


Abstract
Neuropathic pain remains a significant clinical problem. Current understanding implicates the spinal cord dorsal horn N-methyl-d-aspartate (NMDA) receptor apparatus in its pathogenesis. Previous reports have described NMDA antagonist reduction of nerve injury-induced thermal hyperalgesia and formalin injection-related electrical activity. We examined a panel of spinally administered NMDA antagonists in two models: allodynia evoked by tight ligation of the fifth and sixth lumbar spinal nerves (a model of chronic nerve injury pain), and the formalin paw test (a model wherein pretreatment with drug may preempt the development of a pain state). A wide range of efficacies was observed. In the nerve injury model, order of efficacy (expressed as percent of maximum possible effect ± S.E.), at the maximum dose not yielding motor impairment, was memantine (96 ± 5%) = AP5 (91 ± 7%) > dextrorphan (64 ± 11%) = dextromethorphan (65 ± 22%) > MK801 (34 ± 8%) > ketamine (18 ± 6%). For the formalin test, the order of efficacy was AP5 (86 ± 9%) > memantine (74 ± 5%) ≥ MK801 (67 ± 16%) > dextrorphan (47 ± 16%) > dextromethorphan (31 ± 12%) > ketamine (17 ± 15%). In the nerve injury model, no supraspinal action was seen after intracerebroventricular injections of dextromethorphan and ketamine. NMDA antagonists by the spinal route appear to be useful therapeutic agents for chemically induced facilitated pain as well as nerve injury induced tactile allodynia. It is not known what accounts for the wide range of efficacies.
Protracted activation of small primary afferent fibers can induce states of facilitated spinal sensory processing. The behavioral outcome of such facilitation includes decreases in pain thresholds to stimuli that, when intense, are normally painful and may cause tissue damage; this behavior is rationally termed hyperalgesia. Peripheral nerve injuries may similarly lead to a facilitated state. In addition to hyperalgesia, the finding that low intensity mechanical stimuli such as brushing the skin (typically adequate to activate only large diameter low threshold mechanoreceptors) can induce pain, is a prominent component of nerve-injury evoked pain syndromes (Wahren and Torebjörk, 1992). Such pain after light touch appears to represent a qualitative change in perception, rather than merely an increase in sensitivity; hence, the term “allodynia” or tactile allodynia is preferable to describe this phenomenon.
The mechanisms underlying the alterations in function that mediate those various pain components are not completely understood. However, several lines of investigation have highlighted the involvement of spinal dorsal horn glutamatergic systems in the development and perpetuation of centrally facilitated pain conditions. 1) Iontophoretic application of glutamate produces both bursting activity in second-order neurons and facilitation of the response of the neuron to A∂ and C fiber intensity electrical stimulation (Chapman et al., 1994) as well as facilitation of responses to low and high intensity mechanical stimulation of the skin (Dougherty and Willis, 1991). Delivery of lumbar intrathecal NMDA in the unanesthetized animal yields spontaneous agitation and exaggerated behavioral responses to thermal and low intensity mechanical stimuli, interpreted as thermal hyperalgesia and tactile allodynia (Coderre and Melzack, 1992; Malmberg and Yaksh, 1992b; Bach et al., 1994). 2) The progressive augmentation in response (windup) caused by repetitive small primary afferent fiber stimulation is reversed by spinal NMDA antagonists (Davies and Lodge, 1987; Dickenson and Sullivan, 1987). Similarly, behavioral models such as the knee arthritis models or the s.c. injection of irritant (e.g., formalin) lead to a hyperalgesic state involving protracted afferent input that is diminished by spinally delivered NMDA antagonists (Neugebauer et al., 1993; Coderre and Van Empel, 1994). Moreover, in models of nerve injury, spinal delivery of several NMDA antagonists can diminish the hyperalgesic state (Yamamoto and Yaksh, 1992b). 3) After the injection of an irritant or the initiation of a chronic inflammatory arthritis or nerve injury, there is an increase in the spinal release of excitatory amino acids such as aspartate and glutamate in the spinal cord (Sluka and Westlund, 1992; Malmberg and Yaksh, 1995b; Yanget al., 1995). These observations provide support for the hypothesis that spinal glutamate receptors, particularly those of the NMDA subtype, play an important role in regulating spinal encoding of afferent information and that after tissue or nerve injury, there is increased spinal glutamate release that may induce a hyperalgesic and allodynic state.
Despite the compelling evidence for the role of a spinal glutamatergic site, there are few systematic studies seeking to define the NMDA receptor pharmacology for these two pain states. Such characterization requires the comparison of the structure activity series of a family of glutamatergic receptor antagonists delivered spinally in the several behavioral models. To accomplish these goals, we have examined a panel of spinally delivered glutamate antagonists in both the rat formalin paw model (Wheeler-Aceto et al., 1990) and a chronic nerve ligation model (Kim and Chung, 1992). We compared the effects of both noncompetitive (open-channel) NMDA antagonist drugs (dextromethorphan, dextrorphan, MK801, memantine, ketamine) and a competitive NMDA antagonist in the two models by the intrathecal route. Three of these drugs are in clinical usage for diverse indications: ketamine (dissociative anesthetic agent), dextromethorphan (antitussive: parent drug of more potent metabolite, dextrorphan) and memantine (antiparkinsonian). MK801 and AP5 are commonly used in electrophysiological and behavioral studies of NMDA receptor function. We also examined a limited selection of NMDA antagonists in the Chung model by the intracerebroventricular route to look for supraspinal effects. In addition, we compared the effects of an intrathecal non-NMDA antagonist, DNQX, in the two models, and examined the effects of an antagonist at a metabotropic glutamate receptor site, AP3, in the Chung model.
Methods
All studies were conducted in accordance with the guidelines of the Institutional Animal Care Committee of the University of California, San Diego.
Animals
Male Harlan Sprague-Dawley rats (weights below) were housed in cages with solid bottoms and sawdust bedding, with a 12/12 hr light cycle (0600–1800), and allowed free access to food pellets and water. Animals were housed singly after surgical interventions.
Surgical Preparation
Neuropathy.
A surgical neuropathy was created in 100 to 200 g rats as follows, to create a model commonly referred to as the Chung or tight nerve ligation model (Kim and Chung, 1992). Under halothane/oxygen anesthesia, a dorsal midline incision was made from approximately L3-S2. Using a mixture of sharp and blunt dissection, the left L6/S1 posterior interarticular process was exposed and resected to permit adequate visualization of the L6 transverse process, which was gently removed. Careful teasing of the underlying fascia exposed the left L4 and L5 spinal nerves distal to their emergence from the intervertebral foramina. The nerves were gently separated, and the L5 nerve firmly ligated with 6-0 silk suture material. The left L6 spinal nerve was then located just caudal and medial to the sacroiliac junction, and similarly ligated with 6-0 suture. The wound was then inspected for hemostasis and closed in two layers with 4-0 vicryl sutures. Five ml of lactated Ringer’s solution were administered i.p., and the animal was allowed to emerge from anesthesia in an observation chamber under a warming light. Animals with inability to flex the left hind limb postoperatively, indicating damage to the L4 nerve, were discarded; those with thresholds of more than 4 g were considered unsuccessful preparations (Chaplan et al., 1994).
Intrathecal cannulation.
Lumbar IT cannulation was carried out immediately after, or up to 17 days after neuropathy surgery, using a modification of the method described by Yaksh and Rudy (1976). The IT catheter was a length of PE-10 prepared with a loose knot secured with a drop of dental acrylic. The tubing was cut 3 cm from the knot (peripheral end) and stretched on the other end to reduce the diameter by approximately half, and was cut to a length of 9 cm (for rats ≥250 g) or 7 cm (rats ≤250 g), and occluded with a 1-cm 28-gauge wire obturator. Under halothane anesthesia, this end was inserted through an incision in the dura over the cisterna, and threaded caudally to the vicinity of the lumbar enlargement. Rats with discernible neurological deficits after IT implantation were discarded. One group of rats underwent IT implantation before nerve ligation surgery to observe for differences in thresholds attributable to IT cannulation alone. Catheters were considered usable for 21 days after implantation based on previous experience.
For ICV drug delivery, guide cannulae were fashioned of thin walled 23-gauge stainless steel tubing, according the method described byMoron et al. (1990). The head was immobilized in a stereotaxic frame, and the guide cannula was inserted through a calvarial burr hole to a depth of 4 mm, and affixed to the skull using cranioplastic cement and three stabilizing calvarial screws (0–80, 3/16 inch, stainless steel wood screws, Small Parts Inc., Miami, FL) using dental cement. ICV injection was accomplished using a 29-gauge cannula 1 mm longer than the guide cannula. All drugs were delivered in a volume of 10 μl. Ventricular placement was confirmed at the conclusion of the experiment by methylene blue injection (10 μl), followed by sacrifice and dissection of the cannula path to inspect for ventricular spread of dye.
Testing
Tactile allodynia.
Rats were tested only during the daylight portion of their circadian cycle. Rats were placed in a plastic cage (4.25 × 10 × 6 inch high) with a coated, thermally neutral wire mesh bottom and allowed to adapt for approximately 15 min or until explorative behavior ceased. The 50% probability thresholds of paw withdrawal to mechanical stimulus were determined prior to drug injection, and at 30, 60 and 120 min thereafter; for drugs with more delayed onset, testing was carried out to 300 to 360 min. These data were determined by applying von Frey hairs to the mid-plantar left hindpaw in sequential ascending or descending order, as necessary, to hover as closely as possible around the threshold of response, as previously described (Chaplan et al., 1994). In brief, a withdrawal response was cause to present the next weaker stimulus, and lack of withdrawal led to presentation of the next stronger stimulus. Interpolation of the 50% threshold was carried out according to the method of Dixon (1980). Results are presented as either the raw threshold, or as efficacy, represented as a fraction of MPE (% MPE). The threshold value of 15 g indicating 100% MPE was chosen based on studies of responses in sham operated rats. The following formula was used to compute % MPE: % MPE = (result − baseline predrug)/(15 g − baseline predrug) × 100.
Rats were tested no more frequently than every 3 days, for a maximum of six times total and were kept for a maximum of 50 days after neuropathy surgery. Motor dysfunction was evaluated by testing the righting reflex, the ability to stand and ambulate in a normal posture and to place and step with the hind paws. Generalized cutaneous allodynia was considered to be present when the animal reacted to stroking of the body fur with vocalization.
Formalin tests.
Preliminary data showed no significant differences between pretreatment with dextromethorphan (one of the drugs with a later peak of effect in the Chung model) at 120, 30, or 15 min before formalin injection (see table1). Therefore, rats (300–350 g) with previously implanted IT catheters were formalin tested beginning 15 min after drug administration. Under a brief halothane anesthetic, the dorsum of the right hindpaw was injected s.c. with 50 μl of 5% formalin. Animals were recovered in clear Lucite observation cylinders backed by mirrors, permitting observation from every angle. The number of flinches with the injected hindpaw was counted over 60-sec periods, beginning 1 min after emergence (regaining of the righting reflex) and repeated at 5, 10 and every 5 min thereafter for a total of 1 hr, after which time they were killed. Phase 2A was defined as the period from 10 to 40 min inclusive. Results are expressed as mean paw flinches ± S.E., or as efficacy, stated as % MPE ± S.E. % MPE was computed as: % MPE = (no. flinches, saline control − no. flinches, test drug)/(no. flinches, saline control) × 100.
Effect of time of pretreatment with intrathecal dextromethorphan, maximum usable dose (250 μg) on the formalin test phase 2A
Drugs
All drugs were dissolved in 0.9% sterile preservative-free saline, with subsequent dilutions using the same, with the exception of DNQX, for which a stock solution was prepared using bicarbonate 25 mM (pH 8.0) and subsequent dilutions were made with saline as above. Drug doses were injected IT over approximately 15 sec in a volume of 10 μl, followed by 10 μl of saline flush, using calibrated PE-90 tubing attached to a Hamilton glass syringe seated in a geared microinjector. Awake rats were gently restrained by swaddling in a towel during injection and catheters were immediately replugged, to prevent leakage of solutions. No rat received the same dose of drug twice. The following drugs were used: dizocilpine maleate (MK801; FW = 337) (RBI), dextrorphan tartrate; FW = 407 (RBI), dextromethorphan hydrobromide; FW = 352 (RBI), and memantine; FW = 216 (Merz, Frankfurt-am-Main, Germany), and ketamine hydrochloride; FW = 546, (Parke-Davis, Morris Plains, NJ), all noncompetitive NMDA antagonists; ±-2-amino-5 phosphonopentanoic acid (AP5, FW 197) (RBI), and DNQX (FW 252) (RBI), competitive non-NMDA antagonist ±-2-amino-3-phosphonopropionic acid (AP3, FW 169) (RBI) an antagonist at the metabotropic glutamate receptor, and morphine sulfate (FW 669) (Merck Sharpe & Dohme, West Point, PA).
Statistics
All statistical comparisons were computed using Statview 4.0 and SuperAnova software for the Macintosh (Abacus Concepts Inc, Berkeley, CA). Multiple comparisons (MPE) were performed using one-way analysis of variance followed by Fisher’s Protected Least Significant Difference post hoc analysis. The small groups of pre- and postintrathecal catheter implantation paw thresholds were compared using the Wilcoxon signed-rank test. ED50 and ED33 were calculated using the analysis of Tallarida and Murray (1987). Results are presented as mean ± S.E., as ED50 or ED33 as indicated. Significance was chosen at P < .05.
Results
Neuropathy model.
The tight segmental nerve ligation reliably produced allodynia, as defined by a marked reduction in the tactile stimulus required to evoke organized withdrawal of the paw ipsilateral to the nerve lesion; we have previously reported that 93% of rats with tight nerve ligations have thresholds <4 g, vs. unoperated and sham-operated means of 15 g (Chaplan et al., 1994). Because rats were used multiple times to exclude the possibility of cumulative drug or testing effects, baseline thresholds before each drug testing session were analyzed and compared, and showed no statistically significant trend (table 2). No differences were seen between rats with IT implantation before or after nerve ligation (see table 3).
Base-line paw withdrawal thresholds in a cohort of 30 Chung neuropathy rats exposed to repeated testing
Base-line paw withdrawal thresholds in nine Chung neuropathy rats pre and post intrathecal catheter implantation
General observations.
No specific behavioral effects were observed (sedation/agitation) after spinal delivery. Motor weakness was a frequent factor in limiting drug dosing: at high doses of most agents, beyond the range of doses used for allodynia testing, hindlimb flaccidity was noted. The doses at which motor dysfunction was noted are presented in Tables 4 and 5. In all cases, such weakness appeared almost immediately, within a few minutes, and typically resolved within 1 to 3 hr. At the doses used, there was no loss of the righting reflex. Although not systematically examined, there was no evidence of an impairment of bladder or bowel function. No drug had effects that were still detectable after the 3 day poststudy period.
Tight segmental nerve ligation (Chung model)
Formalin test, phase 2A
Tactile allodynia: spinal glutamate antagonists.
Tactile allodynia was reliably reduced by the IT administration of NMDA antagonists in doses that did not cause motor dysfunction. The different agents tested displayed different efficacies as well as potencies in this regard. Therapeutic effect was limited in most cases by motor impairment, but in the case of memantine and dextromethorphan, by solubility in an aqueous vehicle. Peak effect for dextromethorphan, memantine and AP5 was at approximately 120 min, and for dextrorphan, MK801 and ketamine at approximately 60 min (see fig. 1 A and B). The order of IT potency (ED50, μg) was AP5 (2)>dextrorphan (26)>memantine (71)>dextromethorphan (115)>MK801≫ketamine≥0, as shown in figure 2 (and see table 4). ED50 could not be calculated for MK801 and ketamine due to limited efficacy in this model. The order of efficacy (i.e., effects seen at the maximum usable dose, % ± SE) was memantine (96 ± 5%) = AP5 (91 ± 7%) = > dextrorphan (64 ± 11%) = dextromethorphan (65 ± 22%) > MK801 (34 ± 8%). These values were significantly different from vehicle injections (6 ± 3%) (P < .0001). Ketamine, alone in this class, did not have significant efficacy (18 ± 6%, P = .4).

A, Paw withdrawal thresholds (g) to von Frey hairs in Chung neuropathy rats at predrug baseline and for 120 min after IT application of the maximum usable dose of ketamine, MK801, dextrorphan and saline (mean ± S.E.). N = six to nine per group. B, Paw withdrawal thresholds (g) to von Frey hairs in Chung neuropathy rats at predrug baseline and for 360 min after IT application of the maximum usable dose of NMDA antagonists with prolonged activity: dextromethorphan, AP5 and memantine (mean ± S.E.). N = five to six per group.

Dose-response curves for suppression of allodynia in Chung neuropathy rats (normalization of paw withdrawal thresholds to von Frey hairs) by IT NMDA antagonists. Y-axis, Percent of maximum possible effect (% MPE); x-axis, log (dose, μg). N = five to nine per group. See text and tables for ED50calculations.
Blockade of non-NMDA receptors with DNQX produced a profound, but less sustained reduction in allodynia, with a peak effect at 30 min (fig.3). ED50 was 1.2 μg; peak effect was 85 ± 10% (P < .01, see table 4). Efficacy was also limited by motor dysfunction.

Dose-response curves for suppression of (top) allodynia in Chung neuropathy rats (normalization of paw withdrawal thresholds to von Frey hairs) and (bottom) paw flinches in the formalin test, by IT non-NMDA antagonist DNQX and (Chung model) metabotropic glutamate receptor antagonist AP3. Y-axis, Percent of maximum possible effect (% MPE); X-axis, log (dose, μg). N = six to nine per group. See text and tables for ED50/ED33calculations.
Blockade of one of the metabotropic glutamate receptors by AP3 had no statistically significant effect on allodynia. The maximum usable dose was limited by generalized cutaneous drug-induced algogenic behavior (fig. 3).
Supraspinal NMDA antagonism.
ICV administration of one of the NMDA antagonists with greatest therapeutic/toxic ratio, dextrorphan, at a dose of 100 μg (four times the ED50 for antiallodynic effect by lumbar spinal administration) had no effect on allodynia (% MPE = 5 ± 4%), and was not significantly different from ICV saline control (8 ± 6%) over a 60-min observation period in the tight nerve ligation model. ICV administration of the maximum usable spinal dose of ketamine, 100 μg, also had no significant effect (5 ± 4%). Lumbar IT morphine 10 μg had a modest but significant effect (26 ± 6%); a higher dose produced sedation that precluded allodynia assessment. In contrast, ICV morphine 10 μg resulted in significant allodynia suppression of 76 ± 15% (P < .0001, fig. 4).
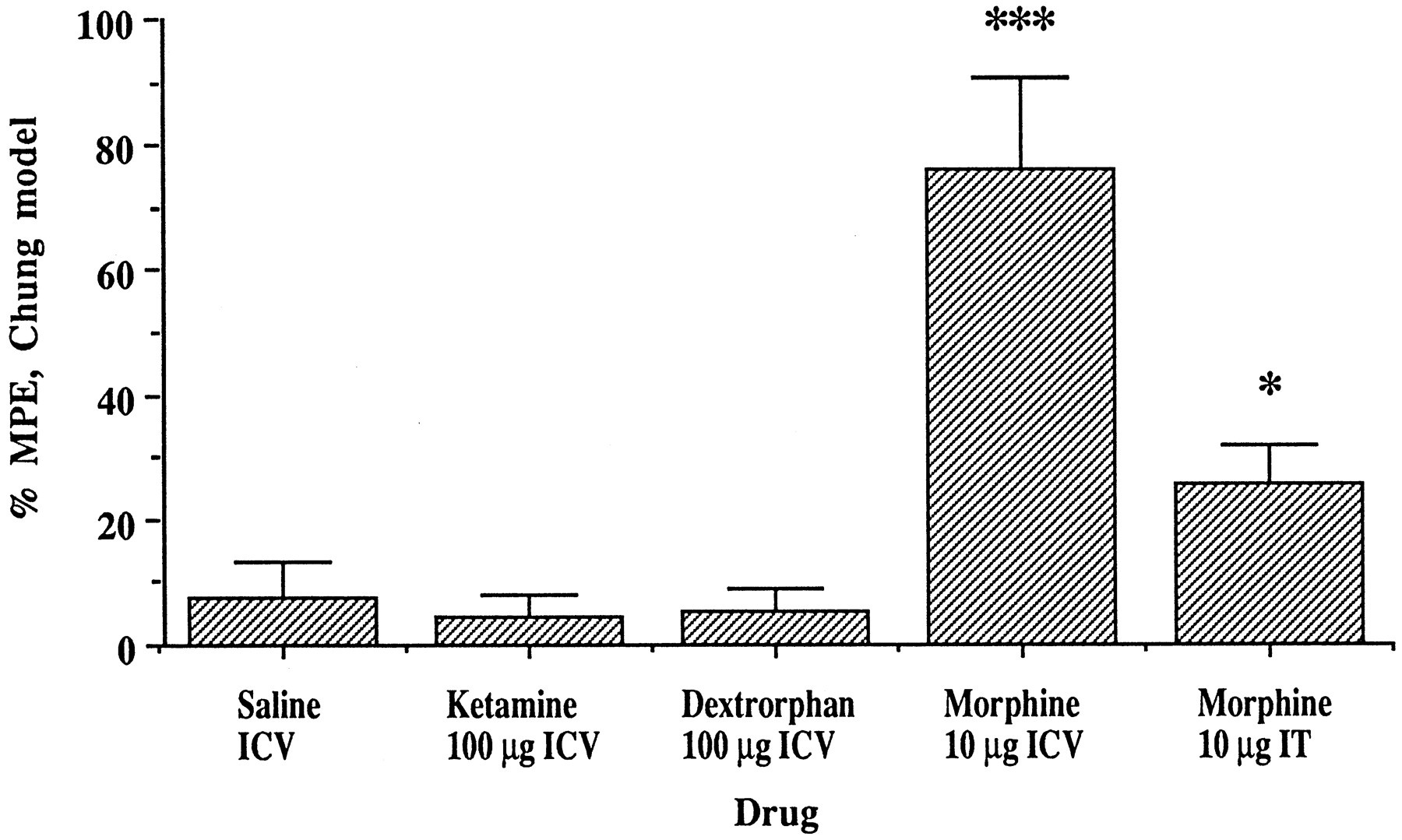
Effect of ICV administration of ketamine and dextrorphan, compared with ICV morphine and IT morphine, on allodynia in the Chung neuropathy model. *** Indicates P < .0001vs. all other groups; * indicates P < .05vs. dextrorphan and ketamine (one-way ANOVA, Fisher’s PSLD, F = 10.69).
Formalin test.
We have previously published our observation that some drug effects are most prominent in the early period of the formalin test phase 2 (Malmberg and Yaksh, 1992a). As with the nonsteroidal anti-inflammatory drugs, we found the greater effects of the drugs analyzed in our study in this early period. IT NMDA antagonists significantly decreased spontaneous flinching behavior in phase 2A of the formalin paw test. Animals receiving saline displayed a mean of 105.2 ± 7.6 flinches, compared with memantine (18.4 ± 3.8), MK801 (26.8 ± 12.5), AP5 (32.8 ± 19.5), dextrorphan (56 ± 17.3), dextromethorphan (72.3 ± 13.0) and ketamine (87.2 ± 15.8). Except in the case of ketamine, these values were significantly different from saline (P = .0001). The order of potency (ED50, μg) was AP5 (0.6) > MK801 (4.3) > memantine (24.5) ≫ dextrorphan (234); dextromethorphan; ketamine ≥ 0. ED50for dextromethorphan and ketamine were not determinable due to limited efficacy; ED33 were calculated for these two agents (see table 5). The order of maximum efficacy was AP5, 86 ± 9% > memantine, 74 ± 5% ≥ MK801, 67 ± 16% ≫ dextrorphan, 47 ± 16% > dextromethorphan, 31 ± 12% ≫ ketamine, 17 ± 15% as shown in figure5. Figure 6 presents the time course of flinching for the agents listed. No agent completely suppressed flinching in phase 2A at doses not causing motor dysfunction.

Dose response curves for suppression of flinching behavior by NMDA antagonists, phase 2A of the formalin test. Y-axis, Percent of maximum possible effect (% MPE); X-axis, log (dose, μg). N = four to eight per group. See text for calculation of ED50/ED33.

Time course of paw flinching behavior after spinal administration of maximum usable doses of NMDA antagonists in the formalin test: N = four to eight per group.
Blockade of non-NMDA receptors with DNQX produced variable results, with limited efficacy below 50% (fig. 3). Peak effect was 46 ± 9% (P < .005, F = 6.022, one-way analysis of variance with Fisher’s Protected Least Significant Difference: see table 5). Efficacy was limited by motor dysfunction.
Therapeutic ratio.
To estimate a TI, we divided the lowest dose at which motor effects were seen by the ED50 for the antihyperalgesic effect in both the tight segmental nerve ligation and formalin tests. A therapeutic ratio of ≤2 was considered to represent a drug with no useful therapeutic window in this model. Higher numbers represent higher margins of discrimination between antihyperalgesia/allodynia and motor effects. These results are summarized in tables 4 and 5.
Discussion
In this study we systematically extended previous observations regarding the role of NMDA receptors in the mediation of pathological sensory states. There are four principal observations: 1) the spinal structure activity relationship emphasizes the role of a spinal NMDA site, without minimizing the importance of other glutamatergic receptors; 2) the nerve injury-induced tactile allodynia displays a similar structure activity relationship to the formalin test with regard to blockade of the NMDA receptor, suggesting the importance of a similar receptor complex; 3) the NMDA receptor plays a major role in spinal motor function, but significant and sometimes complete normalization of aberrant sensory thresholds can be readily observed at spinally doses that do not impair motor function and, also importantly, have no apparent effect on behavior; 4) in these models, effects appear to be mediated by a spinal, but not a supraspinal action; and 5) consistencies in the order of efficacies of the NMDA antagonists tested in the two models suggest a pharmacological profile specific to NMDA receptors in the lumbar dorsal horn.
The effect of DNQX, a non-NMDA receptor antagonist, was significant but brief in the Chung model, and again significant although to a lesser extent in the formalin test. Although these data imply that blockade of non-NMDA receptors is insufficient to produce sustained antihyperalgesia/antiallodynia, further investigation of this class of drugs is warranted before such a conclusion. The observation that the metabotropic receptor antagonist AP3 was ineffective and indeed produced algogenic behavior is consistent with current understanding of the multiple (both negative and positive) modulatory roles played by the metabotropic glutamate receptor.
Of note, although the effects of the various agents studied were qualitatively similar in both the tight nerve ligation and the formalin models, there were quantitative differences (see fig. 7 for comparisons). Maximum efficacy of dextrorphan and dextromethorphan and memantine appeared to be somewhat higher in the nerve ligation model, whereas efficacy of MK801 appeared to be greater in the formalin test. These differences may reflect subtle distinctions 1) between the neurochemistry of evoked behavior (with von Frey hairs) and spontaneous pain behavior (flinching), or 2) between the models themselves, futher addressed below. An additional significant point of difference between the two models is that the Chung model represents a paradigm of an established pain state, where drug administration amounts to posttreatment, whereas the formalin test as performed represents drug treatment before the noxious stimulus, or basically a “preemptive” intervention. This difference may be important with regard to the presence or absence of a delay in drug effect onset in the respective models (see “Time course of NMDA receptor antagonism effect”).

Comparison of NMDA antagonist efficacy in the Chung model and the formalin test. Percent of maximum possible effect (% MPE) attained with spinal administration of the maximum usable dose of each drug is illustrated in the upper panel for the Chung model and in the lower panel for the formalin test. Inset shoes the same data plotted as a correlation between formalin test and Chung model results: R = .74. * P < .05; ** P < .01; *** P < .001, within each model, vs. saline (not shown). N = four to nine per group. (One-way ANOVA, Fisher’s PSLD; F value = 11.779, Chung model, 10.55 formalin test.)
Comparison of models.
The role of C fibers in the initial barrage and maintenance of the hyperalgesic state appears to be greater in the formalin test: neonatal capsaicin treatment sufficient to depopulate unmyelinated fibers (C fibers and to some extent Aδ) only mildly reduces the development of allodynia after subsequent nerve injury (Shir and Seltzer, 1990; S. R. Chaplan, X.-Y. Hua, B. P. Scott and T. L. Yaksh, unpublished data) whereas it significantly reduces responsiveness in the formalin test (Nagy and van der Kooy, 1983; Haraet al., 1984).
Glutamatergic basis.
In the formalin test, acute activation of small primary afferents is followed by a low level of discharge sustained nevertheless over the subsequent usual 60-min period of observation. A burst of glutamate release in the lumbar spinal cord in phase 1 has been identified by dialysis in the unanesthetized rat, followed by a low tonic level of release, paralleling the electrical activity of primary afferents. These phase 1 effects appear to activate cyclooxygenase and nitric oxide synthase, leading to facilitated release of prostanoids and nitric oxide (Malmberg and Yaksh, 1995, a and b). The typical behavior pattern (flinching and licking of the injected paw) corresponds more closely to the activity pattern of dorsal horn wide dynamic range neurons: these both exhibit a characteristic biphasic response, with an initial burst of activity such as seen in the primary afferent population, followed by a brief quiescent period, then a sustained period of intense activity from about 10 to 60 min. This second phase of the formalin response is believed to exemplify the exaggerated state of processing that occurs secondary to the initial glutamate effects in phase 1.
Immediately after nerve injury, a barrage of neuronal activity is seen (Devor and Govrin-Lippmann, 1983). Blockade of this barrage by a number of techniques, including local anesthetic and spinal application of either an NMDA antagonist (Davar et al., 1991) or a tachykinin antagonist alone (Luo and Wiesenfeld-Hallin, 1995), reduces the subsequent development of a hyperalgesia syndrome. In the Chung model, spinal glutamate levels rise during the 2- to 3-day interval after nerve ligation (M. Marsala, L.-C. Yang, Y.-W. Lee and T. L. Yaksh, unpublished data). However, unlike the formalin test, a structural alteration takes place with reorganization of the systems that encode low threshold information: sprouting of large afferents occurs into laminae I and II (Woolf et al., 1992), dorsal horn cells, possibly serving to regulate dorsal horn activity, are lost (Sugimoto et al., 1990) and peripheral postganglionic sympathetic fibers sprout into the neuroma and the DRG cells of the injured axon (Chung et al., 1993; Mclachlan et al., 1993). For days to weeks after injury, the neuroma, DRG and dorsal horn display spontaneous activity; it has been suggested that this ongoing discharge may itself be sufficient to sustain a facilitated state of processing.
Thus, the appearance of increased spinal glutamate release appears to be a common component of both posttissue and postnerve injury pain states, albeit with differing time courses; other models also evidence the same phenomenon, e.g., in the rat arthritic knee joint model sustained glutamate release and hyperalgesia are seen for a period in excess of 24 hr (Yang et al., 1995). In addition, it is important to note that spinal NMDA receptor activation can lead to facilitated electrophysiological responses to low threshold and high threshold input after iontophoretic delivery, and behavioral responses during continuous IT delivery (Biella et al., 1993; Bachet al., 1994).
Mechanism of action of spinal NMDA receptors in aberrant processing.
Identification of the location of NMDA sites has emphasized that receptors exist preterminally on small primary afferents (Liu et al., 1994), on the cell bodies of dorsal horn neurons in lamina III and IV, and likely postsynaptic to corticospinal tracts neurons in the ventral horn (Valtschanoff et al., 1993). Evidence suggests that NMDA receptors are located postsynaptic to excitatory interneurons that are the initial targets of activation by primary afferents (Davies and Watkins, 1983).
NMDA receptors.
The most current formulation of the role of NMDA receptors in the afferent pathways involved in hyperalgesia/allodynia is that intense stimulation of primary afferents initially activates non-NMDA type glutamate receptors on postsynaptic neurons. Activation exceeding a defined threshold leads to removal of the Mg++ ion block (voltage gating) of the NMDA receptor, likely located at one postsynaptic remove from the primary afferent, in a step involving phosphorylation, permitting the opening of the NMDA receptor ion channel and the influx of Ca++ ions. The entry of Ca++ into the cytoplasm triggers a second messenger cascade activating a number of enzymes and ultimately immediate-early genes. Retrograde second messengers resulting from this cascade have been posited to cause presynaptic amplification of neurotransmitter release; candidates include the neuromediators nitric oxide, and prostaglandins, which may diffuse back to the primary afferent terminal. These events are modulated by the concurrent binding of glutamate to the G-protein linked metabotropic receptor, with either augmentation or diminution as a result of the particular characteristics of the mGluR (and modulatory neuropeptides) involved (Zheng and Gallagher, 1995) and the concurrent binding of neuromodulatory peptides coreleased from the primary afferent with glutamate (see Coderre et al., 1993 for review).
In our study, it is consistent with this model that blockade of NMDA receptor sites (including the glycine site) as well as non-NMDA glutamate receptors produced normalization of paw withdrawal thresholds. Previous reports have documented that the NMDA block is different from others in that it is selective for the modality of hyperalgesia, whereas blockade of AMPA sites appears to produce pansensory hypoesthesia (diminution of sensory perception in all modalities). The pharmacology of metabotropic receptors has yet to be fully defined. In our study activity was limited by the observation of a potent algogenic effect. The mechanism of this effect is unknown but may relate to the loss of a tonic inhibition of glutamate-releasing terminals.
Time course of NMDA receptor antagonism effect.
The ability to reverse the behavioral manifestations of this chain of events during a time period of blockade of the NMDA receptor, indicates the importance of ongoing activation of a glutamate receptor of the NMDA class. Cases where such reversal is slow in onset may indicate that NMDA occupancy initiates processes that are sustained despite the presence of NMDA receptor blockade. In the formalin test, the release of glutamate appears to be limited to phase 1 (Malmberg and Yaksh, 1995a) and it has been shown that NMDA antagonists delivered between phase 1 and 2 have little effect on phase 2 activity (Yamamoto and Yaksh, 1992a). Hence, in this case, the release of glutamate during phase 1 appears to serve a triggering function. Blockade of glutamatergic effects by drug administration before formalin administration, as in our study, however, serves a “preemptive” role. Our observation that there was routinely a measurable delay before normalization of the threshold in the nerve injury model (in contrast to the formalin model), thus suggests that the normalization of threshold requires the presence of an ongoing blockade for an extended period. We speculate that the products of the various second messenger cascades initiated by NMDA receptor-mediated Ca++ influx continue to exert effects for some time after interruption of the inciting signal (Coderre and Yashpal, 1994) in a manner analogous to the potentiation and temporal prolongation of activation seen in hippocampal slices after application of glutamatergic agents. Significantly, the return of neuropathy behavior within hours after drug administration indicates that such a relatively brief period of receptor blockade is inadequate to terminate the central facilitation process. In any case, it is clearly suggested by our dextrorphan data that the delay in analgesia, at the very least for this particular compound, does not reflect local kinetics in the need for spread to supraspinal sites: ICV dextrorphan had no effect, whereas IT dextrorphan did suppress allodynia.
NMDA antagonist drugs.
Our finding that dextrorphan was significantly effective by spinal but not by intracerebral injection illustrates the likelihood that the antiallodynic effect is a spinal cord effect and does not depend on redistribution to higher centers, despite the delay in onset of the effect. The finding of allodynia suppression by ICV morphine may indicate an important descending inhibitory system (Lee et al., 1995); the observation that IT morphine had a very limited effect serves to emphasize the specific relevance of the NMDA receptor in the spinal processing of tactile allodynia.
The lack of antihyperalgesic effect of spinally injected ketamine is remarkable in context with the immediate onset of motor blockade, clearly illustrating a spinal action on motor neurons, and with the complete lack of effect after ICV injection. These results, albeit not comprehensive of the drug class, argue strongly that the locus of antihyperalgesic effect of NMDA antagonists on hyperalgesia is in the spinal cord, not in supraspinal structures (e.g., thalamus). ICV injection of these relatively lipophilic agents should be an adequate method of delivery to adjacent brain structures likely to be relevant to afferent processing. Indeed, others have shown that stereotaxic injections of glutamate antagonists at supraspinal sites antagonize a glutamatergic pathway generating descending inhibitory modulation, likely opioidergic (Jensen and Yaksh, 1992a; Jensen and Yaksh, 1992b; Bach and Yaksh, 1995).
Our observation that ketamine was without effect in both the models examined in this study was surprising in view of the extensive literature supporting the NMDA antagonist efficacy of ketamine. Because the motor effects clearly indicated distribution to the spinal cord, and because spinal ketamine has been reported to have antihyperalgesic effects in other reports (Ren et al., 1992; Yamamoto and Yaksh, 1992b; Mao et al., 1993), it is not convincing to argue that pharmacokinetic factors prevented activity. Studies examining the behavioral effects of spinally applied ketamine in other models of hyperalgesia appear to have uniformly examined thermal hyperalgesia, whether evoked by paw carrageenan injection or chronic sciatic nerve constriction: we are not aware of previous behavioral assessments of IT ketamine effects in the rat formalin paw test, or of IT ketamine effects on tactile allodynia. The pharmacology of thermal hyperalgesia may differ in important ways from other hyperalgesic phenomena. This result is intriguing in context with the range of efficacies displayed by the panel of NMDA antagonists examined in this study. For example, MK801, although a potent/efficacious NMDA antagonist in other studies, showed some limitation of effect in the formalin test and markedly limited activity in the tight segmental nerve ligation model, a test that appeared more sensitive to the effects of other NMDA antagonists. Cloning of the NMDA receptor has revealed that there are a number of subtypes, and in situhybridization has mapped a differential geography for several of the subtypes in the brain (Brose et al., 1993; Watanabe et al., 1993; Bockers et al., 1994; Buller et al., 1994; Laurie and Seeburg, 1994; Monyer et al., 1994). Functional diversity of NMDA receptor subtypes has been described pharmacologically in the brains of both mouse (Kutsuwadaet al., 1992) and rat (Porter and Greenamyre, 1995). It is thus possible that the described subtypes that exist in the spinal cord (Tolle et al., 1993; Watanabe et al., 1994, a, b and c) have a distinctive pharmacology from the supraspinal types and that the activity of ketamine is lower at the predominant functional subtype in the lumbar dorsal horn mediating the anomalous pain states assessed. Alternatively, the selectivity of the antagonists studied for sensory over motor receptors may vary, and in the case of ketamine, may not differ significantly, in which case motor effects may be eclipsing sensory effects, hindering sensory evaluation in these purely behavioral models.
Clinical significance of NMDA antagonism.
A number of recent studies have attempted to define a clinical role for NMDA antagonists in neuropathic pain states. Such studies are typically limited by the prominent cognitive effects of these agents, usually psychotomimetic in nature, as exemplified by i.v. injection of the dissociative anesthetic agent ketamine. Two recent studies of systemically administered NMDA antagonists noted significant adverse cognitive effects amongst subjects (McQuay et al., 1994; Max et al., 1995). The spinal administration of NMDA antagonists thus represents an effort to increase drug concentrations at the presumed site of action although limiting spread to the higher central nervous system. Despite the documented cognitive side effects, there is increasing evidence that NMDA antagonists may serve a useful role in managing certain aberrant pain states. With particular reference to spinal delivery, Kristensenet al. (1992) have shown that the I.T. administration of the competitive antagonist CPP can diminish a major component of a post nerve injury pain state. Further investigations of the spinal role of the NMDA receptor antagonists requires considerable work and the development of an appropriate preclinical safety background. Recent studies examining the effects of dextrorphan, ketamine and memantine in a well-defined rat model of toxicology have shown that doses in excess of those used in our studies were without effect on function or spinal pathology (T. L. Yaksh, unpublished data). Subsequent studies confirming this lack of toxicity will require additional large animal investigations. Although it is possible that these agents may ultimately be systemically effective, such an outcome may hinge on the development of agents with receptor selectivity for afferent spinal cord systems, should this be demonstrated to be achievable. Pending such an advance, an appropriate alternative may in fact be spinal delivery for the management of otherwise refractory neuropathic states.
Acknowledgment
The technical assistance of Damon McCumber is gratefully acknowledged.
Footnotes
-
Send reprint requests to: Dr. Sandra R. Chaplan, Anesthesiology Research Laboratory, 0818, 9500 Gilman Drive, University of California, La Jolla, CA 92093-0818.
-
↵1 This work was supported by NIH Grants DA02110 (T.L.Y.) and THNS T-32-NS-07407 and the RSD Foundation (S.R.C.). Portions of this work were presented at the following meetings: CSA 4/93, IASP 8/93, ASA 10/93, SASP 3/94).
- Abbreviations:
- % MPE
- percent of maximum possible effect
- ANOVA
- analysis of variance
- AP3
- ±-2-amino-3-phosphonopropionic acid
- AP5
- ±-2-amino-5 phosphonopentanoic acid
- Ca++
- calcium
- CI
- confidence interval
- DNQX
- 6,7,dinitroquinoxaline-2,3-dione
- ICV
- intracerebroventricular
- IT
- intrathecal
- L2 (3
- 4,5,6), second (third, fourth, fifth, sixth) lumbar nerve(s)
- MD
- motor dysfunction
- Mg++
- magnesium
- mGluR
- metabotropic glutamate receptor
- MK801
- dizocilpine maleate
- MPE
- maximum possible drug effect
- NMDA
- N-methyl d-aspartate
- PE
- polyethylene
- S1 (2)
- first (second) sacral nerve
- TI
- therapeutic index
- Received February 27, 1996.
- Accepted October 21, 1996.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}