


Abstract
Hyperaldosteronism and hypertension were unexpected side effects observed in trials of torcetrapib, a cholesteryl ester-transfer protein (CETP) inhibitor that increases high-density lipoprotein. Given that CETP inhibitors are lipid soluble, accumulate in adipose tissue, and have binding sites for proteins involved in adipogenesis, and that adipocytes are a source of aldosterone, we questioned whether CETP inhibitors (torcetrapib, dalcetrapib, and anacetrapib) influence aldosterone production by adipocytes. Studies were performed using human adipocytes (SW872), which express CETP, and mouse adipocytes (3T3-L1), which lack the CETP gene. Torcetrapib, dalcetrapib, and anacetrapib increased expression of CYP11B2, CYP11B1, and steroidogenic acute regulatory protein, enzymes involved in mineralocorticoid and glucocorticoid generation. These effects were associated with increased reactive oxygen species formation. Torcetrapib, dalcetrapib, and anacetrapib upregulated signal transducer and activator of transcription 3 (STAT3) and peroxisome proliferation-activated receptor-γ, important in adipogenesis, but only torcetrapib stimulated production of chemerin, a proinflammatory adipokine. To determine mechanisms whereby CETP inhibitors mediate effects, cells were pretreated with inhibitors of Nox1/Nox4 [GKT137831; 2-(2-chlorophenyl)-4-[3-(dimethylamino)phenyl]-5-methyl-1H-pyrazolo[4,3-c]pyridine-3,6(2H,5H)-dione], Nox1 (ML171 [2-acetylphenothiazine]), mitochondria (rotenone), and STAT3 (S3I-201 [2-hydroxy-4-(((4-methylphenyl)sulfonyloxy)acetyl)amino)-benzoic acid]). In torcetrapib-stimulated cells, Nox inhibitors, rotenone, and S3I-201 downregulated CYP11B2 and steroidogenic acute regulatory protein and reduced aldosterone. Dalcetrapib and anacetrapib effects on aldosterone were variably blocked by GKT137831, ML171, rotenone, and S3I-201. In adipocytes, torcetrapib, dalcetrapib, and anacetrapib inhibit enzymatic pathways responsible for aldosterone production through Nox1/Nox4- and mitochondrial-generated reactive oxygen species and STAT3. CETP inhibitors also influence adipokine production. These processes may be CETP independent. Our findings identify novel adipocyte-related mechanisms whereby CETP inhibitors increase aldosterone production. Such phenomena may contribute to hyperaldosteronism observed in CETP inhibitor clinical trials.
Introduction
Cardiovascular disease (CVD) is a leading cause of death worldwide (Alwan et al., 2010). Dyslipidemia is one of the many risk factors associated with CVD is dyslipidemia, particularly low levels of high-density lipoprotein (HDL) and high levels of low-density lipoprotein (LDL) (Miller and Miller, 1975; Gordon et al., 1977; Roheim, 1986). Low HDL is a risk factor for coronary heart disease independent of LDL levels (Gordon et al., 1977). Factors implicated in low HDL include genetics (single-nucleotide polymorphisms of genes regulating HDL) and environment (diet, exercise, obesity, etc.) (Voight et al., 2012; Luscher et al., 2014). HDL reduces cholesterol by transferring it from peripheral cells to the liver for excretion (reverse cholesterol transport). On the other hand, cholesteryl ester-transfer protein (CETP) transfers cholesterol ester from antiatherogenic HDL to proatherogenic LDL or very-low-density lipoprotein (Tall, 1986; Barter et al., 2003). As such, HDL has been considered a potential therapeutic target for the prevention and treatment of CVDs. Various lipid-modulating drugs are currently available for clinical use, including statins, niacin, and fibrates, but their HDL-elevating effects are modest. Based on genetic studies demonstrating that CETP deficiency is associated with markedly increased HDL levels (Inazu et al., 1990), drugs have been developed to inhibit CETP as a strategy to increase HDL, which may have cardiovascular protective effects, at least in some patients (Voight et al., 2012). Small-molecule CETP inhibitors used clinically include torcetrapib, dalcetrapib, anacetrapib, and evacetrapib, all of which increase HDL (Johns et al., 2012).
The Investigation of Lipid Level Management to Understand Its Impact in Atherosclerotic Events (ILLUMINATE) trial, a major phase 3 secondary prevention morbidity and mortality trial in more than 15,000 subjects, compared the CETP inhibitor torcetrapib plus atorvastatin with atorvastatin alone. The ILLUMINATE trial was terminated prematurely because of higher all-cause morbidity and mortality, despite elevated HDL. Reasons for this remain unclear, although elevated blood pressure and an associated increase in plasma aldosterone have been implicated (Barter et al., 2007). Aldosterone can cause cardiovascular injury independently of blood pressure–elevating effects. Data from dalcetrapib trials demonstrated a modest blood pressure–elevating effect (Schwartz et al., 2012), whereas anacetrapib does not seem to influence blood pressure, although clinical trials are still ongoing (Robinson and Frishman, 2014). Variable effects on plasma aldosterone have been demonstrated for these CETP inhibitors (Kontush et al., 2008; Johns et al., 2012).
Mechanisms underlying torcetrapib-induced hyperaldosteronism are unclear, but in vitro studies demonstrated that in adrenal cell lines, torcetrapib stimulated aldosterone production by increasing expression of aldosterone synthase (CYP11B2) (Forrest et al., 2008; Hu et al., 2009). We previously demonstrated that adipocytes possess the enzymatic machinery to synthesize mineralocorticoids and glucocorticoids, namely CYP11B2, CYP11B1, and steroidogenic acute regulatory protein (StAR), and that they produce aldosterone and corticosteroids in basal and stimulated conditions (Briones et al., 2011, 2012). Adipocyte-derived aldosterone involves generation of reactive oxygen species (ROS) through Nox-dependent processes and plays a role in adipogenesis, adipocyte maturation, and adipokine production (Briones et al., 2012).
Considering the fact that CETP inhibitors are lipophilic and may accumulate in adipose tissue (Dalvie et al., 2008; Gotto et al., 2014), we hypothesized that adipocytes may be an extra-adrenal source of CETP inhibitor–induced aldosterone production.
Materials and Methods
Cell Culture.
Human SW872 and murine 3T3-L1 cell lines were obtained from the American Type Culture Collection (Manassas, VA). SW872 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) high glucose, supplemented with 10% fetal bovine serum (FBS). 3T3-L1 cells were cultured in DMEM low glucose, supplemented with 10% calf bovine serum (Life Technologies, Paisley, UK). Both cell lines were supplemented with antibiotics (0.1 mg/ml streptomycin and 100 U/ml penicillin) in 5% CO2, at 37°C. Medium was changed every 2 days until confluence; 2 days after confluence, cells were differentiated to adipocytes. Differentiation for both cell lines was performed in DMEM supplemented with 10% FBS, 0.5 mM 3-isobutyl-1-methylxanthine, 0.25 µM dexamethasone (Sigma-Aldrich, St. Louis, MO), and 1 µM insulin (Cell Applications, San Diego, CA) for 2 days. Medium was replaced by DMEM/10% FBS, and 1 µM insulin for an additional 2 days and then replaced by insulin-free DMEM/10% FBS. Ten days after the differentiation process, cells exhibited adipocyte morphology. One day before experiments, the medium was changed to DMEM 1% FBS or 1% calf bovine serum, for SW872 or 3T3-L1, respectively. V79 hamster cells stably transfected with human CYP11B1 or human CYP11B2, and H295R cells that express hCYP11B1 and hCYP11B2 were used as positive controls (gift from Dr. Eleanor Davis, University of Glasgow, Glasgow, Scotland, UK).
Experimental Protocols.
To evaluate adipocyte effects of CETP inhibitors, cells were treated with torcetrapib, dalcetrapib, or anacetrapib (0.1–10 µM) (Santa Cruz Biotechnology, Santa Cruz, CA) for 5 minutes to 5 hours. To determine molecular mechanisms involved in cell activation by CETP inhibitors, cells were pretreated for 30 minutes with N-acetyl-l-cysteine (ROS scavenger; Sigma-Aldrich), ML171 (2-acetylphenothiazine; Nox1 inhibitor; Calbiochem-EMD Millipore, Billerica, MA), rotenone (mitochondrial inhibitor; Sigma-Aldrich), GKT137831 [2-(2-chlorophenyl)-4-[3-(dimethylamino)phenyl]-5-methyl-1H-pyrazolo[4,3-c]pyridine-3,6(2H,5H)-dione; Nox1/Nox4 inhibitor; Genkyotex, Geneva, Switzerland], and S3I-201 [2-hydroxy-4-(((4-methylphenyl)sulfonyloxy)acetyl)amino)-benzoic acid; Stat3 inhibitor; Santa Cruz Biotechnology].
NAD(P)H Oxidase Activity and Hydrogen Peroxide Production.
Stimulated adipocytes were washed with cold phosphate-buffered saline and homogenized in lysis buffer (20 mM KH2PO4, 1 mM EGTA, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, and 1 mM phenylmethylsulfonyl fluoride). Activity of NADPH oxidase was measured by chemiluminescence with lucigenin (5 μM) as the electron acceptor and NADPH (100 μM) as the substrate as we previously described (Briones et al., 2011). Luminescence was measured every 1.8 seconds for 5 minutes in a luminometer (AutoLumat LB 953; Berthold Technologies, Bad Wildbad, Germany). A buffer blank was subtracted from each reading.
Hydrogen peroxide was evaluated by the Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Molecular Probes/Life Technologies) according to the manufacturer’s instructions. Obtained values were normalized by protein concentration in the cell lysate.
Western Blot.
Cells were lysed in radioimmunoprecipitation assay buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.1% SDS) supplemented with 1 mM phenylmethylsulfonyl fluoride, 1 µg/ml pepstatin A, 1 µg/ml leupeptin, 1 µg/ml aprotinin (Sigma-Aldrich), 10 mM sodium fluorate (AnalaR Normapur; VWR, Leuven, Belgium), and 1 mM sodium orthovanadate (Alfa Aesar, Ward Hill, MA). Protein concentrations were determined using the DC protein assay kit (Bio-Rad, Hercules, CA). Equal amounts of protein were separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad). The nonspecific binding was blocked with nonfat dry milk. Membranes were incubated overnight at 4°C in constant agitation with primary antibodies specific for phospho-signal transducer and activator of transcription 3 (STAT3), STAT3, phospho-cAMP response-element binding protein (CREB), and CREB (Cell Signaling Technology, Beverly, MA); Nox2, Nox4, and CYP11B2 (Abcam, Cambridge, UK); Nox1 and Nox5 (Santa Cruz Biotechnology); and β-actin (Sigma-Aldrich). The anti-StAR was a kind gift from Dr. David Stocco (Department of Cell Biology and Biochemistry, Texas Tech University Health Sciences Center, Lubbock, TX) and used 1:500 dilution. As secondary antibodies, we used anti-rabbit IgG horseradish peroxidase (1:2000) and anti-mouse horseradish peroxidase (1:5000) (Jackson ImmunoResearch, West Grove, PA). Protein expression was visualized using the chemiluminescent substrate SuperSignal West Pico (Thermo Scientific, Rockford, IL). The resulting autoradiograms were analyzed using the ImageJ 1.44p package (http://imagej.nih.gov/ij; Wayne Rasband, NIH, Bethesda, MD).
Real-Time Reverse-Transcription Polymerase Chain Reaction.
Total RNA was isolated using the Trizol reagent (Life Technologies) according to the manufacturer’s instructions and diluted in nuclease-free H2O (Ambion/Life Technologies, Paisley, UK). cDNA was generated from total RNA using the High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Warrington, UK). Real-time polymerase chain reaction was performed with the Applied Biosystems 7900HT Fast Real-Time PCR system, using Power SyBr Green Master Mix (Applied Biosystems) and specific human primers, as follows: glyceraldehyde-3-phosphate dehydrogenase, CYP11B1, CYP11B2, StAR, mineralocorticoid receptor (MR), glucocorticoid receptor (GR), peroxisome proliferation-activated receptor-γ (PPARγ), Nox1, Nox2, Nox4, and Nox5 (Supplemental Table 1). Relative gene expression was calculated by the 2−ΔΔ cycle threshold method as previously described (Livak and Schmittgen, 2001). Data are shown as the fold change in expression of the target gene relative to the internal control gene (glyceraldehyde-3-phosphate dehydrogenase).
Enzymatic Immunoassays.
Commercially available enzyme-linked immunosorbent assay kits were used to measure the concentration of aldosterone, cortisol, and corticosterone (Cayman Chemical, Ann Arbor, MI), and chemerin (R&D Systems, Abingdon, UK) in cell supernatant according to the manufacturer’s instructions.
Statistical Analysis.
Results are presented as means ± S.E.M. Statistical differences between mean values were determined by one-way analysis of variance followed by the Newman–Keuls test or t test, as appropriate using GraphPad Prism 5.0 software (GraphPad Software, Inc., San Diego, CA). P < 0.05 values were considered as significant.
Results
Torcetrapib, Dalcetrapib, and Anacetrapib Increase Aldosterone Production in Human Adipocytes.
Human differentiated adipocytes were treated with torcetrapib, dalcetrapib, and anacetrapib. All drugs increased aldosterone concentration in the supernatant after 5 hours of stimulation (Fig. 1A). Significant effects were evident at doses as low as 0.1 µM. Similar responses were observed in mouse 3T3-L1 adipocytes (Supplemental Fig. 1), which lack the CETP gene. To avoid any interference that could result in a false positive effect due to cross-reactivity by glucocorticoids, we evaluated the cortisol concentration in the same samples. Cortisol levels were not significantly increased by any of the drugs (Fig. 1B).

Torcetrapib, dalcetrapib, and anacetrapib induce aldosterone production by human SW872 adipocytes. Cells were treated with 0.1, 1, and 10 µM torcetrapib (torc), dalcetrapib (dalc), or anacetrapib (anac) for 5 hours. (A and B) Aldosterone (A) and cortisol (B) concentration in the cell supernatant was evaluated by enzyme-linked immunosorbent assay and normalized by total RNA or protein concentration (n = 12). Data are expressed as the mean ± S.E.M. *P < 0.05 versus control vehicle group.
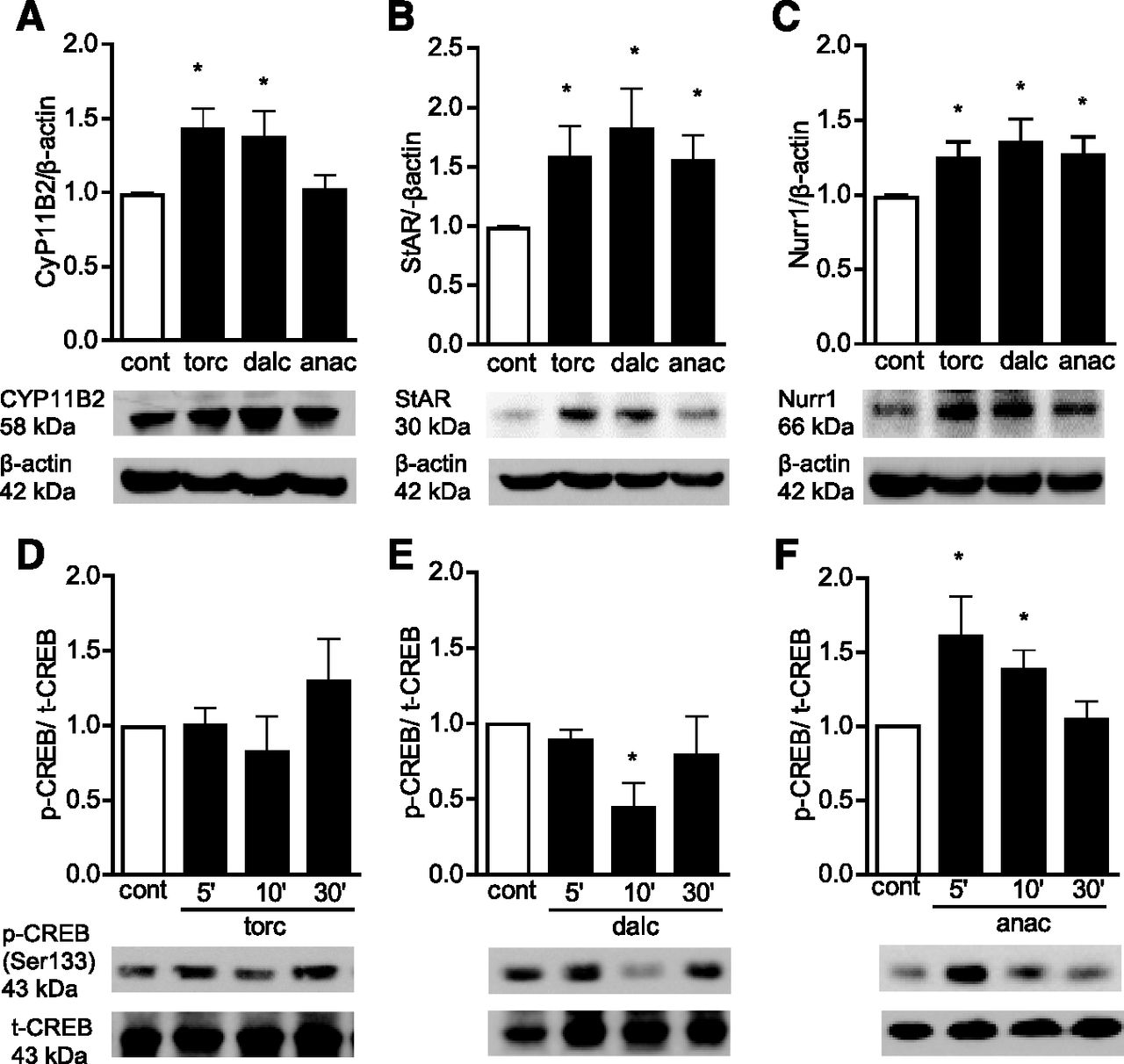
Torcetrapib, dalcetrapip, and anacetrapib increased mRNA and protein expression of CYP11B2 and CYP11B1 (Fig. 2A; Supplemental Fig. 2), enzymes responsible for aldosterone and cortisol production, respectively. The specificity of the antibody was verified using V79 cells stably transfected with either human CYP11B1 or CYP11B2, and H295 cells expressing both enzymes (Supplemental Fig. 2C).

Torcetrapib, dalcetrapib, and anacetrapib induce the expression of CYP11B2, StAR, and Nurr1, and CREB phosphorylation in human SW872 adipocytes. Cells were treated with 1 µM torcetrapib (torc), dalcetrapib (dalc), or anacetrapib (anac). (A–C) The expression of CYP11B2 (A), StAR (B), and Nurr1 (C) was analyzed in the cell lysate after 5 hours by Western blot and normalized by β-actin. (D–F) Phospho-CREB (p-CREB) was evaluated by Western blot after 5, 15, and 30 minutes and normalized by total CREB (t-CREB). Autoradiographs show one representative experiment. Protein expression was quantified using ImageJ software. Graph data are presented as the mean ± S.E.M. (n = 6). *P < 0.05 versus control vehicle group.
StAR transports cholesterol into the mitochondria and plays an important role in aldosterone production (Hattangady et al., 2012). Expression of StAR at the mRNA and protein levels, was increased by CETP inhibitors (Fig. 2B; Supplemental Fig. 2D). These drugs also increased protein levels of the transcription factor Nurr1 (Fig. 2C) associated with enhanced activity of the CYP11B2 promoter. The StAR promoter has a binding sequence for CREB/ATF-1 (activating transcription factor-1) that acts synergically with Nurr1 to induce CYP11B2 expression and aldosterone production (Spyroglou et al., 2009). Phosphorylation of CREB was increased in adipocytes stimulated with anacetrapib (Fig. 2, D–F).
To evaluate whether CETP inhibitors influence adipocyte receptors through which aldosterone signals, we investigated expression of MRs and GRs. We found that both receptor types are increased in treated cells (Supplemental Fig. 3).
CETP Inhibitors Influence Adipocyte Function.
The STAT3 pathway plays an important role in adipogenesis (Zhang et al., 2011). STAT3 was rapidly phosphorylated in treated adipocytes (Fig. 3A). This was associated with increased expression of PPARγ, an adipocyte differentiation marker (Fig. 3B). Torcetrapib stimulated production of chemerin, a proinflammatory adipokine (Fig. 3C).

STAT3 phosphorylation, PPARγ expression, and chemerin production by human SW872 adipocytes stimulated with torcetrapib, dalcetrapib, and anacetrapib. Cells were treated with 1 µM torcetrapib (torc), dalcetrapib (dalc), or anacetrapib (anac). (A) Phospho-STAT3 (p-STAT3) was evaluated by Western blot after 5, 15, and 30 minutes and normalized to total STAT3 (t-STAT3). Autoradiographs show one representative experiment. Protein expression was quantified using ImageJ software. (B) The PPARγ mRNA expression was evaluated after 5 hours and normalized to GAPDH mRNA. (C) Chemerin production was evaluated after 5 hours in the cell supernatant by enzyme-linked immunosorbent assay and normalized to total RNA. Graph data are presented as the mean ± S.E.M. (n = 6). *P < 0.05 versus control vehicle group. glyceraldehyde-3-phosphate dehydrogenase
CETP Inhibitor Effects Are Mediated through ROS.
Studies in adrenal cortical cells showed that ROS contributes to aldosterone production (Rajamohan et al., 2012). As such, we questioned whether CETP inhibitors influence aldosterone production by adipocytes through ROS-dependent processes. All three drugs increased generation of O2− and H2O2 in human adipocytes (Fig. 4) and mouse adipocytes (Supplemental Fig. 4). In SW872 adipocytes pretreated with the ROS scavenger N-acetyl-cysteine, CETP inhibitor effects on aldosterone production and mRNA expression for CYP11B2, CYP11B1, StAR, MR, and GR were inhibited (Supplemental Fig. 5).

Torcetrapib, dalcetrapib, and anacetrapib induce ROS generation in human SW872 adipocytes. Cells were treated with 1 µM torcetrapib (torc), dalcetrapib (dalc), or anacetrapib (anac). (A and B) ROS production was evaluated in the cell lysate by lucigenin chemiluminescence assay after 5 and 30 minutes (A) and Amplex Red assay after 5, 30, and 60 minutes (B). Values were normalized by protein concentration of the cell lysate. Data are presented as fold change of the control vehicle values and expressed as the mean ± S.E.M. (n = 5). *P < 0.05 versus control vehicle group.
Nox isoform expression (mRNA and protein) was variably increased by the different CETP inhibitors (Fig. 5; Supplemental Fig. 6). To determine whether Noxs play a role in the effects observed for CETP inhibitors, human SW872 adipocytes were pretreated with ML171 (Nox1 inhibitor), GKT137831 (Nox1/Nox4 inhibitor), rotenone (mitochondrial oxidase inhibitor), and S3I-201 (STAT3 inhibitor). PPARγ expression induced by torcetrapib was inhibited by GKT137831 and ML171 (Supplemental Fig. 7, A–C). Torcetrapib-stimulated chemerin production was reduced by GKT137831 and S3I-201 (Supplemental Fig. 7D). In torcetrapib-treated adipocytes, GKT137831, ML171, rotenone, and S3I-201 decreased aldosterone production and mRNA levels of CYP11B2, StAR (Fig. 6), MR, and GR (Supplemental Fig. 8). In dalcetrapib-stimulated cells, aldosterone biosynthesis was inhibited by ML171 and S3I-201 (Fig. 6; Supplemental Fig. 9) without effect of GKT137831 or rotenone. In anacetrapib-stimulated adipocytes, aldosterone production and biosynthetic enzyme expression were reduced by GKT137831, ML171, and rotenone (Fig. 6; Supplemental Fig. 10).

Nox isoforms expression are induced in human SW872 adipocytes stimulated by torcetrapib, dalcetrapib, and anacetrapib. Cells were treated with 1 µM torcetrapib (torc), dalcetrapib (dalc), or anacetrapib (anac). (A–D) The expression Nox1 (A), Nox2 (B), Nox4 (C), and Nox5 (D) was analyzed in the cell lysate after 5 hours by Western blot. β-Actin was used as a load control. The autoradiographs show one representative experiment. Protein expression was quantified using ImageJ software. Graph data are expressed as the mean ± S.E.M. (n = 6). *P < 0.05 versus control vehicle group.

Effects of torcetrapib, dalcetrapib, and anacetrapib on aldosterone production are affected by inhibitors of ROS production and STAT3 phosphorylation. Cells were pretreated with GKT137831 (GKT; Nox1/Nox4 inhibitor; 10 µM), ML171 (ML; Nox1 inhibitor; 1 µM), rotenone (Rot; mitochondrial oxidase inhibitor; 10 µM), or S3I-201 (S3I; STAT3 inhibitor; 50 µM) for 30 minutes, then treated with 1 µM of torcetrapib (A–C), dalcetrapib (D–F), or anacetrapib (G–I) for 5 hours. Aldosterone concentration in the cell supernatant was evaluated by enzyme-linked immunosorbent assay and normalized by total RNA. The mRNA expression for CYP11B2 and StAR was evaluated and normalized to GAPDH mRNA. Graph data are expressed as the mean ± S.E.M. (n = 7). *P < 0.05 versus control vehicle group; +P < 0.05 versus the torcetrapib-, dalcetrapib-, or anacetrapib-treated group.
Discussion
Blockade of transfer of cholesterol esters from HDL to proatherogenic lipoproteins LDL and very-low-density lipoprotein with CETP inhibitors has been considered a promising therapeutic strategy in the prevention and management of atherosclerosis (Tall, 1986; Barter et al., 2003). However, the ILLUMINATE trial, which evaluated cardiovascular effects in patients treated with torcetrapib plus atorvastatin versus atorvastatin alone, was terminated prematurely because of increased events in the torcetrapib group (Barter et al., 2007). Exact reasons for this remain unclear, but torcetrapib caused hypertension and hyperaldosteronism. Mechanisms whereby torcetrapib increases plasma aldosterone are elusive, although increased adrenal secretion through processes involving endogenous ouabain have been implicated (Funder, 2010). Here we provide evidence for another source of torcetrapib-induced aldosterone production by showing that adipocytes generate aldosterone in response to CETP inhibitors. Reasons for probing adipocytes were 3-fold. First, we previously demonstrated that adipocytes have a functional renin angiotensin aldosterone system and produce aldosterone in a highly regulated manner (Briones et al., 2011, 2012). Second, CETP inhibitors are lipid soluble and accumulate, to variable degrees, in adipocytes (Dalvie et al., 2008; Gotto et al., 2014). Third, by computational analysis, these drugs were found to bind to several endogenous proteins related to adipogenesis, such as PPARα, PPARβ, PPARγ, and LXR (Xie et al., 2009). We studied human and mouse adipocytes and found similar responses in both cell types, indicating that CETP inhibitor–induced adipocyte aldosterone production is independent of CETP, since mice lack the CETP gene. The biosynthesis of aldosterone has been very well characterized in adrenal cells and is mediated by StAR and the rate-limiting enzyme CYP11B2 (Hattangady et al., 2012). Both enzymes are expressed in adipocytes and the three CETP inhibitors studied here significantly increased their expression. StAR, which is regulated by CREB, plays an important role in the acute generation of aldosterone through processes dependent on cholesterol flux into the mitochondria and conversion to pregnenolone. Aldosterone is also controlled chronically by CYP11B2 (Hattangady et al., 2012). Our results show differential regulation of these enzymes by CETP inhibitors. Torcetrapib and dalcetrapib increased expression of Nurr1, a transcription factor that regulates CYP11B2 (Spyroglou et al., 2009), and increased protein content of CYP11B2, whereas anacetrapib increased expression of CREB and StAR. Hence, torcetrapib and dalcetrapib influence pathways associated with chronic aldosterone production, whereas anacetrapib influences processes associated with acute biosynthesis. Whether increased adipocyte-derived aldosterone translates into changes in plasma aldosterone levels still remains unknown. Our ongoing studies in adrenalectomized mice and in mice stimulated with CETP inhibitors will address this.
We previously showed that angiotensin II induced adipocyte-derived aldosterone involves ROS. Other studies demonstrated that aldosterone production by adrenal cortical cells occurs in a ROS-dependent manner by regulating Nurr1 protein expression (Rajamohan et al., 2012). Here we extend those findings to show that CETP inhibitors modulate expression of Noxs and increase production of superoxide and hydrogen peroxide in adipocytes. Aldosterone production was reduced by N-acetyl-cysteine, a ROS scavenger, in CETP inhibitor–treated cells. To dissect the source of ROS required for aldosterone production, we used the pharmacologic inhibitors GKT137831, ML171, and rotenone, inhibitors of Nox1/Nox4, Nox1, and mitochondrial oxidases, respectively. GKT137831, ML171, and rotenone inhibited torcetrapib- and anacetrapib-induced aldosterone production, suggesting a role for Nox1, Nox4, and mitochondria in ROS-sensitive aldosterone production by these CETP inhibitors. For dalcetrapib, the source of ROS is less clear, because ML171, but not GKT137831, reduced aldosterone synthesis, indicating a potential partial role for Nox1.
Downregulation of enzymes involved in aldosterone production, including CYP11B2 and StAR, was associated with changes in aldosterone biosynthesis. Together these findings indicate that CETP inhibitors induce ROS generation, which stimulate aldosterone-synthesizing enzymes to produce aldosterone in adipocytes. Exactly how these drugs regulate ROS-generating enzymes is unclear, but it may be possible that they have binding sites that target oxidases, as suggested by computational analysis (Chang et al., 2010).
In addition to influencing mineralocorticoid biosynthesis in adipocytes, CETP inhibitors affect adipocyte function as evidenced by effects on differentiation processes and production of adipokines. In drug-treated adipocytes, STAT3 phosphorylation and PPARγ expression, important in adipogenesis and differentiation, respectively (Chang et al., 2010) were increased and production of chemerin, a proinflammatory adipokine was stimulated. STAT3 also plays some role in aldosterone biosynthesis in CETP inhibitor–treated cells, because STAT3 inhibition blunted aldosterone production by torcetrapib and dalcetrapib.
From a functional view point, our data have important clinical implications. We show that CETP inhibitors, which are lipid soluble, stimulate production of aldosterone by adipocytes through highly regulated processes. These drugs also influence adipocyte function by promoting adipocyte differentiation and production of adipokines, processes associated with the proinflammatory phenotype of adipose tissue, important in CVD. Finally, we demonstrate that adipocyte effects of torcetrapib, dalcetrapib, and anacetrapib occur through CETP-independent processes, because mouse adipocytes, which lack the CETP gene, responded in a similar manner to human adipocytes, which possess functionally active CETP.
In conclusion, our study provides insights into novel molecular mechanisms whereby CETP inhibitors increase aldosterone production by adipocytes. These findings may explain, in part, the unexpected side effect of hyperaldosteronism in the large torcetrapib clinical studies. The exact contribution of adipocyte- versus adrenal-derived aldosterone is unknown, but our ongoing studies in adrenalectomized mice should provide information to address this. Our observations are important because other CETP inhibitors are in clinical development and potential injurious effects on adipocyte biology should be considered.
Acknowledgments
The authors thank Dr. David Stocco (Department of Cell Biology and Biochemistry, Texas Tech University Health Sciences Center) for the anti-StAR antibody, Dr. Eleanor Davis (Institute of Cardiovascular and Medical Sciences, BHF Glasgow Cardiovascular Research Centre, University of Glasgow) for the H295R cells, and C. Jenkins and A. Carswell for technical assistance.
Authorship Contributions
Participated in research design: Rios, Nguyen Dinh Cat, Montezano, Touyz.
Conducted experiments: Rios, Neves, Nguyen Dinh Cat, Even, Palacios.
Contributed new reagents or analytic tools: Touyz.
Wrote or contributed to the writing of the manuscript: Rios, Montezano, Touyz.
Footnotes
- Received October 29, 2014.
- Accepted January 22, 2015.
This research was supported by the British Heart Foundation [Grants 30099 and 30087]. R.M.T. is funded through a British Heart Foundation Chair.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- CETP
- cholesteryl ester-transfer protein
- CREB
- cAMP response-element binding protein
- CVD
- cardiovascular disease
- DMEM
- Dulbecco’s modified Eagle’s medium
- FBS
- fetal bovine serum
- GKT137831
- 2-(2-chlorophenyl)-4-[3-(dimethylamino)phenyl]-5-methyl-1H- pyrazolo[4,3-c]pyridine-3,6(2H,5H)-dione
- GR
- glucocorticoid receptor
- HDL
- high-density lipoprotein
- ILLUMINATE
- Investigation of Lipid Level Management to Understand Its Impact in Atherosclerotic Events
- LDL
- low-density lipoprotein
- ML171
- 2-acetylphenothiazine
- MR
- mineralocorticoid receptor
- PPAR
- peroxisome proliferation-activated receptor
- ROS
- reactive oxygen species
- S3I-201
- 2-hydroxy-4-(((4-methylphenyl)sulfonyloxy)acetyl)amino)-benzoic acid
- StAR
- steroidogenic acute regulatory protein
- STAT3
- signal transducer and activator of transcription 3
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}