibition of NMDA responses in rat hippocampal neurons 5 to 15 days in vitro (table 2) (lower panel). Correlation between potency of NR1a/2B receptor inhibition and affinity for the NMDA receptor PCP site (table 2).
All the ς site ligands tested would be expected to affect the function of ς receptors, either as agonists or antagonists. Some of the compounds show selectivity for the ς1 receptor subtype although others are nonselective, or have uncharacterized patterns of selectivity (Rothman et al., 1991; Quirionet al., 1992). It follows that common mechanisms of action at ς receptors should result in common patterns of selectivity for inhibition of NMDA receptor subtypes. This raises serious complications in explaining our results. For example, our experiments indicate that there at least two distinct mechanisms by which ς ligands inhibit NMDA receptors; resulting in selective and the nonselective patterns antagonism. If all this inhibition is mediated by ς sites (Hayashiet al., 1995; Yamamoto et al., 1995), it becomes necessary to postulate two types of ς receptors each coupled to a distinct inhibitory pathway. Obvious difficulties are that carbetapentane, (+)-pentazocine and (+)-SKF 10,047 are selective ς1 ligands and are nonselective NMDA receptor antagonists, whereas haloperidol and its analogue 4-PPBP are nonselective ς site ligands but are selective for NR1a/2B. If haloperidol, for example, interacts with ς1 sites, then why doesn’t it inhibit NR1a/2A and NR1a/2C receptors? The facile answer would be that haloperidol is a ς1 site antagonist, although the other ligands are agonists (Walker et al., 1990; Debonnel, 1993). However, if this were the case, haloperidol should reduce the inhibitory effects of (+)-SKF 10,047 on NR1a/2C receptors. Our experiments indicate that there is no such interaction.
Correlations between potency of NMDA receptor inhibition and ς site affinity.
Plotting potency for inhibition of any of the three NMDA receptor subtypes against affinity for ς1 sites gives no positive correlation between these two activities. In particular, there is no positive relationship where the correlation curve is displaced to the right, which might have been anticipated considering that we are comparing affinities from binding assays with potencies in a functional assay (Bowen et al., 1988; Walkeret al., 1990). Indeed, the only relationship is a negative trend that does not achieve significance (e.g.,r = 0.47, P = .092, for NR1a/2A, n= 14; r = 0.14, P = .61, for NR1a/2B,n = 16) (tables 1 and 2) (fig. 9). The negative trend is probably trivial, arising because the second generation of ς ligands were specifically designed to have high ς potency and weak activity at PCP sites. Some of the more obvious disparities in the data are: 1) The stereoselectivity of benzomorphans for ς1 sites is not evident for the inhibition of NMDA responses. (+)-Pentazocine and (+)-SKF 10,047 have 15- to 30-fold higher affinity for ς1 sites than their (−)-enantiomers (Hellewell and Bowen, 1990; Rothman et al., 1991), whereas (+)-pentazocine is 6-fold weaker for inhibition of NMDA responses than the (−)-enantiomer. The two SKF 10,047 enantiomers have comparable potency. 2) The stereoselectivity of 3-PPP for ς1 sites is reversed for inhibition of NMDA responses; R(+)-3-PPP has 6-fold higher affinity for ς1 sites than S(−)-3-PPP but is 5-fold weaker in the NMDA receptor assays. 3) A number of high potency ς1 ligands are either weak or inactive in terms of NMDA receptor inhibition; these include carbetapentane, BD 1008, 4-IBP and R(+)-3-PPP. For these compounds the discrepancy between ς1 affinity and the IC50 for NMDA responses ranges between 9000- to 60,000-fold.

Upper panel, Correlation between potency of NR1a/2B receptor inhibition and affinity for ς1 receptors. Lower panel, correlation between potency of NR1a/2A receptor inhibition and affinity for ς1 receptors.
Making similar plots of NMDA receptor inhibition vs.affinity for ς2 sites again gives only nonsignificant negative correlations (e.g., r = 0.34, P = .31, for NR1a/2A, n = 11; r = 0.25, P = .41, for NR1a/2B, n = 13) (tables 1 and2) (not illustrated). In this case, some of the more obvious discrepancies are: 1) The benzomorphans (+)-pentazocine and (+)-SKF 10,047 have low affinity for ς2 sites but are among the most potent antagonists of NMDA receptors. 2) 4-IBP is a potent ligand for ς2 receptors but does not antagonize NMDA receptors. Moreover, 4-IBP does not reduce inhibition of NR1a/2A receptors by DTG, an effect that would be predicted if it is a ς2antagonist and if inhibition by DTG is mediated by ς2activation. 3) R(+)-3-PPP has high affinity for ς2 sites but is a very weak inhibitor of NMDA responses.
We had thought that potency of the NR1a/2B-selective inhibitors might correlate with one or other of the ς binding sites. However, within this group, potency follows an inverse correlation with ς1 affinity; haloperidol is at least 10-times more potent than ifenprodil at ς1 sites but is 10-fold less potent as an inhibitor of NMDA receptors. Similarly, haloperidol and 4-PPBP are 15- to 50-fold more potent than trifluperidol and eliprodil as ligands for ς2 receptors, but are appreciably weaker at inhibiting NMDA responses (table 2). Affinities for this group of compounds at ς3 sites is not available.
Voltage-dependence and use-dependence of NMDA receptor inhibition.
All the ς ligands tested are noncompetitive NMDA receptors antagonists with respect to glutamate and glycine. Thus the compounds are not ligands for the agonist or coagonist sites. The small changes in apparent potency seen with rimcazole and ifenprodil are presumably due to allosteric interactions between sites (Williams, 1993; Kew et al., 1996). Inhibition by all the nonselective or weakly selective antagonists is voltage dependent. For the high potency antagonists it is also possible to demonstrate use-dependence for washout of the inhibition. The straightforward explanation for the voltage-dependence and use-dependence is that these ς ligands bind to sites in the NMDA receptor channel pore (Huettner and Bean, 1988;MacDonald et al., 1991). For the inhibition to be indirect, mediated by ς receptors, one would have to propose that activation of ς receptors causes release of a diffusible messenger molecule that then binds to a site in the pore. No such molecules or mechanisms are known. Moreover, for the nonselective NMDA antagonists, if you plot IC50 for NMDA responses versus affinity for the PCP site there is a good correlation (r = 0.97, P < .0001,n = 8) (fig. 10) (table 2). Therefore, for the majority of the nonselective antagonists, proposing ς-mediated inhibition of NMDA receptors is simply unnecessary.
Inhibition of NR1a/2B receptors by the selective antagonists is independent of voltage. This mechanism is, therefore, quite distinct from inhibition at the PCP site, or at other sites deep in the channel pore (Williams, 1993; Ilyin et al., 1996). In contrast, the low potency inhibition of NR1a/2C receptors, and also NR1a/2A receptors (E. R. Whittemore, V. I. Ilyin and R. M. Woodward, unpublished data), by ifenprodil, trifluperidol and 4-PPBP does show voltage-dependence. It would appear that these ligands bind with low affinity to a second site that is indeed located in the pore (Williams, 1993). We suspect that the second site is well conserved between the various subunit combinations, although it is difficult to assay for NR1a/2B because most of the response has been inhibited via the high potency, voltage-independent, mechanism.
Ifenprodil binding studies.
Radiolabeled ifenprodil shows multicomponent binding to rat brain membranes (Mercer et al., 1993; Hashimoto and London, 1993, 1995; Coughenour and Cordon, 1997). In the presence of a saturating concentration of a nonselective ς site ligand, a high affinity component of ifenprodil binding remains and is modulated by polyamines in a manner consistent with a site located on the NMDA receptor complex (Mercer et al., 1993; Coughenour and Cordon, 1997). Eliprodil, haloperidol and trifluperidol all displace this component of ifenprodil binding, and the potency and rank order of inhibition correspond closely to potency for inhibition of NR1a/2B receptors (Coughenour and Cordon, 1997). These studies provide further evidence that the subtype-selective inhibitors interact directly with NMDA receptors. In addition, the binding studies suggest that either the sites of interaction for butyrophenones such as haloperidol overlap with those for ifenprodil, or that there is strong negative allosteric coupling between the two sites.
Previous studies proposing indirect inhibition of NMDA receptors.
Discrepancies between our data and the previously reported [3H]-TCP binding studies are most striking the two enantiomers of SKF 10,047 and (−)-pentazocine, which are between 20- to 100-fold weaker in the binding assays, and for R(+)-3-PPP, which is ∼15-fold more potent (table 2) (Yamamoto et al., 1995). Reasons for the differences in potency are unclear. In particular, it is difficult to account for SKF 10,047 and (−)-pentazocine having such weak activity in the whole cell [3H]-TCP binding studies when our experiments and previous studies indicate that the drugs are potent channel blockers (e.g., Tam and Zhang, 1988; Lockhartet al., 1995; Fletcher et al., 1995; Hayashiet al., 1995). From our perspective, any proposed role for ς receptors in the [3H]-TCP binding data for SKF 10,047 and (−)-pentazocine would have to be to reduce sensitivity of NMDA receptors to the direct channel blocking effects of the molecules, rather than to mediate indirect inhibition! We find little discrepancy between IC50 values of ς ligands tested in our study and steady-state potencies reported previously from NMDA-induced Ca++ mobilization assays (Hayashi et al., 1995) (table 2). For these compounds we think that inhibition ascribed to ς receptors can be readily explained by direct antagonism of NMDA receptors.
Positive modulation of NMDA receptor function by ς site ligands.
The only compound that potentiated NMDA receptor responses was haloperidol. This effect was not seen in all oocytes, was specific to the NR1a/2A subunit combination, and only occurred at micromolar concentrations of haloperidol. In the short term, we did not detect potentiation of steady-state NMDA responses with nanomolar concentrations of DTG. Our experiments suggest that the increase in sensitivity of CA3 hippocampal neurons to NMDA induced by ς site ligands in vivo is not due to direct facilitation of NMDA receptor currents (Monnet et al., 1990; Debonnel, 1993). The same holds for effects of ς ligands on NMDA-induced release of catecholamines from hippocampal and striatal slices (Gonzales-Avear and Werling, 1994, 1995; Monnet et al., 1996). For both types of experiments we would suggest that ς receptors affect processes at some stage downstream from the NMDA receptors. In addition, our experiments failed to provide any support for a recent study suggesting that low nanomolar, or subnanomolar, concentrations of haloperidol directly potentiate NMDA receptor function in rat forebrain (Banerjeeet al., 1995).
Neuroprotective properties of ς ligands.
Drugs such as DTG, eliprodil, ifenprodil, haloperidol and 4-PPBP have neuroprotective properties in models of excitotoxicity and acute cerebral ischemia (e.g., MacDonald and Johnston, 1990; Carter et al., 1991; Takahashi et al., 1995; Takahashi et al., 1996). All these drugs antagonize NMDA receptors. In conjunction with previous studies (Fletcher et al., 1993;Kirk et al., 1994), our results raise the issue whether the neuroprotective effects of ς ligands are solely due to NMDA receptor antagonism. Recent in vitro studies indicate that the explanation is probably more complex (Lockhart et al., 1995). Specifically, protection against hypoxia-induced neurotoxicity by ς ligands does not correlate with NMDA receptor antagonism, suggesting that more than one neuroprotective mechanism is involved. What role ς site activation or inhibition actually plays in these additional mechanisms remains uncertain. Still, the surprising potency of drugs such as eliprodil and 4-PPBP in animal models of focal ischemia raises the possibility that a combination of NMDA receptor antagonism and appropriate interactions at ς sites may be a favorable profile for a neuroprotectant (Carter et al., 1991;Takahashi et al., 1995; Takahashi et al., 1996).
Conclusion.
Antagonism of NMDA receptors by the ς site ligands tested in this study is due to direct actions at two, or more, sites on the receptor channel complex. Nonselective antagonism is due to blockade at the PCP-site, or at other sites in the channel pore. Subtype-selective antagonism is mediated by allosteric modulatory sites associated with the NR2B subunit.
Acknowledgments
The authors thank Dr. P. H. Seeburg for the gift of cDNAs encoding NMDA receptor subunits, and Drs. J. Guastella and J. A. Drewe for synthesis cRNA. We extend especial thanks to Dr. W. D. Bowen for critical reading of the manuscript, helpful suggestions, and for providing much of the ς ligand binding data presented in this paper, some of which has not been published previously.
Footnotes
-
Send reprint requests to: Dr. Richard Woodward, Cocensys Pharmaceuticals Inc., Hitachi Chemical Research Center, 1003, Health Sciences Road West, Irvine, CA 92715.
- Abbreviations:
- BD 1008
- N-[2-(3,4-dichlorophenyl)-ethyl]-N-methyl-2-(1-pyrrolidinyl)ethylamine
- DTG
- 1,3-di(2-tolyl)guanidine
- 4-IBP
- N-(N-benzyl-piperidin-4yl)-4-iodobenzamide
- IPAG
- 1-(4-iodophenyl)-3-(1-adamantyl)guanidine
- NMDA
- N-methyl-d-aspartate
- PCP
- phencyclidine
- 4-PPBP
- 4-phenyl-1-(4-phenylbutyl)-piperidine
- R(+)- and S(−)-3-PPP
- R(+)- and S(−)-3-(3-hydroxyphenyl)-N-propylpiperidine
- (+)- and (−)-SKF 10
- 047, (+)- and (−)-N-allylnormetazocine
- HEPES
- N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid
- Received October 24, 1996.
- Accepted March 7, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}