


Abstract
Buprenorphine is known as a μ-opioid peptide (MOP) receptor agonist, but its antinociception is compromised by the activation of nociceptin/orphanin FQ peptide (NOP) receptors in rodents. The aim of this study was to investigate the roles of MOP and NOP receptors in regulating buprenorphine-induced physiological responses in primates (rhesus monkeys). The effects of MOP antagonist (naltrexone), NOP antagonist [(±)-1-[(3R*,4R*)-1-(cyclooctylmethyl)-3-(hydroxymethyl)-4-piperidinyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one (J-113397)], and NOP agonists [(1S,3aS)-8-(2,3,3a,4,5,6-hexahydro-1H-phenalen-1-yl)-1-phenyl-1,3,8-triaza-spiro[4.5] decan-4-one (Ro 64-6198) and 3-endo-8-[bis(2-methylphenyl)methyl]-3-phenyl-8-azabicyclo[3.2.1]octan-3-ol (SCH 221510)] on buprenorphine were studied in three functional assays for measuring analgesia, respiratory depression, and itch in primates. Over the dose range of 0.01 to 0.1 mg/kg, buprenorphine dose-dependently produced antinociception, respiratory depression, and itch/scratching responses, and there was a ceiling effect at higher doses (0.1–1 mg/kg). Naltrexone (0.03 mg/kg) produced similar degrees of rightward shifts of buprenorphine's dose-response curves for all three endpoints. Mean pKB values of naltrexone (8.1–8.3) confirmed that MOP receptors mediated mainly buprenorphine-induced antinociception, respiratory depression, and itch/scratching. In contrast, J-113397 (0.1 mg/kg) did not change buprenorphine-induced physiological responses, indicating that there were no functional NOP receptors in buprenorphine-induced effects. More importantly, both NOP agonists, Ro 64-6198 and SCH 221510, enhanced buprenorphine-induced antinociception without respiratory depression and itch/ scratching. The dose-addition analysis revealed that buprenorphine in combination with the NOP agonist synergistically produced antinociceptive effects. These findings provided functional evidence that the activation of NOP receptors did not attenuate buprenorphine-induced antinociception in primates; instead, the coactivation of MOP and NOP receptors produced synergistic antinociception without other side effects. This study strongly supports the therapeutic potential of mixed MOP/NOP agonists as innovative analgesics.
Introduction
Buprenorphine is a μ-opioid peptide (MOP) receptor agonist commonly used in clinics. Subutex (Reckitt Benckiser Healthcare Ltd., Hull, UK), trade name of buprenorphine, is used for the transition from opioid withdrawal to the treatment of addiction; when used in an injectable formulation [Buprenex (Reckitt Benckiser Healthcare Ltd.)], it is used for analgesia (Rosenblum et al., 2008; Kress, 2009; Pergolizzi et al., 2010). Pharmacological studies indicate that buprenorphine has relatively high binding affinity at MOP, κ-opioid peptide, and δ-opioid peptide receptors (Huang et al., 2001), and it has partial agonist activity at MOP receptors measured by both in vitro and in vivo assays (Cowan et al., 1977; Dum and Herz, 1981; Huang et al., 2001; Clark et al., 2006). Given its favorable safety profile, such as the ceiling effect on respiratory depression and other side effects, buprenorphine is considered an effective and realistic option for treating patients for a variety of pain conditions (Walsh et al., 1995; Johnson et al., 2005; Pergolizzi et al., 2010).
The unique pharmacological profile of buprenorphine was further documented by the involvement of nociceptin/orphanin FQ (N/OFQ) peptide (NOP) receptor. NOP receptor knockout mice showed an enhanced antinociceptive effect produced by buprenorphine, but morphine-induced antinociception was not changed in mice lacking NOP receptors. After pretreatment with the NOP antagonist (±)-1-[(3R*,4R*)-1-(cyclooctylmethyl)-3-(hydroxymethyl)-4-piperidinyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one (J-113397), buprenorphine-induced antinociception was enhanced in wild-type mice, but not in NOP receptor knockout mice (Lutfy et al., 2003). In particular, the ascending portion of the dose-response curve for systemic buprenorphine-induced antinociception was enhanced by the NOP antagonists (Lutfy et al., 2003; Ding and Raffa, 2009; Khroyan et al., 2009). These findings suggest that in mice buprenorphine-induced antinociception is compromised by concomitant activation of NOP receptors. However, ligand-receptor binding studies reveal that buprenorphine has extremely low binding affinity at NOP versus MOP receptors (Ki 285 versus 0.08 nM), and the functional assay of G-protein activation indicates that buprenorphine is much less potent in stimulating [35S]GTPγS binding to membranes of Chinese hamster ovary cells transfected with NOP and MOP receptors with percentages of stimulation less than half that of MOP receptors (EC50 35 versus 0.08 nM) (Huang et al., 2001). It is puzzling how buprenorphine activates NOP receptors at the antinociceptive dose range. Nevertheless, it is important to investigate the receptor mechanisms underlying buprenorphine-induced antinociception, particularly in other species, such as nonhuman primates.
Activation of NOP receptors in rodents at the peripheral, spinal, and systemic levels produces pronociception, antinociception, or no effect depending on the doses and assays (Inoue et al., 1999; Calo' et al., 2000; Jenck et al., 2000). In contrast, in primates after peripheral, spinal, and systemic administration NOP agonists produce only antinociception regardless of doses and assays applied (Ko, 2004; Ko et al., 2006b, 2009; Ko and Naughton, 2009; Hu et al., 2010). More importantly, there are two independent components (MOP and NOP receptors) that can equally contribute to antinociceptive effects in primates (Ko et al., 2009; Hu et al., 2010). Given that the physiological functions of opioid receptor subtypes are similar between monkeys and humans (Bailey et al., 1993; Palmer et al., 1999; Lee et al., 2007; Ko and Husbands, 2009) pharmacological studies using nonhuman primates may establish a translational bridge to the therapeutic profiles of drugs in humans. Therefore, it is valuable to pharmacologically investigate buprenorphine-induced physiological responses in primates to improve our understanding of receptor mechanisms underlying such effects in humans.
To date, the effects of NOP antagonists and agonists on buprenorphine-induced antinociception in primates are totally unknown. Given that some pharmacological bases of MOP- and NOP-related ligands such as active dose ranges in primates have been established (Ko et al., 2006a,b, 2009; Hu et al., 2010), it would facilitate the study of buprenorphine's receptor mechanisms. Therefore, the aim of this study was to compare the effects of MOP and NOP antagonists on buprenorphine-induced antinociception, respiratory depression, and itch/scratching in primates. More importantly, a NOP antagonist and agonist were used to elucidate the role of NOP receptors in buprenorphine-induced antinociception.
Materials and Methods
Subjects
Twenty-four adult female and male rhesus monkeys (Macaca mulatta) ranging in body weight from 7.4 to 12.2 kg were used. The monkeys were individually housed, and their daily dietary intake included approximately 25 to 30 biscuits (Purina Monkey Chow; Purina, St. Louis, MO), fresh fruit, and water ad libitum. For 1 month before the present study, the monkeys were not exposed to any opioid compound. All monkeys had previously been trained in the warm-water tail-withdrawal assay and acclimated to being video- recorded in cages. Eighteen monkeys participated in the first two parts of the study; six subjects were consistently used in a single assay, and there were three assays: antinociception, respiratory depression, and itch/scratching responses. The remaining six monkeys were used in the third part of the study for all three assays. The monkeys were housed in facilities accredited by the American Association for the Accreditation of Laboratory Animal Care. The studies were conducted in accordance with the University Committee on the Use and Care of Animals at the University of Michigan (Ann Arbor, MI) and the Guide for Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health (Bethesda, MD) (Institute of Laboratory Animal Resources, 1996).
Procedures
Nociceptive Responses.
The warm-water (50°C) tail-withdrawal assay was used to assess thermal antinociceptive effects of the test compound (Ko et al., 2006a). Monkeys were seated in primate restraint chairs, and the lower parts of their shaved tails (approximately 15 cm) were immersed in a thermal flask containing water maintained at either 42, 46, or 50°C. Water at 42 and 46°C was used as non-noxious stimuli, and 50°C water was used as an acute noxious stimulus. Tail-withdrawal latencies were measured by using a computerized timer by an experimenter who was blinded to experimental conditions. If the monkeys did not remove their tails within 20 s, the flask was removed and a maximum time of 20 s was recorded. Test sessions began with control determinations at each temperature. Subsequent tail-withdrawal latencies were determined at multiple time points after systemic administration of the test compound.
Respiratory Function.
The apparatus was similar to that described previously (Butelman et al., 1993). The monkey was seated in a primate restraint chair, enclosed within a sound-attenuating chamber. A rectangular helmet (13.5 × 17.0 × 13.5 cm) was placed over the head of the monkey and sealed around its neck by a latex shield. Gas (either air or a mixture of 5% CO2 in air) flowed into the helmet and was pumped out at a rate of 8 l/min. The monkeys' breathing produced changes in pressure inside the helmet that were measured with a pressure transducer connected to a polygraph (Grass model 7; Grass Instruments, Quincy, MA). The data were recorded on a polygraph trace and in a microprocessor (IBM computer; IBM, White Plains, NY) through an analog-to-digital converter. The polygraph integrator was connected to a computer, which analyzed the data collected over a 3-min period. The rate of breathing (ƒ; respiratory frequency) was determined directly. The minute volume (VE), the number of liters of air inspired per minute, was determined from the integration of the plethysmograph system. Each test cycle was 30 min, which included the first 23-min exposure to air alone and the remaining 7-min exposure to 5% CO2 mixed in air. Responses in the first two cycles were averaged as a control value. Subsequent respiratory parameters were determined at multiple time points after intramuscular administration of the test compound.
Scratching Responses.
Monkeys were recorded in-cage for their scratching behavior, which has been associated previously to an itch sensation after administration of opioid-related ligands (Ko et al., 2004). A scratch was defined as one brief (<1 s) episode of scraping contact of the forepaw or hind paw on the skin surface or other body parts. Each recording session was conducted in 15-min intervals and scored by individuals blinded to experimental conditions after intramuscular administration of the test compound.
Experimental Design
The first part of the study was to determine the monkeys' physiological responses to buprenorphine, along with corresponding dose-dependent effects. The dose range (0.01–1 mg/kg) was selected based on previous human and monkey studies illustrating active dosing conditions (Walker et al., 1995; Zacny et al., 1997; Kishioka et al., 2000; Pergolizzi et al., 2010). Buprenorphine-induced physiological responses were characterized mainly in vivo by three distinct measurements: antinociception, respiratory depression, and itch/scratching activity. These endpoints have been used previously to document the roles of MOP and NOP in regulating physiological functions in monkeys (Butelman et al., 1993; Ko et al., 2004, 2009). Buprenorphine was administered systemically by using a single dosing procedure, and each measurement was conducted per 30 min during a 3-h test session.
The second part of the study was to elucidate the receptor mechanisms underlying buprenorphine-induced physiological responses by using the MOP and NOP antagonists. The dose-response curve of buprenorphine for each endpoint was established by using a cumulative dosing procedure with a 30-min interinjection interval. A single dose of the MOP antagonist naltrexone (0.03 mg/kg) or the NOP antagonist J-113397 (0.1 mg/kg) was used based on previous studies, demonstrating that these dosing regimens produced selective, same degree of MOP versus NOP antagonism in monkeys (Ko et al., 2009; Hu et al., 2010). The dose-response curve of buprenorphine for antinociception or itch/scratching was redetermined 15 min after pretreatment with either naltrexone or J-113397. For measuring respiratory depression, the pretreatment time was 30 min because of the setting of 30-min cycles.
The third part of the study was to determine how the activation of NOP affected buprenorphine-induced physiological responses. The dose-addition analysis was used to evaluate the drug interaction between buprenorphine and two selective NOP agonists, (1S,3aS)- 8-(2,3,3a,4,5,6-hexahydro-1H-phenalen-1-yl)-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one (Ro 64-6198) and 3-endo-8-[bis(2-methylphenyl)methyl]-3-phenyl-8-azabicyclo[3.2.1]octan-3-ol (SCH 221510) (Jenck et al., 2000; Varty et al., 2008). Both NOP agonists have been studied in primates, and their antinociceptive effects were antagonized by the NOP antagonist J-113397, but not by the MOP antagonist naltrexone (Ko et al., 2009; Wladischkin et al., 2012). Initially, dose-response curves for buprenorphine- and NOP agonist-induced antinociception were determined. Depending on the potency of both NOP agonists, dose-response curves for three mixtures of either Ro 64-6198 or SCH 221510 in combination with buprenorphine were redetermined to elucidate the nature of the interaction between MOP- and NOP-mediated functions (subadditive, additive, or supra-additive effects) (Tallarida, 2000). After drug combination studies, the dose-response curve of buprenorphine alone was redetermined. Dose-response curves for all three endpoints (antinociception, respiratory depression, and itch/scratching) were established to verify the therapeutic profiles of all three mixtures. All experiments including systemic buprenorphine were conducted once per 2 to 3 weeks.
Data Analysis
Mean values (mean ± S.E.M.) were calculated from individual values for all endpoints. Comparisons were made across all test sessions in the same experiment. Data were analyzed by a two-way analysis of variance followed by the Newman-Keuls test for multiple (post hoc) comparisons. The criterion for significance was set at p < 0.05. For analyzing the dose-response curves for antinociception, individual tail-withdrawal latencies were converted to the percentage of maximum possible effect. The formula of the percentage of maximum possible effect is defined as [(test latency − control latency)/(cutoff latency, 20 s − control latency)] × 100. ED50 values were calculated by least-squares regression with the portion of the dose-response curves spanning the 50% maximum possible effect. For analyzing the dose-response curves for respiratory depression, ED30 values were calculated because of the ceiling effects of buprenorphine on this endpoint. The 95% confidence limits were also determined. Mean ED30/ED50 values were considered to be significantly different when their 95% confidence limits did not overlap.
For in vivo apparent pKB analysis, a dose ratio was determined for a single dose of the antagonist by using a modified equation, pKB = −log [B/(dose ratio − 1)], where B equals the antagonist dose in moles per kilogram. Mean pKB values ± 95% confidence limits were averaged from individual pKB values for the antagonist. The dose-addition analysis using isobolograms has been widely used to provide a more quantitative evaluation of drug-drug interactions through various endpoints measured in different laboratories (Tallarida 2001; Stevenson et al., 2003; Fischer and Dykstra, 2006; Ko and Husbands, 2009). Statistical evaluation of drug interactions between NOP agonists and buprenorphine was conducted by comparing the experimentally determined ED50 values for each mixture (Zmix) with predicted additive ED50 values (Zadd) that were calculated by the software developed by Tallarida (2000). Zadd values were calculated individually for each monkey by using the equation Zadd = fA + (1 − f) B, where A is the ED50 for the NOP agonist alone and B is the ED50 for buprenorphine alone (Tallarida, 2001). The proportion of the NOP agonist in each mixture was determined by the equation fA/[fA + (1 − f)B]. The present study investigated effects produced by mixtures with f = 0.25, 0.5, and 0.75 representing the fractional multiplier for three mixtures of the NOP agonist in combination with buprenorphine (Tallarida, 2001; Stevenson et al., 2003; Fischer and Dykstra, 2006; Ko and Husbands, 2009). When f = 0.25, the mixture contains a proportion of [A/(A + 3B)] NOP agonist and a mixture ratio of [(A/B)/3] parts NOP agonist to one-part buprenorphine; f = 0.5 contains a proportion of [A/(A + B)] NOP agonist in the mixture and a mixture ratio of (A/B) parts NOP agonist to one-part buprenorphine; and f = 0.75 contains a proportion of [A/(A + B/3)] NOP agonist in the mixture and a mixture ratio of [(A/B) ×3] parts NOP agonist to one-part buprenorphine. Mean experimentally determined ED50 values (Zmix) and predicted additive ED50 values (Zadd) for each mixture were compared with a t test. The criterion for significance was set at p < 0.05.
Drugs
Naltrexone HCl (National Institute on Drug Abuse, Bethesda, MD) and J-113397 (Tocris Bioscience, Ellisville, MO) were dissolved in sterile water. Ro 64-6198 (Amgen, Thousand Oaks, CA) and SCH 221510 (Tocris Bioscience) were dissolved in a solution of dimethyl sulfoxide/Tween80/sterile water in a ratio of 1:1:8. Buprenorphine HCl (National Institute on Drug Abuse) was dissolved in sterile water with the addition of a few drops of lactic acid. Doses are presented in the compound forms listed above. For systemic administration, all compounds were administered at a volume of 0.1 ml/kg.
Results
Figure 1 illustrates the time course and dose dependence of buprenorphine-induced antinociceptive effects against an acute nociceptive stimulus (50°C water) in primates. After subcutaneous administration, buprenorphine produced antinociception in both dose-dependent (F5,25 = 30.7; p < 0.05) and time-dependent (F5,25 = 13.4; p < 0.05) manners. Over low doses (0.03–0.1 mg/kg) buprenorphine-induced antinociception peaked at the first observation period (30 min after administration), then decreased 2 to 3 h after administration (Fig. 1A). Over higher doses (0.3–1 mg/kg) similar magnitudes and durations of buprenorphine-induced antinociception were observed; higher doses of buprenorphine did not further increase antinociceptive effects under this condition (Fig. 1B).

Antinociceptive effects of subcutaneously administered buprenorphine over a wide dose range against an acute noxious stimulus (50°C water) in primates. A, low doses. B, high doses. Each data point represents a mean ± S.E.M. (n = 6). *, a significant difference from the vehicle condition from the time point 30 min to the corresponding time point for each dose (p < 0.05). The same data of buprenorphine (0.1 mg/kg) are presented in A (●) and B (▼) for comparison with other doses.
Figure 2 illustrates the time course and dose dependence of buprenorphine-induced respiratory depression in primates breathing 5% CO2 mixed in air. Because the magnitudes and durations of buprenorphine-induced respiratory depression were similar between cycles of air versus 5% CO2 in air, only data during the cycle of 5% CO2 in air are presented herein. After intramuscular administration, buprenorphine-induced respiratory depression was dose-dependently manifested by both parameters, ƒ (F5,25 = 10.1; p < 0.05) and VE (F5,25 = 15.0; p < 0.05) (Fig. 2, A and B). There was no time dependence in changes of both parameters, ƒ (F5,25 = 1.4; p < 0.05) and VE (F5,25 = 0.6; p > 0.05), during the first 3 h after administration. Over low doses (0.03–0.1 mg/kg) buprenorphine-induced respiratory depression peaked at 30 min and continued throughout the 3-h test period (Fig. 2, A and B). Over higher doses (0.3–1 mg/kg) buprenorphine produced similar effects as 0.1 mg/kg did in suppressing respiratory parameters, ƒ and VE (Fig. 2, C and D).

Respiratory depressant effects of intramuscularly administered buprenorphine over a wide dose range in primates breathing air mixed with 5% CO2. A and C, f values for low doses (A) and high doses (C) of buprenorphine. B and D, Ve values for low doses (B) and high doses (D) of buprenorphine. Each data point represents a mean ± S.E.M. (n = 6). *, a significant difference from the vehicle condition for all time points (p < 0.05). The same data of buprenorphine (0.1 mg/kg) are presented in A and B (●) and C and D ▼) for comparison with other doses.
Based on data shown in Figs. 1 and 2, multiple doses of buprenorphine (0.01–0.3 mg/kg) were selected to study the effects of buprenorphine on eliciting itch/scratching responses. Figure 3 shows the duration and magnitude of buprenorphine-induced scratching activity. After intramuscular administration, buprenorphine dose-dependently elicited scratching responses (F4,20 = 9.7; p < 0.05). Effects of buprenorphine (0.1–0.3 mg/kg) peaked at the first observation period (30 min after administration) and continued throughout the 3-h observation period.
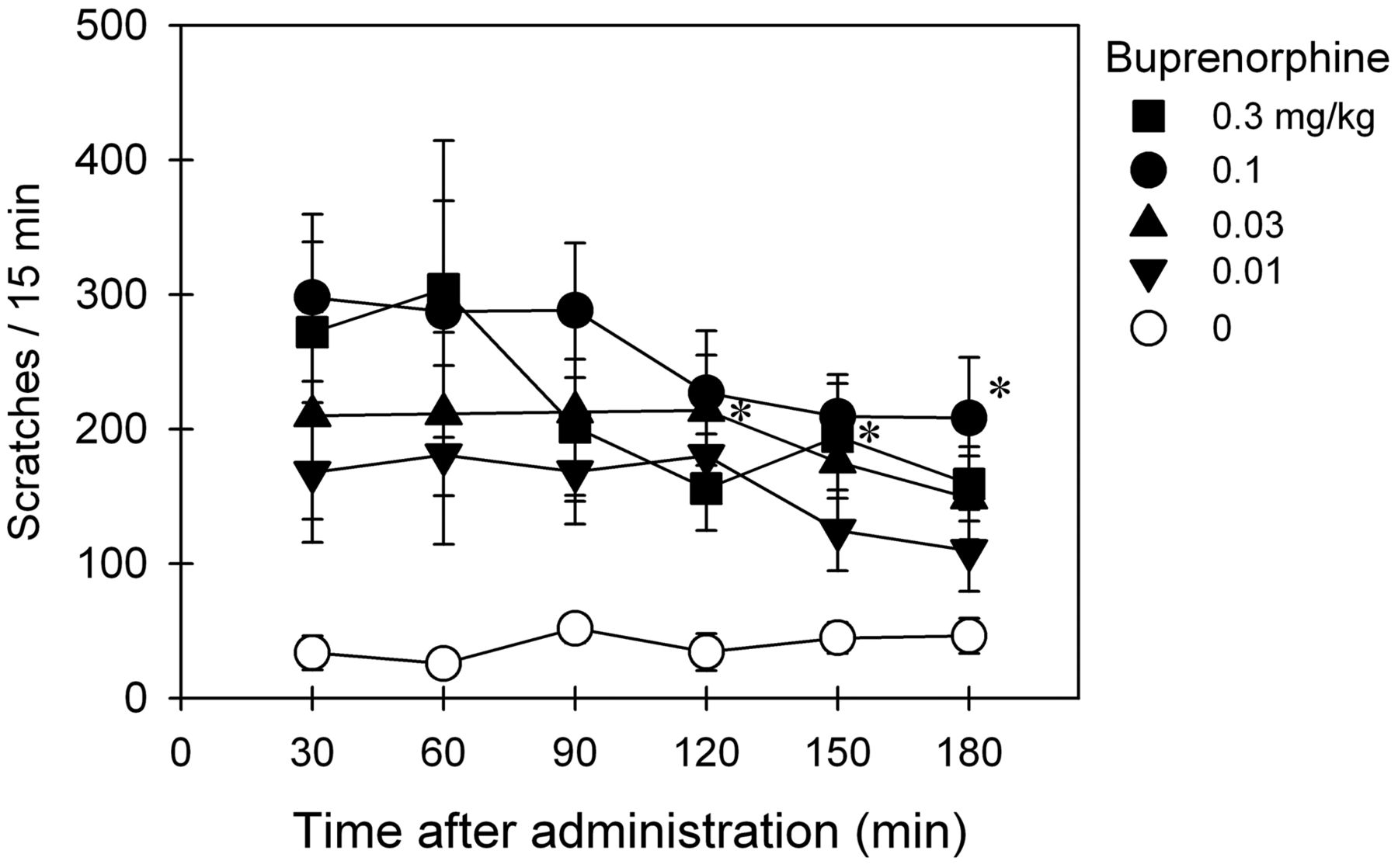
Itch/scratching eliciting effects of intramuscularly administered buprenorphine over a wide dose range in primates. Each data point represents a mean ± S.E.M. (n = 6). *, a significant difference from the vehicle condition from the time point 30 min to the corresponding time point for each dose (p < 0.05).
Figure 4 compares the antagonist effects of the MOP antagonist naltrexone and NOP antagonist J-113397 on buprenorphine-induced antinociceptive effects. After a 15-min pretreatment, a single dose of subcutaneous naltrexone (0.03 mg/kg) produced a large rightward shift of the dose-response curve of buprenorphine-induced antinociception, and its mean naltrexone pKB value was 8.2. In contrast, subcutaneous pretreatment with J-113397 (0.1 mg/kg) failed to block buprenorphine-induced antinociception. The ED50 value of buprenorphine dose response for vehicle pretreatment (0.02 mg/kg) was similar to that for J-113397 pretreatment (0.03 mg/kg) (see details in Table 1).

Effects of MOP and NOP antagonists on buprenorphine-induced antinociception in primates. The MOP antagonist naltrexone (0.03 mg/kg) or the NOP antagonist J-113397 (0.1 mg/kg) was administered subcutaneously 15 min before determination of buprenorphine's dose-response curve. Each data point represents a mean ± S.E.M. (n = 6).
Antagonist studies of systemic buprenorphine-induced physiological responses in primates
Figure 5 compares the effects of the MOP antagonist naltrexone and NOP antagonist J-113397 on blocking the respiratory depressant effects of buprenorphine. After a 30-min pretreatment intramuscular naltrexone (0.03 mg/kg) produced a large rightward shift of the dose-response curve of buprenorphine-induced respiratory depression in the VE parameter, and its mean naltrexone pKB value was 8.1. In contrast, J-113397 (0.1 mg/kg) did not change the dose-response curves of buprenorphine for both the ƒ and VE parameters. The ED30 value of buprenorphine dose response for vehicle pretreatment (0.032 mg/kg) was similar to that for J-113397 pretreatment (0.045 mg/kg) (Table 1). As noted, there were no changes in both ƒ and VE parameters after administration of either naltrexone or J-113397 compared with the control value under this condition (data not shown).

Effects of MOP and NOP antagonists on buprenorphine-induced respiratory depression in primates. A single dose of the MOP antagonist naltrexone (0.03 mg/kg) or the NOP antagonist J-113397 (0.1 mg/kg) was administered intramuscularly 30 min before administration of the first dose of buprenorphine. Data represent the changes of both parameters, ƒ (A) and VE (B), in primates breathing air mixed with 5% CO2. Each data point represents a mean ± S.E.M. (n = 6).
Figure 6 compares the antagonist effects of the MOP antagonist naltrexone and NOP antagonist J-113397 on buprenorphine-induced scratching responses. After a 15-min pretreatment, intramuscular naltrexone (0.03 mg/kg) produced a large rightward shift of the dose-response curve of buprenorphine-induced scratching, and its mean naltrexone pKB value was 8.3. In contrast, J-113397 (0.1 mg/kg) failed to block buprenorphine-induced scratching. The ED50 value of buprenorphine dose response for vehicle pretreatment (0.015 mg/kg) was similar to that for J-113397 pretreatment (0.016 mg/kg) (Table 1).
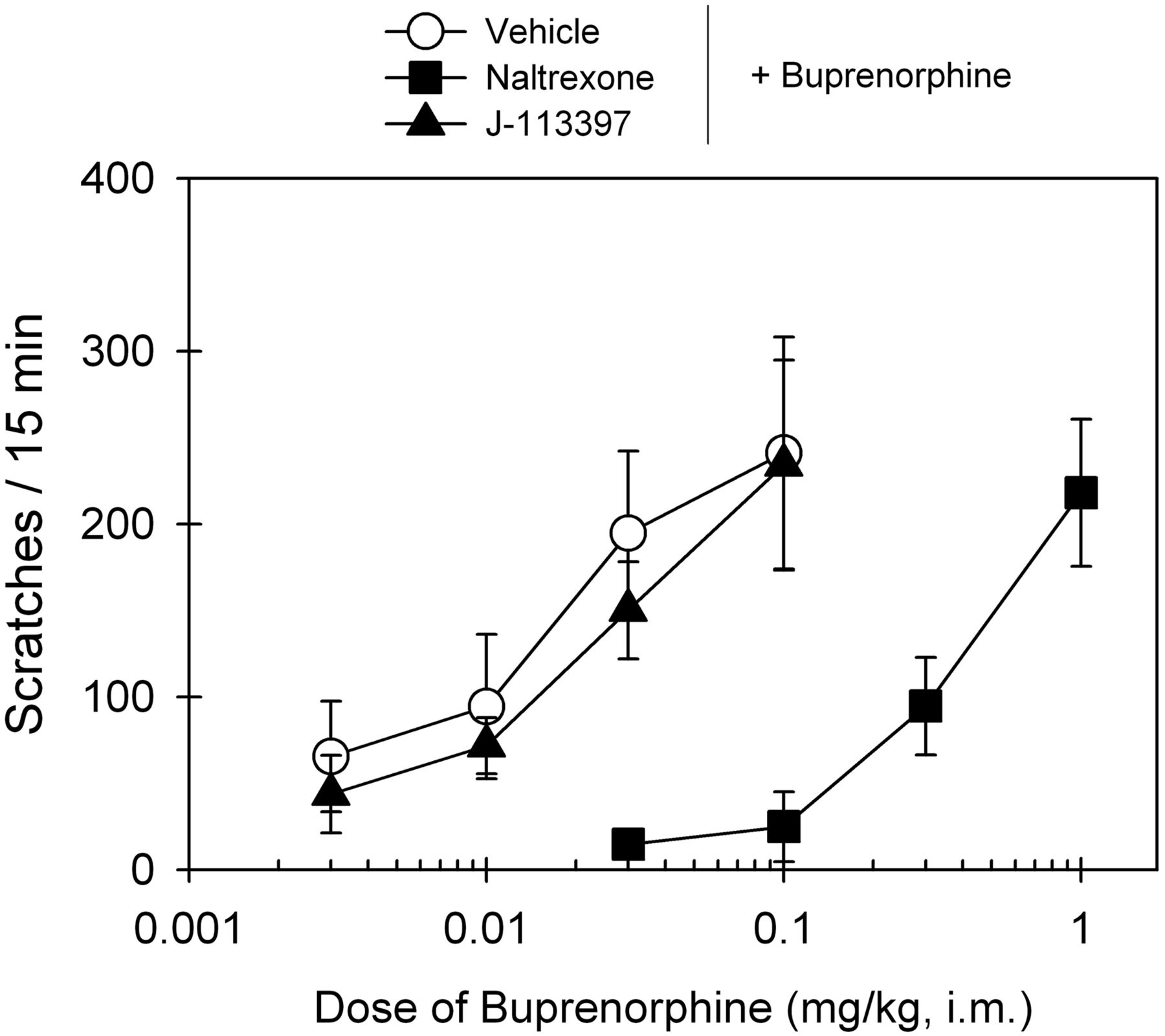
Effects of MOP and NOP antagonists on buprenorphine-induced itch/scratching activity in primates. A single dose of the MOP antagonist naltrexone (0.03 mg/kg) or the NOP antagonist J-113397 (0.1 mg/kg) was administered intramuscularly 15 min before determination of buprenorphine's dose-response curve. Each data point represents a mean ± S.E.M. (n = 6).
Figure 7 illustrates the effects of buprenorphine alone or in combination with either Ro 64-6198 or SCH 221510. The addition of each NOP agonist dose-dependently produced leftward shifts in the dose-response curve of buprenorphine-induced antinociception (Fig. 7, A and B). In contrast, when the ratio of the NOP agonist versus buprenorphine increased the combination of each NOP agonist with buprenorphine did not significantly produce respiratory depression (Fig. 7, C and D) or elicit itch/scratching responses (Fig. 7, E and F) along with the corresponding doses that produced full antinociceptive effects.

Effects of the NOP agonists Ro 64-6198 and SCH 221510 in combination with buprenorphine on antinociception, respiratory depression, and itch/scratching responses. A and B, dose-response curves for antinociception produced by buprenorphine alone and in mixtures with either Ro 64-6198 (A) or SCH 221510 (B). C and D, dose-response curves for respiratory depression produced by buprenorphine alone and in the same dose combination, as presented in A and B, with either Ro 64-6198 (C) or SCH 221510 (D). E and F, dose-response curves for itch/scratching responses elicited by buprenorphine alone or in the same dose combination, as presented in A and B, with either Ro 64-6198 (E) or SCH 221510 (F). All drugs were administered intramuscularly in the same subjects. Each data point represents a mean ± S.E.M. (n = 6). *, a significant difference from the dosing condition of buprenorphine (0.003 mg/kg) alone (p < 0.05).
Figure 8 displays isobolograms for both mixtures of Ro 64-6198 + buprenorphine (Fig. 8A) and SCH 221510 + buprenorphine (Fig. 8B). The experimentally determined ED50 values (Zmix) and predicted additive ED50 values (Zadd) are shown in Tables 2 and 3. The statistical analysis confirmed that Zmix values were significantly different from Zadd values for each mixture, indicating that there was a synergistic interaction for antinociceptive effects produced by buprenorphine in combination with either Ro 64-6198 or SCH 221510 in primates.

Effects of the NOP agonists Ro 64-6198 and SCH 221510 in combination with buprenorphine on antinociceptive effects against 50°C water. Isobolograms for the mixture of buprenorphine with either Ro 64-6198-induced antinociception (A) or SCH 221510-induced antinociception (B) are displayed. . Each data point represents a mean ± S.E.M. (n = 6). See Tables 2 and 3 and Data Analysis for other details.
ED50 values for agonists alone in the primate antinociceptive assay
Experimentally determined ED50 values (Zmix) and predicted additive ED50 values (Zadd) of mixtures of NOP agonists administered in combination with buprenorphine in the primate antinociceptive assay
Discussion
The first part of the study showed that systemic administration of buprenorphine dose-dependently produced antinociception, respiratory depression, and itch/scratching at the dose range of 0.01 to 0.1 mg/kg (Figs. 1⇑–3). Up to a 10-fold higher dose 1 mg/kg of buprenorphine retained its antinociception without aggravating respiratory depression. This ceiling effect of buprenorphine-induced respiratory depression clearly supports its wide safety margin in clinical use (Rosenblum et al., 2008; Kress, 2009; Pergolizzi et al., 2010). Buprenorphine has been estimated to be 30 to 50 times more potent than morphine in humans (Jasinski et al., 1978; Kress, 2009; Pergolizzi et al., 2010). This potency difference between systemic buprenorphine and morphine maintains the same ratio in the primate nociceptive assay, based on either ED50 values (0.02–0.04 versus 0.9 mg/kg) or the minimum doses (0.1 versus 3 mg/kg) producing full antinociception (Walker et al., 1995; Lee et al., 2007; Ko and Husbands, 2009). More importantly, a single dose of buprenorphine (0.1 mg/kg) in primates produced antinociceptive effects accompanied by a mild to moderate degree of respiratory depression and itch, a similar profile as reported previously in human studies (Zacny et al., 1997; Kress, 2009; Pergolizzi et al., 2010). These findings indicate that the therapeutic profile of buprenorphine in humans can be manifested and studied in different physiological functional assays in nonhuman primates.
The second part of the study demonstrated that there were distinct antagonist effects between MOP and NOP antagonists on buprenorphine-induced physiological responses (Figs. 4⇑–6). Pretreatment with the MOP antagonist naltrexone (0.03 mg/kg) produced large rightward shifts (∼20- to 30-fold) of buprenorphine's dose-response curves for antinociception, respiratory depression, and itch/scratching. The mean naltrexone pKB values for these endpoints ranged between 8.1 and 8.3. In previous studies using primates, this dose of naltrexone has been shown to selectively block MOP agonist-induced effects, and it has similar naltrexone pKB values ranging from 8.2 to 8.6 (Ko et al., 2004, 2006a,b, 2009). These findings clearly suggest that buprenorphine-induced antinociception, respiratory depression, and itch/scratching in primates are mediated mainly by MOP receptors. Such a conclusion may be expected because buprenorphine has relatively high binding affinity and measurable intrinsic activity at MOP receptors (Traynor and Nahorski, 1995; Huang et al., 2001; Zaveri et al., 2001; Clark et al., 2006), and rodent studies also indicate that buprenorphine-induced antinociception is mediated mainly by MOP receptors (Kögel et al., 2005; Yamamoto et al., 2006).
Unlike findings in rodent studies (Lutfy et al., 2003; Ding and Raffa, 2009; Khroyan et al., 2009), the NOP antagonist J-113397 did not enhance buprenorphine-induced antinociception in primates. A single dose (0.1 mg/kg) of J-113397 has been demonstrated to selectively block NOP agonist-induced effects without interfering with MOP receptor-mediated effects (Ko et al., 2006b, 2009; Hu et al., 2010; Podlesnik et al., 2011). In particular, this dosing regimen produced approximately a 30-fold rightward shift of the dose-response curve for the NOP agonist Ro 64-6198-induced antinociception in primates (Ko et al., 2009). Pretreatment with the same dose of J-113397 did not produce any shifts of buprenorphine's dose-response curves, indicating that there are no detectable functional NOP receptors involved in buprenorphine-induced physiological responses in primates under this condition.
Buprenorphine has relatively low binding affinity at NOP receptors; its Ki values range from 77 to 285 nM, and its binding selectivity for MOP versus NOP receptors varies from 50- to 3500-fold (Huang et al., 2001; Zaveri et al., 2001; Spagnolo et al., 2008; Khroyan et al., 2009). In the functional assay of [35S]GTPγS binding for NOP receptors, buprenorphine produced no stimulation (Spagnolo et al., 2008) or mild to moderate stimulation (16–60% compared with N/OFQ) as a partial agonist action (Huang et al., 2001; Khroyan et al., 2009). It seems unlikely that buprenorphine activates NOP receptors within the antinociceptive dose range. There are several newly developed compounds such as 1-(1-(bicyclo[3.3.1]nonan-9-yl)piperidin-4-yl)indolin-2-one (SR16435) and 2-(N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxymorphinan-7-yl)-3,3-dimethylpentan-2-ol (BU08028) that have high binding affinity for both MOP and NOP receptors (Ki values 2–8 nM) with 20 to 50% of [35S]GTPγS binding stimulation (Spagnolo et al., 2008; Khroyan et al., 2009, 2011). It is noteworthy that in rodent studies pretreatment with the NOP antagonist enhanced all of the ascending portions of the dose-response curves for antinociception produced by SR16435, BU08028, and buprenorphine (Khroyan et al., 2009, 2011). These findings may suggest that the binding “selectivity” for MOP versus NOP receptors (50- to 3500-fold versus no selectivity) does not prevent the compound's susceptibility to the NOP antagonism in rodents. It will be important to further investigate whether compounds without the MOP/NOP binding selectivity (mixed MOP/NOP agonists) have different therapeutic profiles compared with buprenorphine in primates.
The third part of the study examined the effects of the NOP agonists on buprenorphine-induced physiological functions by using the dose-addition analysis and isobolograms (Figs. 7 and 8). What is exciting is that the combined administration of buprenorphine with either Ro 64-6198 or SCH 221510 resulted in synergistic antinociceptive effects in primates. These results provide novel functional evidence that the activation of NOP receptors does not attenuate MOP agonist-induced antinociception; instead, there is a potentiated antinociception produced by coactivation of MOP and NOP receptors at the systemic level in primates. What is more stimulating is that by increasing the ratio of the NOP agonist in combination with buprenorphine, the specific mixture produced full antinociception without respiratory depression and itch/scratching (dosing conditions presented in Fig. 7). When N/OFQ was combined with a single dose of intrathecal morphine, this combination dose-dependently enhanced morphine-induced antinociception without compromising the primates' motor function (Ko and Naughton, 2009). When an inactive dose of the NOP agonist [(pF)Phe4Aib7Arg14Lys15]N/OFQ-NH2 (UFP-112) was combined intrathecally with an inactive dose of morphine, such a mixture significantly produced antinociception against capsaicin-induced allodynia (Hu et al., 2010). Considering that simultaneous activation of two receptors to a small degree produces therapeutic effects with fewer side effects (a wider therapeutic window), these findings strongly indicate that mixed MOP/NOP agonists may represent a novel strategy for pain management.
Collectively, this study demonstrates that MOP receptors mediate mainly buprenorphine-induced antinociception, respiratory depression, and itch/scratching in primates. Unlike rodent studies showing that the NOP antagonists enhanced buprenorphine-induced antinociception (Lutfy et al., 2003; Ding and Raffa, 2009; Khroyan et al., 2009), the NOP antagonist J-113397 did not change buprenorphine-induced antinociception in primates, indicating that NOP receptors are not involved in buprenorphine-induced physiological responses in primates. More importantly, it is the NOP “agonists” that actually potentiated MOP-mediated antinociception in primates. Physiological functions of opioid receptor subtypes and the pharmacological profiles of synthetic opioid-related compounds may vary between rodents and primates. For example, a buprenorphine-like compound, BU72, was characterized as a MOP agonist with a wide therapeutic window in rodents, but 17-methyl-3-hydroxy-[5β, 7β, 3′, 5′]-pyrrolidino-2′[S]-phenyl-7α-methyl-6,14-endoetheno morphinan (BU72) was very potent in producing respiratory depression in primates and displayed a very narrow window between antinociceptive doses and doses producing side effects (Neilan et al., 2004). The present study is the first to provide functional evidence that systemic coadministration of MOP and NOP agonists produced synergistic antinociceptive effects without other side effects in primates. Given that NOP agonists have low abuse liability and/or antiaddiction property (Ko et al., 2009; Khroyan et al., 2011; Zaveri, 2011), it is worth developing bifunctional MOP/NOP agonists as analgesics or antiaddiction drugs (Spagnolo et al., 2008; Cami-Kobeci et al., 2011; Khroyan et al., 2011; Zaveri, 2011). If NOP agonists have as yet undescribed side effects that limit their use as analgesics, adding them to MOP agonists may allow doses of both drugs to be reduced, analgesia enhanced, and side-effects of both reduced. In other words, by reserving most functional receptor pools, simultaneous activation of two receptor components to a small degree may produce desirable therapeutic effects with fewer side effects. Not only producing a wider therapeutic window, this approach may also provide a reduced tolerance liability when two receptors are repeatedly activated because of a large reservoir for both receptor populations. Future studies are warranted to compare the rates and degrees of tolerance development after chronic administration of bifunctional MOP/NOP agonists versus selective agonists targeting one receptor and validate their therapeutic potential in primate models.
Authorship Contributions
Participated in research design: Kyle and Ko.
Conducted experiments: Cremeans and Gruley.
Performed data analysis: Cremeans, Gruley, and Ko.
Wrote or contributed to the writing of the manuscript: Cremeans, Gruley, Kyle, and Ko.
Acknowledgments
We thank Steve Pouliot and Angela Bodinus for technical assistance.
Footnotes
This study was supported by the National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases [Grant R01-AR-059193] and Purdue Pharma L.P. [Grant UM-DRDA-N010183].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
ABBREVIATIONS:
- MOP
- μ-opioid peptide
- NOP
- nociception/orphanin FQ peptide
- N/OFQ
- nociceptin/orphanin FQ
- J-113397
- (±)-1-[(3R*,4R*)- 1-(cyclooctylmethyl)-3-(hydroxymethyl)-4-piperidinyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one
- Ro 64-6198
- (1S,3aS)-8-(2,3,3a,4,5,6-hexahydro-1H-phenalen-1-yl)-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one
- SCH 221510
- 3-endo-8-[bis(2-methylphenyl)methyl]-3-phenyl-8-azabicyclo[3.2.1]octan-3-ol
- GTPγS
- guanosine 5′-O-(3-thio)triphosphate
- CL
- confidence limit
- SR 16435
- 1-(1-(bicyclo[3.3.1]nonan-9-yl)piperidin-4-yl)indolin-2-one
- BU08028
- 2-(N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxymorphinan-7-yl)-3,3-dimethylpentan-2-ol
- UFP-112
- [(pF)Phe4Aib7Arg14Lys15]N/OFQ-NH2
- BU72
- 17-methyl-3-hydroxy-[5β, 7β, 3′, 5′]-pyrrolidino-2′[S]-phenyl-7α-methyl-6,14-endoetheno morphinan.
- Received March 13, 2012.
- Accepted June 20, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}