


Abstract
In an effort to correlate the recently cloned MOR-1 receptor with the pharmacological actions of morphine and morphine-6β-glucuronide (M6G), we have used an antisense paradigm. Rats were injected intracerebroventricularly (i.c.v.) with antisense oligodeoxynucleotides on days 1, 3 and 5 and tested for analgesia on day 6 after administration of morphine or M6G i.c.v. or after microinjection of morphine directly into either the periaqueductal gray or the locus coeruleus. When given i.c.v., the antisense oligodeoxynucleotide targeting the 5′-untranslated region of exon 1 significantly decreased the analgesic actions of morphine administered i.c.v. or microinjected directly into the periaqueductal gray or locus coeruleus, with the most profound inhibition occurring in the periaqueductal gray. Thus, antisense oligodeoxynucleotides administered into the lateral ventricle can diffuse into the brainstem and interfere with morphine actions. A mismatch antisense oligodeoxynucleotide with the same base composition in which the sequence of four bases was changed was inactive. This same exon 1 antisense oligodeoxynucleotide, which was active against morphine analgesia, failed to block M6G analgesia. In contrast, antisense sequences from exons 2 and 3 decreased M6G, and not morphine, analgesia. The antisense oligodeoxynucleotide against exon 4 slightly decreased both morphine and M6G antinociception. These results confirm the antisense mapping studies on exons 1, 2 and 3 of MOR-1 in mice, which implied the presence of a novel μ receptor subtype responsible for M6G analgesia that may represent a splice variant of MOR-1. Unlike in mice, the probe against exon 4 had a small effect on M6G analgesia.
Morphine is a potent analgesic when administered into any one of a number of brainstem structures (Tsou and Jang, 1964; Jacquet and Lajtha, 1973; Pert and Yaksh, 1974;Jensen and Yaksh, 1986; Smith et al., 1988; Bodnar et al., 1988, 1990). Further studies have established complex interactions among many of these regions, with profound synergy when morphine is given into more than one brainstem structure (Rossiet al., 1993, 1994b). For example, multiplicative interactions are observed between the PAG, the LC and the rostro-ventral medulla. Mapping of these brainstem regions has been done primarily with morphine, a selective μ-opioid (for review, seePasternak, 1993). Other opioid receptors also can elicit analgesia within the brainstem. Although κ1 agonists are inactive in the PAG, the δ2 agonist [d-Ala2,Glu4]deltorphin can produce modest analgesia (Rossi et al., 1994b).
M6G is an exceptionally potent analgesic in the brain, with an activity >100-fold greater than that of morphine (Shimomura et al., 1971; Pasternak et al., 1987; Abbott and Palmour, 1988;Sullivan et al., 1989; Paul et al., 1989). However, this extraordinary activity in vivo contrasts with its potency in binding assays, where M6G labels the traditional μ receptors slightly less potently than does morphine (Paul et al., 1989). Similar results are seen in Chinese hamster ovary cells stably expressing the μ-opioid receptor clone MOR-1 (G. Brown and G. W. Pasternak, unpublished observations). This discrepancy between functional activity and binding affinity cannot be explained by differences in efficacy between the two drugs. In human neuroblastoma cell lines expressing μ receptors, both morphine and M6G inhibit cyclase, with potencies corresponding to results from the binding assays (G. Brown and G.W. Pasternak, unpublished observations). In these studies the two agents had similar maximal effects, suggesting similar efficacies. Understanding the receptor mechanisms underlying the actions of these two analgesics would be a major advance in the design and use of opioid analgesics.
The recent cloning of the δ-opioid receptor (Evans et al., 1992; Kieffer et al., 1992) was soon followed by clones encoding μ (MOR-1) (Chen et al., 1993; Fukuda et al., 1993; Thompson et al., 1993; Reisine and Bell, 1993; Wang et al., 1993, 1994b; Uhl et al., 1994;Zastawny et al., 1994; Kozak et al., 1994; Minet al., 1994), κ1 (KOR-1) (Yasuda et al., 1993; Uhl et al., 1994; Zhu et al., 1995; Yakovlev et al., 1995; Knapp et al., 1995) and κ3-related (KOR-3 or ORL-1) (Pan et al., 1994, 1995; Uhl et al., 1994; Chen et al., 1994;Keith et al., 1994; Wick et al., 1994; Wanget al., 1994a; Mollereau et al., 1994; Bunzowet al., 1994; Fukuda et al., 1994; Lachowiczet al., 1995) opioid receptors. Antisense strategies (Wahlestedt, 1994) have proven invaluable in correlating these clones with in vivo opioid receptor pharmacology (Pasternak and Standifer, 1995). Antisense oligodeoxynucleotides against DOR-1, a δ-opioid receptor, selectively block [3H]DPDPE binding in neuroblastoma cells and spinal δ analgesia induced by either DPDPE (δ1) or deltorphin (δ2) (Standifer et al., 1994). These effects correlate well with the down-regulation of mRNA and δ receptor protein levels (Standifer et al., 1995). These initial studies examining DOR-1 have been confirmed and extended by others, and the approach has been applied to the KOR-1 (κ1) and MOR-1 (μ) clones (Chien et al., 1994; Pan et al., 1994; Tseng et al., 1994; Adamset al., 1994; Bilsky et al., 1994; Lai et al., 1994a,b; Rossi et al., 1995a; Chen et al., 1995).
The ability to coordinate the molecular biology of opioid receptors with their pharmacological activity offers a powerful approach to exploring these systems. Antisense approaches have been successfully used against different portions of the mRNA encoding opioid receptors (Standifer et al., 1994), which permits an assessment of the presence of individual exons within the potential receptor subtypes mediating selected responses. Detailed antisense mapping of all four exons of MOR-1 in mice reveal that morphine analgesia is blocked by oligodeoxynucleotides targeting either exon 1 or exon 4 of the μ-opioid receptor, but not those directed against exons 2 or 3 (Rossiet al., 1995a; Pasternak and Standifer, 1995). In contrast, the sensitivity of M6G analgesia shows a very different pattern (Rossiet al., 1995a,b; Pasternak and Standifer, 1995). The antisense probes against exons 2 and 3 of MOR-1, which were inactive against morphine, blocked M6G analgesia, whereas those targeting exons 1 and 4 were ineffective. Differences between morphine and M6G analgesia were also observed using probes targeting distinct Giα subunits (Rossi et al., 1995b). Down-regulation of Giα2 lowered morphine but not M6G analgesia, whereas the loss of Giα1 blocked M6G analgesia but not that seen with morphine. The present study analyzes the extent, specificity and time course by which antisense oligodeoxynucleotides based upon MOR-1 can modulate opioid analgesia in selected brain regions of rats.
Materials and Methods
Adult male Sprague-Dawley rats (250–400 g; Charles River Laboratories, Raleigh, VA) were housed individually and maintained on a 12-hr light/12-hr dark cycle, with food and water available ad libitum. All phosphodiester oligodeoxynucleotides were synthesized by Midland Certified Reagent Co. (Midland, TX). Antisense oligodeoxynucleotides (table 1) were directed against four regions of the MOR-1 clone (exons 1, 2, 3 and 4). The 19- to 22-base oligodeoxynucleotides were dissolved in 0.9% normal saline. A mismatch antisense oligodeoxynucleotide in which the sequence of four bases from the antisense A sequence has been switched, without altering the remaining sequence, was used as a control. All sequences are specific to the MOR-1 clone and are not present in the other opioid receptor cDNAs. Morphine was a gift from the Research Technology Branch of the National Institute on Drug Abuse.
Oligodeoxynucleotide sequences
Rats were anesthetized with chlorpromazine (3 mg/kg i.p.) and ketamine HC1 (100 mg/kg i.m.) and cannulated either in the lateral ventricle alone or in the lateral ventricle and one of two sites, the PAG or the LC. For the ventricular cannulation, a stainless steel guide cannula (22 gauge; Plastics One, Roanoke, VA) was placed stereotaxically (Kopf Instruments) 0.2 mm above the left lateral ventricle, by using the following coordinates: incisor bar (+5 mm), 0.5 mm anterior to the bregma suture, 1.3 mm lateral to the saggital suture and 3.6 mm from the top of the skull. Coordinates for the PAG were as previously described (Rossi et al., 1993, 1994b). With the incisor bar at −5 mm, the coordinates for the LC were 1.5 mm posterior to the lambda suture, 2.0 mm lateral to the saggital suture, 6.9 mm from the top of the skull and angled toward the saggital plane at 8.2 degrees. Cannulae were secured to the skull with three anchor screws and dental acrylic and kept patent with dummy cannulae. All animals were allowed 1 week to recover from cannula surgery and clearing of the anesthetic before the initiation of behavioral testing.
All rats received a test dose of morphine (7.5 μg i.c.v. or 2.5 μg into either the PAG or LC) and assessed in the tail-flick assay to ensure proper cannula placement. Rats were given at least 1 week after this test dose to recover. Groups of rats received vehicle or the chosen antisense oligodeoxynucleotide (i.c.v., 10 μg in 2–5 μl; PAG and LC, 10 μg in 1 μl) on days 1, 3 and 5, and analgesia was tested with the stated dose of morphine or M6G on day 6. Analgesia was assessed in a graded manner in the tail-flick assay (D’Amour and Smith, 1941) using the radiant heat assay (IITC, Woodland Hills, CA). Base-line latencies ranged from 1.8 to 3.2 sec. A maximal cut-off latency of 12 sec was used to minimize tissue damage. All animals displayed reproducible latencies in base-line and vehicle testing. The evaluation of the antinociceptive response of the animals was made by an observer blind to the experimental groups. Cannula placement was further evaluated histologically in all rats not used for biochemical studies, as previously reported (Rossi et al., 1993, 1994b).
Analgesia was assessed using both peak effects and areas under the time-action curve. Peak effects were determined at 30 min. Areas under the curve were calculated as previously described (Rossi et al., 1994b) and the area corresponding to base-line values was subtracted, leaving only the area due to the drug. Statistical significance (P < .05) was assessed with Student’s ttest or analysis of variance followed by Tukey’s protectedt test.
Results
Efficacy and selectivity of an antisense oligodeoxynucleotide to MOR-1 administered into the PAG.
Prior studies from our laboratory have established the ability of an antisense oligodeoxynucleotide targeting the 5′-untranslated region of the MOR-1 clone to block morphine analgesia (Rossi et al., 1994a). [d-Ala2,Glu4]Deltorphin, a highly selective δ receptor ligand, also produces analgesia when administered into the PAG (Rossi et al., 1994b). To explore the selectivity of the antisense treatment, we microinjected the same MOR-1 antisense oligodeoxynucleotide (antisense A, 10 μg) (table 1) into the PAG on days 1, 3 and 5 and examined both morphine and [d-Ala2,Glu4]deltorphin analgesia on day 6 (fig. 1). As previously observed, the antisense treatment virtually eliminated the analgesic actions of morphine administered into the PAG (P < .001). In contrast, the analgesic activity of [d-Ala2,Glu4]deltorphin in these same animals was virtually identical to that in untreated animals (Rossi et al., 1994b). The mismatch antisense oligodeoxynucleotide was inactive. Thus, the actions of antisense A are selective for μ analgesia.

Effect of MOR-1 antisense oligodeoxynucleotide treatment on morphine or deltorphin in the PAG. Groups of rats received saline or antisense A (10 μg) into the PAG on days 1, 3 and 5 and were tested with morphine (2.5 μg, n = 4) or deltorphin, a δ2 agonist (20 μg, n= 7), in the PAG on day 6. Morphine analgesia in the PAG was significantly reduced by antisense A (P < .001), but deltorphin analgesia was not. A mismatch antisense oligodeoxynucleotide was inactive (data not shown). Statistically significant conditions are shown (*P < .01, **P < .001).
Efficacy of i.c.v. antisense A in morphine analgesia.
We next assessed the actions of antisense oligodeoxynucleotides administered directly into the lateral ventricle. Using the same treatment paradigm, antisense A markedly diminished the analgesic activity of i.c.v. morphine, assessed as either peak effects or areas under the curve (P < .001) (fig. 2). The mismatch control failed to reduce the analgesic actions of morphine. The loss in morphine sensitivity after treatment with antisense A gradually returned to control levels over several days (fig. 3). The actions of the i.c.v. antisense A were dose-dependent (fig. 3). The lowest dose, 2.5 μg, had little effect on the analgesic actions of morphine, whereas the highest dose, 25 μg, virtually eliminated the analgesic actions of morphine, lowering the tail-flick latencies to base-line levels. Unlike the 10 μg dose, the 25 μg group failed to show any signs of analgesic recovery over the time period tested.

Effect of i.c.v. MOR-1 antisense oligodeoxynucleotide treatment on morphine analgesia. Groups of rats were administered saline, antisense A (10 μg) or the mismatch sequence i.c.v. on days 1, 3 and 5 and were challenged with morphine (7.5 μg) on day 6. Results are presented as mean ± S.E.M. of the tail-flick latencies (A) or the area under the curve (B). The antisense treatment (n = 6) significantly reduced morphine analgesia (P < .001), shown by either peak effects or areas under the time-action curve (sec·min), whereas the mismatch group (n = 6) showed no significant difference from the saline controls (n = 7). Statistically significant conditions are shown (*P < .001).

Duration and dose-dependence of antisense A treatments against morphine analgesia. Groups of rats were given either saline or antisense A at three different doses (2.5 μg i.c.v.,n = 3; 10 μg i.c.v., n = 6; 25 μg i.c.v., n = 6) on days 1, 3 and 5 and were challenged with morphine (7.5 μg i.c.v.) on day 6 and again on days 8, 10 and 12 to determine analgesic recovery. The lowest antisense oligodeoxynucleotide dose, 2.5 μg, produced no effect. Rats treated with 10 μg showed a significant decrease in morphine antinociception (P < .001) on days 6 and 8, after which their responses returned to normal. The 25 μg antisense oligodeoxynucleotide dose produced a significant decrease (P < .001) in morphine analgesia at all times tested. Statistically significant conditions are shown (*P < .001).
We next examined the ability of i.c.v. antisense A to modulate morphine analgesia in selected brain regions. After treating rats with i.c.v. antisense oligodeoxynucleotide, we assessed the analgesic actions of morphine microinjected directly into either the PAG or the LC (fig.4). Antisense A virtually eliminated the analgesic actions of morphine in the PAG. Indeed, the inhibition in the PAG exceeded that seen after i.c.v. administration of morphine. The i.c.v. antisense oligodeoxynucleotide also significantly decreased morphine analgesia in the LC, but not as effectively as in the PAG. In both regions, analgesic sensitivity recovered over 5 to 7 additional days.
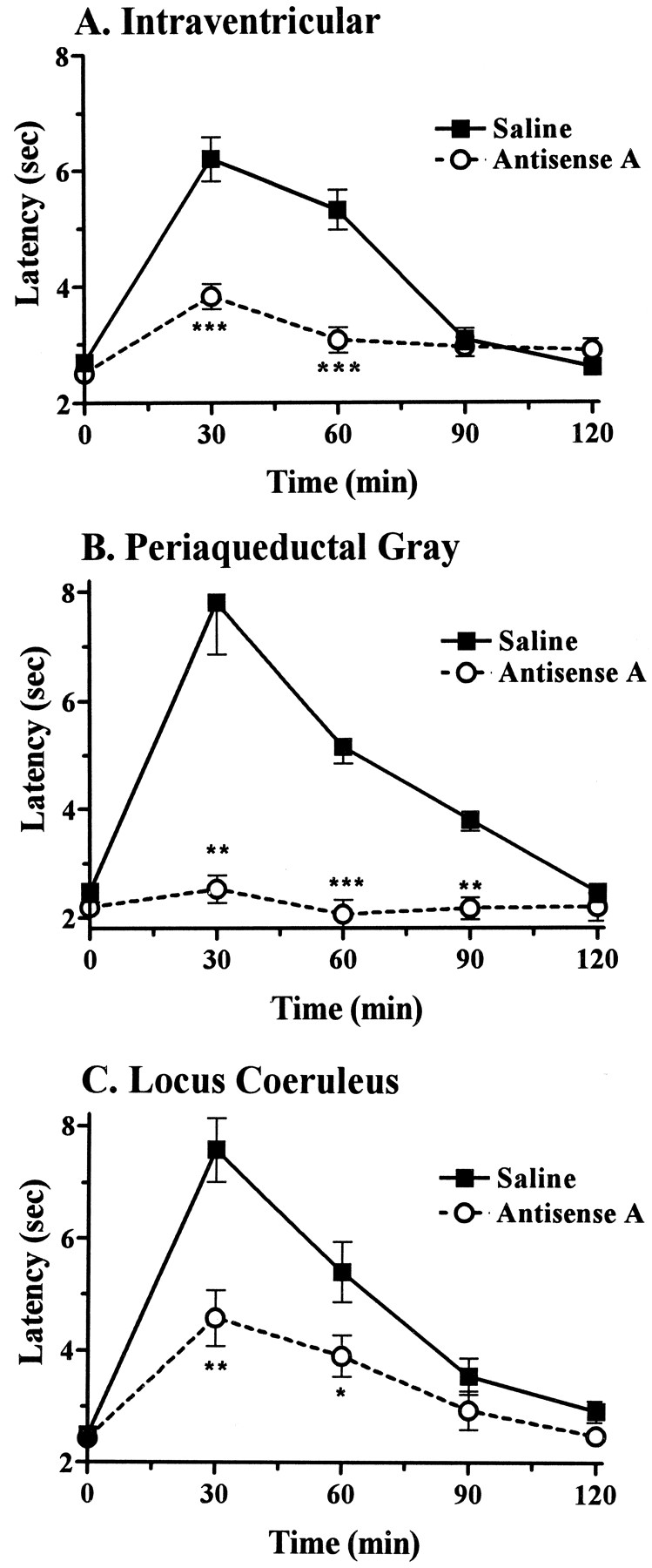
Effect of i.c.v. antisense A treatment on morphine analgesia in the PAG and LC. Groups of rats were given antisense A (10 μg i.c.v.) on days 1, 3 and 5 and challenged with morphine on day 6, given i.c.v. (7.5 μg, n = 6) (A) or directly into either the PAG (2.5 μg, n = 4) (B) or the LC (5 μg, n = 7) (C). Statistically significant conditions are shown (*P < .05, **P < .01, ***P < .001).
Antisense mapping of MOR-1.
Previous studies in mice from our laboratory using antisense oligodeoxynucleotides targeting the various exons of MOR-1 displayed differing activities against morphine and M6G analgesia (Rossi et al., 1995a,b; Pasternak and Standifer, 1995). We therefore examined the actions of i.c.v. antisense oligodeoxynucleotides directed against all four exons of rat MOR-1 against morphine and M6G analgesia (fig. 5). Both antisense A, targeting the untranslated regions of exon 1, and antisense D, directed against the coding region of exon 4, potently blocked i.c.v. morphine analgesia, whereas the oligodeoxynucleotides directed against exons 2 (antisense B) and 3 (antisense C) were inactive. As in our earlier studies in mice (Rossi et al., 1995a) and in the PAG of rats (Rossi et al., 1995b), antisense A was inactive against M6G analgesia. Conversely, the oligodeoxynucleotide antisense probes against exons 2 and 3, which were inactive against morphine analgesia, potently blocked the analgesic actions of M6G. Unlike our studies in mice, antisense D (exon 4) decreased both morphine and M6G analgesia in this rat model.

Antisense mapping of MOR-1 for morphine and M6G analgesia. Groups of rats received the indicated antisense oligodeoxynucleotides (antisense A, n = 6; antisense B, n = 4; antisense C,n = 4; antisense D, n = 4) on days 1, 3 and 5 and were tested for morphine (7.5 μg i.c.v.) (A) or M6G (0.7 μg i.c.v.) (B) analgesia on day 6. Tail-flick latencies were calculated as the maximal percent effect, where maximal percent effect = (experimental score − base-line score)/(maximal score − base-line score), and data are shown as percentage of control values. There was significant inhibition of tail-flick latencies and area under the curve (AUC) values for morphine analgesia by antisense A (P < .001) and antisense D (P < .01). M6G analgesia was lowered by antisense B (P < .001) and antisense C (P < .001) and to a lesser degree by antisense D (P < .01, area under the curve only). *P < .01; **P < .001.
Discussion
The current study continues our studies of the MOR-1 clone in rats. First, we confirmed the selectivity of the antisense treatment. The profound inhibition of morphine analgesia contrasts sharply with the lack of effect on the analgesic actions of the δ drug [d-Ala2,Glu4]deltorphin. We next examined the effectiveness of i.c.v. administration of the oligodeoxynucleotides. Prior studies in mice have shown the utility of this approach and our current studies, along with those of other groups, confirm its applicability to rats in a number of regions. As in the initial study examining DOR-1 (Standifer et al., 1994), the activity of the antisense A (10 μg) gradually resolves over a number of days. This time course is similar to estimates of the turnover of the receptor (Pasternak et al., 1980a,b). The lack of return of analgesic sensitivity with the highest dose tested, 25 μg, is difficult to understand. Although the very high doses of oligodeoxynucleotide used may simply prolong the actions, the possibility of a toxic or nonspecific action cannot be excluded. High doses have been associated with dose-dependent toxicity within the central nervous system, particularly with phosphorothioates (Chiassonet al., 1994; Meeker et al., 1995). By using the minimal doses of antisense oligodeoxynucleotides needed to see an effect, the possibility of nonspecific actions can be minimized.
In an effort to determine the extent of diffusion of the antisense treatment, we treated animals i.c.v. with an oligodeoxynucleotide and looked for morphine analgesia after direct injections into the PAG or LC. The treatment dramatically lowered the tail-flick latencies in the PAG group to base-line levels. Indeed, the inhibition exceeded that seen with i.c.v. morphine. The effects on the LC were significant but more modest, perhaps reflecting the greater distance of this structure from the ventricle. Many of the structures mediating morphine analgesia are periventricular, like the PAG, and appear to be readily influenced by the i.c.v. antisense approach. However, deeper structures may not be as susceptible to the antisense treatments administered i.c.v. This must be considered when designing studies examining other pharmacological actions.
In the initial opioid receptor antisense study, we demonstrated that probes targeting all three exons of DOR-1 lowered [3H]DPDPE binding in NG108–15 cells to similar degrees (Standifer et al., 1994). The ability to down-regulate the receptors by targeting any exon suggested that we could map the importance of each exon in receptor function. Our first studies suggested that the KOR-3 clone and the κ3 receptor shared exons 2 and 3 but not exon 1 (Uhl et al., 1994; Pasternak and Standifer, 1995; Pan et al., 1994, 1995). Our most extensive investigations focused on the mouse MOR-1 clone (Rossiet al., 1995a,b; Pasternak and Standifer, 1995). These antisense mapping studies revealed the importance of exons 1 and 4, but not exons 2 or 3, in supraspinal morphine analgesia. When we explored M6G analgesia, we observed very different results. In contrast to morphine analgesia, M6G analgesia was sensitive to probes targeting exons 2 and 3 and not those against exons 1 and 4. These observations led us to suggest the presence of distinct morphine and M6G receptors in mice, which may represent splice variants of the MOR-1 gene (Rossiet al., 1995a,b; Pasternak and Standifer, 1995). Our current results in rats are similar to those observed in mice. The probe against exon 1 potently blocked morphine analgesia without altering the analgesic actions of M6G. Conversely, exon 2 and 3 probes blocked M6G and not morphine analgesia. The similar effects of the exon 4 probe against the two agents differs from our prior observations in mice and requires further study, particularly with the identification of splice variants of exon 4 (Bare et al., 1994; Zimprich et al., 1994).
In conclusion, antisense approaches have proven valuable in the elucidation of the molecular pharmacology of opioid actions. In addition to confirming the importance of the various opioid receptor clones in mediating opioid pharmacology, antisense strategies suggest the potential importance of splice variants in explaining opioid receptor subtypes. Their selectivity and general utility provide enormous potential in correlating molecular biology and behavior within the central nervous system.
Acknowledgments
We thank Dr. J. Posner for his support and Andre Ragnauth and Wei Zen Yu for their assistance in this study.
Footnotes
-
Send reprint requests to: Dr. Gavril W. Pasternak, Department of Neurology, Memorial Sloan-Kettering Cancer Center, 1275 York Ave., New York, NY 10021.
-
↵1 This work was supported, in part, by research grants from the National Institute on Drug Abuse to G.W.P. (DA07242) and to R.J.B. (DA04191) and a core grant from the National Cancer Institute to Memorial Sloan-Kettering Cancer Center (CA08748). G.C.R. was supported by a training grant from the National Institute on Drug Abuse (DA07274), Y.-X.P. by a fellowship from the Aaron Diamond Foundation and G.W.P. by a Research Scientist Award from the National Institute on Drug Abuse (DA00220).
- Abbreviations:
- DPDPE
- [d-Pen2,d-Pen5]-enkephalin
- LC
- locus coeruleus
- M6G
- morphine-6β-glucuronide
- PAG
- periaqueductal gray
- i.c.v.
- intracerebroventricular
- Received April 9, 1996.
- Accepted December 13, 1996.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}