


Abstract
Opioid-induced hyperalgesia (OIH) is a less-studied phenomenon that has been reported in both preclinical and clinical studies. Although the underlying cause is not entirely understood, OIH is a real-life problem that affects millions of patients on a daily basis. Research has implicated the important contribution of Ca2+/calmodulin-dependent protein kinase IIα (CaMKIIα) to OIH at the level of spinal nociceptors. To expand our understanding of the entire brain circuitry driving OIH, in this study we investigated the role of CaMKIIα in the laterocapcular division of the central amygdala (CeLC), the conjunctive point between the spinal cord and rostro-ventral medulla. OIH was produced by repeated fentanyl administration in the rat. Correlating with the development of mechanical allodynia and thermal hyperalgesia, CaMKIIα activity was significantly elevated in the CeLC in OIH. In addition, the frequency and amplitude of spontaneous miniature excitatory postsynaptic currents (mEPSCs) in CeLC neurons were significantly increased in OIH. 2-[N-(2-hidroxyethyl)-N-(4-methoxy-benzenesulfonyl)]-amino-N-(4-chlorocinnamyl)-N-methylbenzylamine, a CaMKIIα inhibitor, dose dependently reversed sensory hypersensitivity, activation of CeLC CaMKIIα, and mEPSCs in OIH. Taken together, our data for the first time implicate a critical role of CeLC CaMKIIα in OIH.
Introduction
Opioids play an important role in every aspect of modern anesthesia and pain medicine. However, the administration of opioids has sometimes been found to produce opioid-induced hyperalgesia (OIH), defined as a lowered pain threshold caused by opioid exposure (Vanderah et al., 2001; Mao, 2002; Ossipov et al., 2005). Opioids also cause analgesic tolerance that shares similar manifestations of symptoms with OIH since opioid drugs exhibit diminished efficacy with time in both cases (Kim et al., 2014; Lee and Yeomans, 2014). However, OIH differs from opioid tolerance in that increasing opioid dose aggravates pain in OIH, whereas tolerance is generally countered by higher doses (Lee and Yeomans, 2014; Stoicea et al., 2015). The presence of OIH can be a clinical challenge not only in chronic pain management but also in perioperative pain. For the latter, fentanyl is commonly used in routine anesthesia practice given its potent, fast, and short analgesic action. While most OIH studies have focused on the prototype opioid drug morphine, hyperalgesia after fentanyl exposures needs more and urgent scientific and medical attention.
The neurobiology of OIH is complex and several mechanisms have been proposed (Guignard et al., 2000; Ossipov et al., 2005; Lee and Yeomans, 2014). A number of pronociceptive mechanisms proposed thus far include the activation of central glutamatergic pathways, descending facilitation, alteration of endogenous opioid ligands, and others, for inducing OIH (Célèrier et al., 2000; Vanderah et al., 2001; Lee and Yeomans, 2014). The Ca2+/calmodulin-dependent protein kinase IIα (CaMKIIα), colocalized with the μ-opioid receptor in the spinal cord and central nucleus of amygdala [i.e., laterocapcular division of central amygdala (CeLC)] (Brüggemann et al., 2000), is a multifunctional serine/threonine protein kinase that plays a prominent role in glutamate neurotransmission and experience-dependent plasticity at the synaptic level (Lisman et al., 2002; Salling et al., 2016), which makes it a highly significant target in the search for neural mechanisms of OIH. It has been demonstrated that spinal CaMKIIα activity is required for the initiation and maintenance of OIH. Intrathecal CaMKIIα inhibition can attenuate OIH (Chen et al., 2010) and opioid tolerance and dependence (Tang et al., 2006; Yang et al., 2011). CaMKIIα has also been reported to contribute to hyperalgesia priming, a phenomenon implicated in the transition from acute to chronic pain (Ferrari et al., 2013).
However, neither the peripheral nor the spinal mechanisms can completely explain the cause of OIH (Chu et al., 2011). In this study, we investigated the CaMKIIα mechanism in the CeLC, a target of the spino-parabrachio-amygdaloid pain pathway that has been considered as the nociceptive amygdala (Neugebauer et al., 2004; Sarhan et al., 2005, 2013). The CeLC is also the upstream of periaqueductal gray/rostro-ventral medulla/spinal descending pain pathway (Fabry et al., 2003; Neugebauer et al., 2004; Carrasquillo and Gereau, 2007; Hamlin et al., 2007; Tracey and Mantyh, 2007; Martin and Ewan, 2008) that may be involved in OIH (Vanderah et al., 2001; Ossipov et al., 2005). Since spontaneous miniature excitatory postsynaptic currents (mEPSCs), which are observed in the absence of presynaptic action potentials, are frequently used as a parameter to reflect altered synaptic transmission responsible for inflammatory pain (Zhao et al., 2006) and neuropathic pain (Wang et al., 2007; Xu et al., 2008), we determined the changes in CaMKIIα activity and mEPSCs in CeLC neurons in a rat model of fentanyl-induced hyperalgesia.
Materials and Methods
Materials
Fentanyl (fentanyl citrate injection) was obtained from Yi Chang Humanwell Pharmaceutical Co., Ltd (Yi Chang, China). 2-[N-(2-hidroxyethyl)-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine (KN93) and 2-[N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine (KN92) monohydrochloride) were purchased from Cayman (Ann Arbor, MI). Other chemicals were obtained from Beyotime (Shanghai, China) and Boster (Shanghai, China).
Animals
Male Sprague-Dawley rats (50–70 g, from the animal laboratory at Tongji Medical College, Huazhong University of Science and Technology) were provided food and water ad libitum prior to the experiments. All animal experiments were performed under a protocol approved by the Institutional Animal Care and Use Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, and conducted in accordance with the National Institutes of Health’s guide for the care and use of laboratory animals and the policies and recommendations by the International Association for the Study of Pain (Zimmermann, 1983).
OIH Induced by Repeated Subcutaneous Administration of Fentanyl.
OIH was induced in rats by four injections of fentanyl (60 μg/kg per injection, s.c.) at 15-minute intervals, resulting in a cumulative dose of 240 μg/kg/rat (Célèrier et al., 2000). This is an activity-dependent pain model, i.e., signs of spontaneous pain are typically not observed in the absence of external stimulation or movement (Toyoda et al., 2009). This is a model of intermittent fentanyl injections, rather than chronic administration that can also induce opioid tolerance (Zissen et al., 2007). The nociceptive thresholds were evaluated by mechanical and thermal stimulation at different time points, including 0 (baseline), 1, 5, 6, 6.5, 7, and 8 hours and 1, 2, 3, 4, and 5 days after the last injection of fentanyl (time 0).
Assessment of Mechanical Allodynia
Mechanical sensitivity was assayed by the up-and-down paradigm using von Frey filaments (North Coast, San Jose, CA) according to the Dixon method (Luo et al., 2008; Chen et al., 2009). In brief, rats were individually placed into Plexiglas chambers over a mesh table and acclimated for 30 minutes before the test. Beginning with 1.0 g, each von Frey filament was applied to the mid-plantar surface of the left hind paw for 5 seconds or until a withdrawal response occurred. Positive response was defined as paw flinching or brisk withdrawal. The time interval between each test was more than 5 minutes. The 50% probability of paw withdrawal threshold was determined by an up-and-down algorithm described previously (Dixon and Mood, 1948; Luo et al., 2008).
Assessment of Thermal Hyperalgesia
Thermal sensitivity was measured using a radiant thermal stimulator (BME-410C; Biomedical Engineering, Boerni science and technology limited company, Guangzhou, China) according to the Hargreaves’ method (Hargreaves et al., 1988; Luo et al., 2008; Chen et al., 2009). Rats were placed in a clear plastic enclosure and allowed to acclimatize for 30 minutes before assessment. A radiant heat source was focused onto the plantar surface of the left hind paw through the glass floor. Measurement of thermal sensitivity was started by the activation of the heat source and automatically stopped when paw withdrawal occurred. A cutoff time of 15 seconds was applied to prevent tissue damage.
Western Blotting
Immediately after rats were euthanized, the CeLC was quickly dissected out, frozen, and stored at −80°C before western blotting analysis (Chen et al., 2010). In brief, tissues were homogenized in radio-immuno-precipitation assay buffer [20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, sodium pyrophosphate, β-glycerophosphate, EDTA, Na3VO4, and leupeptin] and protein concentration was determined using the bicinchoninic acid method (Beyotime). Samples (20 μg of total protein) were separated by 10% SDS-PAGE and transferred electrophoretically onto a polyvinylidene fluoride (PVDF) membrane. The membranes were blocked with 5% skim milk for 2 hours at room temperature and incubated overnight in 5% skim milk with a rabbit anti-(T286) phosphorylated CaMKIIα (p-CaMKIIα) antibody (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA) or a mouse anti- glyceraldehyde-3-phosphate dehydrogenase antibody (1:500; Boster). After the incubation, blots were washed and incubated at room temperature for 90 minutes with horseradish peroxidase–conjugated anti-rabbit (for p-CaMKIIα) or anti-mouse (for glyceraldehyde-3-phosphate dehydrogenase) secondary antibody IgG (1:10,000; Boster). The p-CaMKIIα antibody detected double bands in the experiments, both of which correspond to p-CaMKIIα (Hu et al., 2016). Signals from both bands were combined for quantification. The immunoreactivity was detected using enhanced chemiluminescence (Thermo Fisher Shanghai, China) and enhanced chemiluminescence signals were detected using a Bio-Rad (Shanghai, China) ChemiDoc system. The p-CaMKIIα immunoreactivity was expressed as the ratio of the optical densities of p-CaMKIIα to those of glyceraldehyde-3-phosphate dehydrogenase.
Intra-CeLC Cannulation and Microinjection
Animals were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and held in a stereotaxic frame (Zenda, Austin, TX), as previously reported (Han et al., 2010; Sarhan et al., 2013; Zhang et al., 2013), A guide cannula (RWD Life Science, Shenzhen, China) was implanted toward the CeLC in the right hemisphere, using the following coordinates (in mm): 2.2 caudal to bregma, 4.2 lateral to midline, and depth 7.5 according to the Paxinos and Watson flat skull coordinate system (Butler et al., 2011). A 33-gauge dummy cannula was inserted in each guide cannula to avoid clogging. Following cannulation, animals were housed singly and allowed to recover for 5 days prior to the experiment. For drug infusion, the dummy cannula was replaced by an injector that was inserted 0.5 mm beyond the guide cannula to target the CeLC. One end of the tubing was connected to the injector and the other to a 10 μl Hamilton syringe. The solution was infused with a pump at 0.25 μl/min for a total of 0.5 μl on the right CeLC. After infusion, the injector was left in the cannula for another minute to allow the chemicals to diffuse into the injected area. At the end of experiments, the placement of the cannula was verified histologically by injecting 0.5 μl of eosin in the same manner as described previously.
Electrophysiology Experiment
Slice Preparation.
Coronal brain slices (350 μm) containing CeLC were obtained from the right hemisphere as described previously (Li et al., 2011) with cutting solution at 4°C. The dissection solution contained (in mM) 213 sucrose, 3 KCl, 1 NaH2PO4, 0.5 CaCl2, 5 MgCl2, 26 NaHCO3, and 10 glucose. After incubation at 25°C for at least 1 hour, a single brain slice was placed in a submerged recording chamber and perfused continuously at a rate of 2 ml/min with artificial cerebrospinal fluid equilibrated with 95% O2 and 5% CO2 at 30°C. The artificial cerebrospinal fluid contained (in mM) 125 NaCl, 5 KCl, 1.2 NaH2PO4, 2.6 CaCl2, 1.3 MgCl2, 26 NaHCO3, and 10 glucose. Only 1 to 2 brain slices per animal were used, and only one neuron was recorded in each slice.
Whole-Cell Patch-Clamp Recording.
Recording of mEPSCs was performed using whole-cell voltage-clamp methods as previously described (Li et al., 2011). The CeLC neurons were easily discerned under light microscopy and visualized in a transparent circular region adjacent to the basal lateral amygdala (Sah et al., 2003; Fu et al., 2008; Watabe et al., 2013). Electrodes (impedance was 4–6 MΩ) made from borosilicate glass capillaries (1.5 mm outer diameter, 1.0 mm inner diameter; WPI, Sarasota, FL) were filled with the following internal solution (in mM): 145 KCl, 5 NaCl, 10 HEPES, 5 EGTA, 4 Mg-ATP, and 0.3 Na3-GTP. A dual four-pole Bessel filter (Warner Instruments, Hamden, CT), a low-noise Digidata 1322 interface (Molecular Devices, Sunnyvale, CA), HEKA EPC-10 amplifier (HEKA, Lambrecht, Germany), a Pentium PC, and PATCHMASTER software (Molecular Devices) were used for data acquisition and analysis. The head stage voltage was monitored continuously on an oscilloscope to ensure precise performance of the amplifier. High (>2 GΩ) seal and low (<20 MΩ) series resistances were checked throughout the experiment [using the pCLAMP10 membrane test function (Molecular Devices)] to ensure high-quality recordings. The mEPSCs were recorded in the presence of 50 μM picrotoxin and 1 μM tetrodotoxin at a holding potential of −70 mV and measured 10 minutes before and 15 minutes after drug application, as described previously (Kiritoshi et al., 2013). A fixed length of traces (5 minutes) was analyzed for frequency and amplitude distributions with Mini Analysis Program 6.0 (Synaptosoft Inc, Fort Lee, NJ) and pCLAMP10 software.
Statistics.
The number of rats for behavioral experiments was estimated by power analyses with the aid of the SSize2021 software (National University of Singapore, Singapore) (version 2). With anticipated P1 = 0.95 and P2 = 0.05, preset analysis power of 0.9, and level of significance of 0.05, the minimal group size was estimate to be four/group. All data are presented as mean ± S.E.M. Comparisons between groups were analyzed using Student’s t test (two groups) or analysis of variance followed by the Bonferroni post hoc test (multiple groups). All graphs and statistical analysis were performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA). A value of P < 0.05 was considered statistically significant.
Results
Fentanyl Induced Mechanical Allodynia and Thermal Hyperalgesia in a Time-Dependent Manner in the Rat.
Fentanyl is a commonly used clinical anesthetic agent with significant problems of OIH. To study fentanyl-induced hyperalgesia, we established a preclinical model using four intermittent injections (s.c.) of fentanyl that resulted in significant increases in mechanical and thermal sensitivities compared with saline-treated rats (Fig. 1). Fentanyl initially increased mechanical thresholds and thermal latencies as expected due to the drug’s acute analgesic action. Five hours after the last dose of fentanyl, the analgesic effect diminished and detectable mechanical allodynia (Fig. 1A) and thermal hyperalgesia (Fig. 1B) developed, which lasted for 3 days (mechanical allodynia) to 4 days (thermal hyperalgesia).

Repeated fentanyl administration induced OIH including mechanical allodynia (A) and thermal hyperalgesia (B). Rats received saline or fentanyl sulfate on day 0 (60 μg/kg, 4 times, 15-minute intervals, s.c.). The paw withdrawal thresholds to von Frey filament probing and withdrawal latencies to radiant heat were determined. *P < 0.05, **P < 0.01, ***P < 0.001, compared with the saline-treated group, two-way analysis of variance followed by Bonferroni post hoc test. Data represent mean ± S.E.M. of six rats per group.
Fentanyl Increased the Level of p-CaMKIIα in the CeLC.
To evaluate the potential effect of fentanyl on CaMKIIα activity, the level of CaMKIIα phosphorylation at Thr-286 (pCaMKIIα) in the CeLC was determined using the western blotting method 24 hours after induction of OIH (Fig. 2). Compared with saline-treated rats, the content of p-CaMKIIα in the CeLC was significantly increased in the rat with fentanyl-induced OIH (P < 0.05, n = 12).

Repeated fentanyl administration induced CaMKIIα activation (p-CaMKIIα) in the CeLC. (A) Representative immunoblots of activated CaMKIIα (p-CaMKIIα) in the CeLC after repeated saline or fentanyl treatment (60 μg/kg, 4 times, 15-minute intervals, s.c.). (B) Histogram of p-CaMKIIα, **P < 0.01 compared with the saline group, Student’s t test (t = 3.58, n = 12).
Fentanyl-Induced Hyperalgesia Was Reversed by KN93 Microinjected into the CeLC.
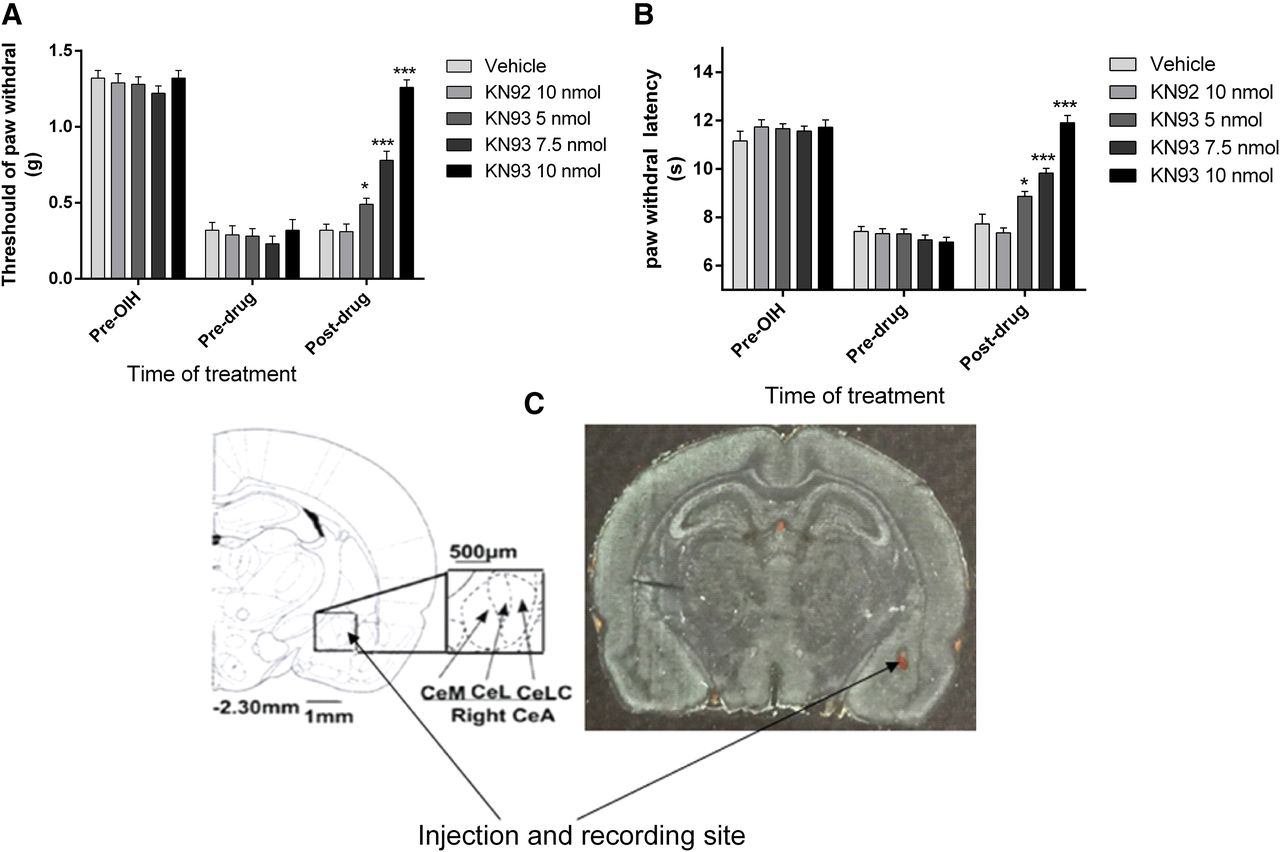
To further investigate the functional role of CeLC CaMKIIα in OIH, rats were implanted with CeLC cannulas before induction of OIH by fentanyl. After OIH was established, KN93 (a CaMKII inhibitor) or KN92 (a kinase-inactive chemical analog of KN93) was microinjected into the CeLC via cannulas 6.5 hours after the last dose of fentanyl administration. The established fentanyl OIH was rapidly attenuated by KN93 in a dose-dependent manner (Fig. 3). KN93 (5–7.5 nmol) partially suppressed mechanical allodynia and thermal hyperalgesia. At the highest dose used, KN93 (10 nmol) completely abolished allodynia and hyperalgesia (Fig. 3) (*P < 0.05, ***P < 0.001, compared with the vehicle-treated group, n = 8–10 per group). As controls, the same treatment with KN92 (10 nmol) or vehicle (equal volume) did not alter the pain threshold. These data suggested a functional role of CeLC CaMKIIα in OIH.
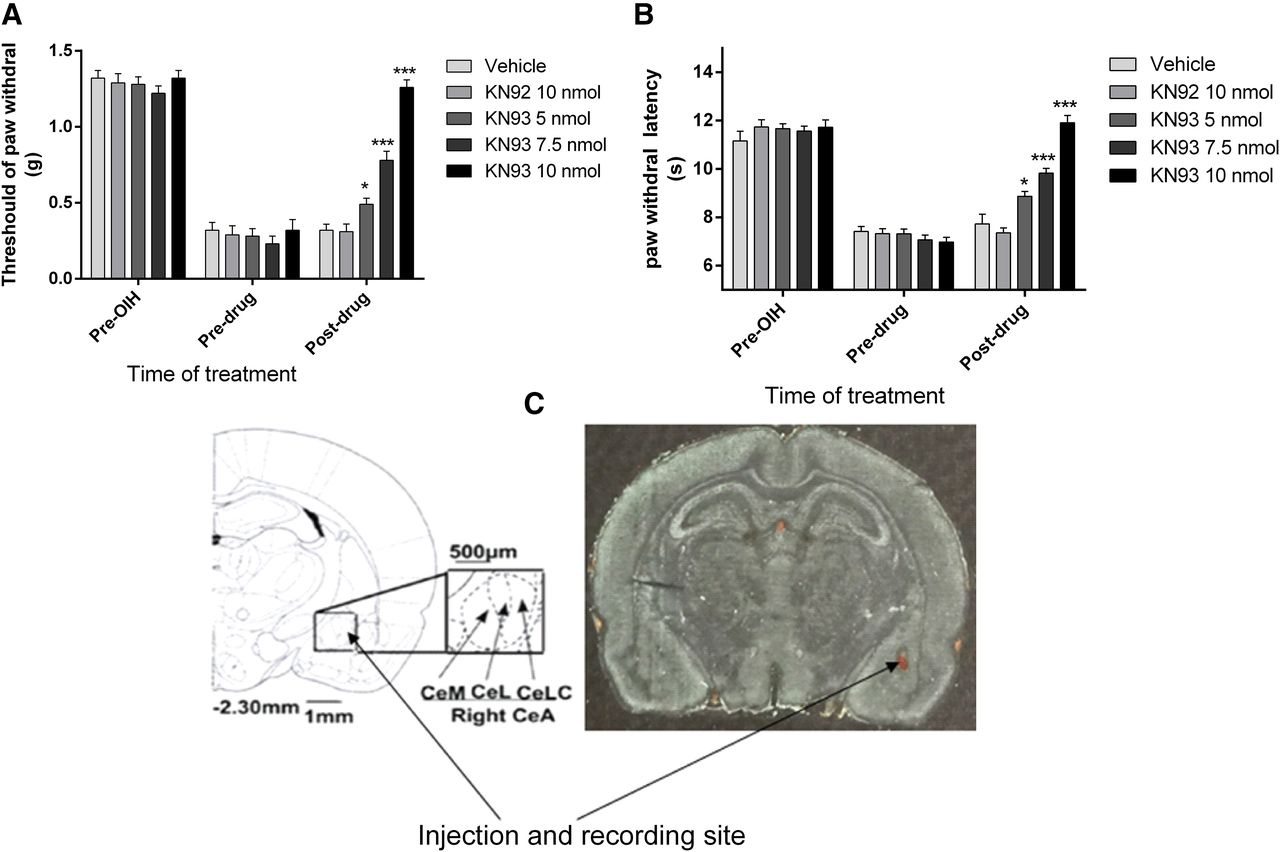
Dose-dependent reversal of fentanyl-induced mechanical allodynia (A) and thermal hyperalgesia (B) by microinjecting KN93 into the CeLC (C). Rats received saline or fentanyl sulfate to induce OIH (see Fig. 1). KN93 (5–10 nmol), KN92 (10 nmol), or vehicle was administered 6.5 hours after the last dose of fentanyl. Mechanical allodynia and thermal hyperalgesia were tested 30 minutes after injection. KN93, but not KN92, reversed the established fentanyl-induced mechanical allodynia (F = 70.16) and thermal hyperalgesia (F = 50.08) in a dose-dependent manner. Data are expressed as mean ± S.E.M. * P < 0.05, *** P < 0.001, compared with the vehicle-treated group, one-way analysis of variance followed by Bonferroni post hoc test, n = 8–10 for each group. The cannula placement in the CeLC was verified at the end of the experiments (C).
Intra-CeLC KN93 Microinjection Inhibited CeLC p-CaMKIIα in OIH.
To correlate the behavioral effects with biochemical changes, CaMKIIα activity (p-CaMKIIα) in the CeLC was determined by analyzing the level of phosphorylation (p-CaMKIIα). The KN93 dose dependently reversed fentanyl-induced activation of p-CaMKIIα. At the highest dose used, KN93 (10 nmol) significantly attenuated p-CaMKIIα (P < 0.001, compared with vehicle group, n = 6). Lower doses of KN93 (5 and 7.5 nmol) had a less potent inhibition effect on P-CaMKIIα. In contrast, fentanyl-induced activation of CaMKIIα was not changed by KN92 (10 nmol) (Fig. 4). These data suggested that repeated fentanyl administration induced CaMKIIα activation, mechanical allodynia, and thermal hyperalgesia, all of which were attenuated by inhibiting the activity of CeLC CaMKIIα with KN93, but not the kinase-inactive chemical analog KN92. Therefore, CeLC CaMKIIα is critical for the persistent of OIH.

Suppression of fentanyl-induced CaMKIIα activation by KN93 in the CeLC. Rats with fentanyl-induced OIH were treated (intra-CeLC) with KN93 (5–10 nmol), KN92 (10 nmol), or vehicle 6.5 hours after the last dose of fentanyl. One hour later, rats were sacrificed and the central amygdala was taken for the analysis of CaMKIIα activation using the immunoblotting method, by determining the degree of CaMKIIα T286 autophosphorylation (p-CaMKIIα). KN93, but not its inactive analog KN92, reversed fentanyl-enhanced CaMKIIα activation. (A) Representative immunoblots for p-CaMKIIα. (B) Histogram of CaMKIIα activation. **P < 0.01, ***P < 0.001, compared with the vehicle-treated group, F = 47.07, one-way analysis of variance followed by Bonferroni post hoc test, n = 6 for each group.
Suppression of mEPSCs in CeLC Neurons from the OIH Rats by KN93.
The analysis of the amplitude and frequency distribution of mEPSCs in the presence of tetrodotoxin can be used to determine pre- versus postsynaptic mechanisms. Presynaptic changes at the transmitter release site affect mEPSC frequency, whereas changes at the postsynaptic membrane alter mEPSC amplitude (quantal size) (Toyoda et al., 2009; Han et al., 2010).We recorded mEPSCs in the CeLC in the presence of 1 μM tetrodotoxin. KN93 (10 μM) (Goforth et al., 2004; Shen et al., 2009; Seto et al., 2013) significantly decreased both the frequency (Fig. 5C) and amplitude (Fig. 5D) of mEPSCs recorded from CeLC neurons in slices from OIH rats (12 hours postinduction), but not those from the vehicle-treated rats (P < 0.05, n = 4–7).

Suppression of mEPSC frequency and amplitude by KN93 in neurons from OIH rats. (A) mEPSC recordings in control and OIH groups at baseline and after the application of KN93. Calibration: 1 second, 20 pA. (B) Individual mEPSCs obtained from respective recordings. Calibration: 10 ms, 20 pA. (C and D) Bar graphs showing the frequency (C) and amplitude (D) of mEPSCs in the CeLC. Note that the frequency (t = 2.9, P < 0.05) and amplitude (t = 3.35, P < 0.05) of mEPSCs were significantly increased in OIH neurons. Also note that the frequency (t = 3.11, P < 0.05) and amplitude (t = 3.74, P < 0.05) of mEPSCs were significantly suppressed by KN93. *P < 0.05, Student’s t test, n = 4–7 neurons from each group.
Discussion
In this study, we examined the role of CaMKIIα in an experimental model of OIH induced by the anesthetic agent fentanyl. Rats received four injections (s.c) of fentanyl at 15-minute intervals, which were highly reproducible to induce OIH. Célèrier et al. (2000) tested different doses (20–100 μg/kg) of fentanyl and found that the higher the dose used, the more pronounced was the fentanyl-induced hyperalgesia. In our pilot experiments, we found that fentanyl at a moderate dose (60 μg/kg) was the optimum dose because of increased mortality after higher doses.
We found that CaMKIIα activity in the CeLC was increased in OIH rats, and this increased CeLC CaMKIIα activity correlates well with increased neuronal activity and behavior hyperalgesia. Moreover, inhibition of CeLC CaMKIIα by microinjection of KN93 reversed established fentanyl-induced hyperalgesia in a dose-dependent manner, correlating with decreased p-CaMKIIα. Meanwhile, the frequency and amplitude of mEPSCs in CeLC cells were reduced by KN93 in rats with OIH. In contrast, the kinase-inactive control compound KN92 did not affect p-CaMKIIα activity or OIH. These data, for the first time, implicated a critical role of CeLC CaMKIIα for the maintenance of fentanyl-induced OIH in young rats. Whether this result can be extrapolated to older rats needs to be further studied.
OIH is an activity-dependent pain model, i.e., signs of pain are typically not observed in the absence of external stimulation or movement (Neugebauer et al., 2007). Indeed, lidocaine or clonidine could not induce conditioned place preference in uninjured animals (He et al., 2012) including animals with OIH (He, Hu, and Wang, unpublished data). The present study demonstrates directly that increased amygdala activity in the absence of tissue or nerve injury can exacerbate physiologic pain responses, such as thermal hyperalgesia evoked by noxious heat stimuli and allodynia induced by otherwise in noxious stimulation.
It has reported that the right amygdala develops pain-related plasticity that is coupled to pain facilitation in the arthritis pain model (Han and Neugebauer, 2005). Because of the strong contralateral projection of the spino-parabrachio-amygdaloid pain pathway (Neugebauer et al., 2004), we chose to perform behavioral tests in the rat left plantar, while analyzing the level of p-CaMKIIα and recording mEPSCs by whole-cell patch-clamp in the right CeLC.
In addition to CaMKIIα, several other potential mechanisms underlying OIH have been suggested (Lee and Yeomans, 2014), such as the β2-adrenergic receptor (Jordan et al., 2003), spinal cycloxygenase (Dunbar et al., 2000), as well as local cytokine production (Liang et al., 2008). To date, central sensitization, providing a mechanistic explanation for how low-threshold A or C fibers can begin to transmit pain, has been generally accepted as an important mechanism in the development of OIH (Chu et al., 2008). Indeed, central sensitization includes both homosynaptic and heterosynaptic facilitations (Sandkühler, 2007), whereas heterosynaptic facilitation alone is responsible for secondary hyperalgesia and allodynia, in which activity in one set of synapses enhances activity in nonactivated synapses, typically by sensitizing the entire neuron (Latremoliere and Woolf, 2009).
The analysis of mEPSCs is a well-established electrophysiological approach to determine pre- versus postsynaptic mechanisms. Miniature postsynaptic currents are assumed to represent the spontaneous release of individual vesicles or quanta of neurotransmitters from the presynaptic membrane (Kaeser and Regehr, 2014). Presynaptic changes at the transmitter release site affect frequency, whereas changes at the postsynaptic membrane would alter amplitude (quantal size) (Wyllie et al., 1994). Based on the analysis of miniature synaptic events in this study, we found that KN93 decreased both the frequency and amplitude of mEPSCs in neurons from the OIH rats. These data suggest that p-CaMKIIα regulate synaptic transmission in CeLC neurons through pre- and postsynaptic mechanisms of action, which is consistent with a previous study indicating that both pre- and postsynaptic CaMKIIα are necessary for the induction of synaptic plasticity (Ninan and Arancio, 2004). Therefore, reduced excitatory synaptic transmission after KN93 injection is attributable to not only a decrease in probability of presynaptic neurotransmitter release but also to a decrease of postsynaptic responsiveness.
CaMKIIα makes up 2% of the total postsynaptic density protein. It has been demonstrated that the N-methyl-D-aspartate receptor (NMDAR) is both a trigger and effector of central sensitization (Woolf and Thompson, 1991; Latremoliere and Woolf, 2009). Activation of the NMDAR is an essential step in both initiating and maintaining activity-dependent central sensitization since its blockade by NMDAR antagonists prevent or reverse OIH (Célèrier et al., 2000; Rivat et al., 2002). The postsynaptic membrane of the NMDAR is activated and drives aside the Mg2+ ion in the receptor channel. With the opening of the NMDAR, Ca2+ ion permeability increases. A large amount of Ca2+ enters into the cell and then further activates intracellular Ca2+-dependent protein kinases, such as CaMKIIα (Lisman et al., 2002). After CaMKIIα phosphorylation and binding to NR2B, the excitability of the NMDAR and Ca2+ influx increases and further activates CaMKIIα, forming a positive feed-forward mechanism (Wang and Wang, 2003; Wilkie et al., 2010).
CaMKIIα is also expressed at the presynaptic nerve terminal where it associates with synaptic vesicles (Wang, 2008). Although presynaptic CaMKIIα appears to have complex functions in synaptic transmission, studies suggest that presynaptic CaMKIIα enhances vesicle motility and facilitates spontaneous neurotransmitter release through promoting synaptic vesicle translocation and increasing Ca2+ entry (Wang, 2008). The transient receptor potential vanilloid type 1 (TRPV1) channel, which has been reported to be an essential peripheral mechanism in the expression of morphine-induced hyperalgesia (Vardanyan et al., 2009), is involved in the regulation of synaptic transmission centrally (Shoudai et al., 2010) or peripherally (Sikand and Premkumar, 2007) by enhancing glutamate release from nerve endings (Kaeser and Regehr, 2014; Ramírez-Barrantes et al., 2016). In view of the fact that both TRPV1 mRNA and protein have been found in the central amygdale (Zschenderlein et al., 2011; Ramírez-Barrantes et al., 2016), and that there is evidence for physiologic and pharmacological interactions between CaMKIIα and TRPV1 receptors (Price et al., 2005; Nakanishi et al., 2010) forming possible feed-forward loops (Wang et al., 2010), it is therefore possible that presynaptic CaMKIIα may also be a regulator of fentanyl-induced hyperalgesia.
In the CeLC, CaMKIIα is colocalized with the μ-opioid receptors (Brüggemann et al., 2000; Carlton, 2002). Cellular and biochemical evidence have supported the possibility that CaMKIIα and the μ-opioid receptor can directly interact with each other, modulating kinase and receptor activity (Tang et al., 2006; Yang et al., 2011).Therefore, it is plausible that the μ-opioid receptor, CaMKIIα, NMDAR, and TRPV1 in the CeLC work in concert to regulate opioid activity and OIH.
Spontaneous activity may adjust synaptic strength by regulating protein synthesis. In cultured hippocampal pyramidal cells, spontaneous glutamate release activates NMDARs and tonically suppresses local protein synthesis in dendrites (Sutton et al., 2004, 2006; Sutton and Schuman, 2006). In this study, the finding of p-CaMKIIα-induced increases in synaptic transmission and excitability in CeLC neurons indicates it maybe one possible mechanism underlying increased nocifensive responses (mechanical allodynia and thermal hyperalgesia) in the absence of tissue injury.
In conclusion, our findings demonstrate that CaMKIIα in the CeLC is involved in the development of fentanyl-induced hyperalgesia that can be disrupted by locally inhibiting CaMKIIα in the CeLC through both pre- and postsynaptic mechanisms. The present study provides insights into superspinal CaMKIIα mechanisms promoting OIH, which may facilitate exploring new pharmacological interventions targeting unsolved opioid side effects in the future.
Authorship Contributions
Participated in research design: Z. Li, Wang, Luo.
Conducted experiments: Z. Li, C. Li, Yin.
Performed data analysis: Z. Li, C. Li, Luo.
Wrote or contributed to the writing of the manuscript: Z. Li, Wang, Luo.
Footnotes
- Received March 23, 2016.
- Accepted July 18, 2016.
↵1 Z.L. and C.L. contributed equally to this work.
This work was supported by grants from the National Science Foundation of the People’s Republic of China [Grants 81328009, 81050023, and 81271234].
Abbreviations
- CaMKIIα
- Ca2+/calmodulin-dependent protein kinase IIα
- CeLC
- laterocapcular division of the central amygdala
- KN92
- 2-[N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine
- KN93
- 2-[N-(2-hidroxyethyl)-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine
- mEPSC
- miniature excitatory postsynaptic current
- NMDAR
- N-methyl-D-aspartate receptor
- OIH
- opioid-induced hyperalgesia
- p-CaMKIIα
- phosphorylated Ca2+/calmodulin-dependent protein kinase IIα
- TRPV1
- transient receptor potential vanilloid type 1
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}