


Abstract
There is considerable interest in understanding the regulation of peripheral opioid receptors to avoid central nervous system side effects associated with systemically administered opioid analgesics. Here, we investigated the regulation of the κ-opioid receptor (KOR) on rat primary sensory neurons in vitro and in a rat model of thermal allodynia. Under basal conditions, application of the KOR agonist trans-(1S,2S)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzeneacetamide hydrochloride hydrate (U50488) did not inhibit adenylyl cyclase (AC) activity nor release of calcitonin gene-related peptide (CGRP) in vitro and did not inhibit thermal allodynia in vivo. However, after 15-min pretreatment with bradykinin (BK), U50488 became capable of inhibiting AC activity, CGRP release, and thermal allodynia. Inhibition of AC by 5-hydroxytryptamine 1 or neuropeptide Y1 receptor agonists and stimulation of extracellular signal-regulated kinase activity by U50488 did not require BK pretreatment. The effect of U50488 in BK-primed tissue was blocked by the KOR antagonist nor-binaltorphimine both in vitro and in vivo. The effect of BK in vitro was blocked by either indomethacin or bisindolylmaleimide, suggesting that an arachidonic acid (AA) metabolite and protein kinase C (PKC) activation mediate BK-induced regulation of the KOR system. Furthermore, the effect of U50488 in BK-treated tissue was blocked by a soluble integrin-blocking peptide (GRGDSP), but not the inactive reverse sequence peptide (GDGRSP), suggesting that, in addition to AA and PKC, RGD-binding integrins participate in the regulation of KOR signaling in response to U50488. Understanding the mechanisms by which peripheral KOR agonist efficacy is regulated may lead to improved pharmacotherapy for the treatment of pain with reduced adverse effects.
Introduction
Drugs that act via opioid receptors, such as morphine and its analogs, are considered a “gold standard” for the treatment of pain. However, the management of pain with opioid analgesics is complicated by adverse effects that limit clinical dosages to below those needed for adequate pain relief. Many of these adverse effects are associated with opioid actions within the CNS and include respiratory depression, dysphoria, and addiction. Consequently, there is considerable interest in understanding the function of opioid receptors located in the periphery on primary sensory neurons. Targeting these receptors with peripherally restricted opioid drugs would avoid centrally mediated adverse effects.
Opioid receptors are expressed on primary sensory neurons; however, the reported analgesic efficacy of peripherally restricted opioid drugs is highly variable. Although in animal studies local, peripheral administration of opioids is antihyperalgesic/antiallodynic when administered to injured or inflamed tissue, peripheral opioids are much less effective, or ineffective, when administered to normal tissue (Joris et al., 1987; Przewlocki and Przewlocka, 2001; Stein and Zöllner, 2009). This has led to the hypothesis that some stimulus derived from inflamed or damaged tissue is required to induce functional competence of quiescent opioid receptor systems. Although many studies with animals demonstrate analgesic efficacy, studies in human indicate that the efficacy of peripherally restricted opioids on inflammatory pain is often mild to moderate, with many studies reporting no efficacy over placebo at all (Gupta et al., 2001; Kalso et al., 2002). This variability may be caused by clinical pain conditions that differ in their ability to evoke and/or maintain opioid receptor system functional competence. Consequently, it is important to understand the mechanisms by which opioid receptor system function in the periphery is regulated because this may yield insight into developing adjunctive agents to maximize peripheral opioid receptor system competence.
We have found that μ- and δ-opioid receptors (MORs and DORs, respectively) are expressed in primary cultures of rat sensory neurons but seem to be functionally quiescent in that opioid agonists are ineffective at inhibiting neuropeptide release and adenylyl cyclase activity. However, brief exposure to an inflammatory mediator, such as bradykinin (BK), rapidly induces functional competence for both inhibition of neuropeptide release and inhibition of adenylyl cyclase activity. This effect of BK is mediated by a cyclooxygenase (COX)-dependent metabolite of arachidonic acid (AA) that is downstream from activation of protein kinase C (PKC) (Patwardhan et al., 2005, 2006; Berg et al., 2007a). Moreover, DOR agonists injected locally into the hindpaw do not alter prostaglandin E2-(PGE2) induced thermal allodynia unless the injection is preceded by an intraplantar injection of BK (Rowan et al., 2009).
Historically, κ-opioid receptors (KORs) have not been considered viable targets for development of analgesic drugs because systemic administration results in CNS-mediated adverse effects including hallucinations and dysphoria (for reviews see Kivell and Prisinzano, 2010; Vanderah, 2010). However, very little is known about analgesic responses mediated by peripheral KOR. Here, we studied regulation of responses to the KOR agonist trans-(1S,2S)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl) cyclohexyl]benzeneacetamide hydrochloride hydrate (U50488) in primary cultures of sensory neurons and in a rat model of thermal allodynia.
Materials and Methods
Drugs and Chemicals.
The following compounds were purchased from Cayman Chemical (Ann Arbor, MI): prostaglandin E2, arachidonic acid, and indomethacin (Indo). The PKC inhibitor bisindolylmaleimide (Bis) was obtained from Calbiochem (San Diego, CA). 125I-cAMP was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). (−)U50488, naltrindole, nor-binaltorphimine (nor-BNI), and 5′-guanidinonaltrindole (5′-GNTI) were purchased from Sigma-Aldrich (St. Louis, MO). Fetal bovine serum was from Atlanta Biologicals (Norcross, GA). Collagenase was from Worthington Biochemicals (Freehold, NJ). All other tissue culture reagents were purchased from Invitrogen (Carlsbad, CA). All other drugs and chemicals (reagent grade) were purchased from Sigma-Aldrich.
Animals.
Adult male Sprague-Dawley rats (Charles River Laboratories, Inc., Wilmington, MA), weighing 250 to 300 g, were used in this study. The animal study protocol was approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio and conformed to International Association for the Study of Pain and federal guidelines. Animals were housed for 1 week, with food and water available ad libitum, before experimentation.
Rat Trigeminal Ganglia Culture.
Primary cultures of rat trigeminal ganglion (TG) cells were prepared as described previously (Patwardhan et al., 2005, 2006; Berg et al., 2007a,b). Fresh TGs were washed with Hanks' balanced salt solution (HBSS; Ca2+, Mg2+ free), digested with 3 mg/ml collagenase for 30 min at 37°C, and centrifuged (1000 rpm; 1 min). The pellet was further digested with 0.1% trypsin (15 min) and 167 μg/ml DNase (10 min) at 37°C in the same solution. Cells were pelleted by centrifugation (2 min at 2000 rpm) and resuspended in Dulbecco's modified Eagle medium (high glucose) containing 100 ng/ml nerve growth factor (Harlan, Indianapolis, IN), 10% fetal bovine serum, 1× penicillin/streptomycin, 1× l-glutamine, and the mitotic inhibitors 7.5 μg/ml uridine and 17.5 mg/ml 5-fluoro-2′-deoxyuridine. After trituration to give a single cell suspension, cells were seeded on polylysine-coated 48-well plates (BD Bioscience, San Jose, CA). Media were changed 24 and 48 h after plating. On the fifth day of culture, cells were refed with serum-free Dulbecco's modified Eagle's medium without nerve growth factor. Cells were used on the sixth day of culture. When present, pertussis toxin (PTx; 400 ng/ml) was added during the 24-h serum-free culture period.
Measurement of Cellular cAMP Accumulation.
Opioid agonist-induced inhibition of PGE2-stimulated adenylyl cyclase activity was measured as described previously (Berg et al., 2007a,b). In brief, TG cultures in 48-well plates were washed twice with HBSS containing 20 mM HEPES, pH 7.4 (wash buffer). Cells were pre-equilibrated in 250 μl of wash buffer per well for 30 min at 37°C (room air). Cells were then incubated with the KOR agonist U50488 (various concentrations) in the presence of the phosphodiesterase inhibitor rolipram (100 μM) for 15 min at 37°C. A maximal concentration of PGE2 (1 μM) was added, and cells were incubated for a further 15 min. As appropriate, BK (10 μM) was added during the pre-equilibration period, 15 min before U50488. Incubations were terminated by aspiration of the wash buffer and addition of 500 μl of ice-cold absolute ethanol. The ethanol extracts from individual wells were dried under a gentle air stream and reconstituted in 100 μl of 50 mM sodium acetate, pH 6.2. The cAMP content of each 100-μl sample was determined by radioimmunoassay (RIA).
Measurement of Immunoreactive Calcitonin Gene-Related Peptide Release.
Opioid agonist-induced inhibition of BK/PGE2-stimulated immunoreactive calcitonin gene-related peptide (iCGRP) release from TG cultures was measured as described previously (Patwardhan et al., 2005; Berg et al., 2007a). In brief, TG cells were harvested and grown in 48-well plates for 5 days as described above. Cells were washed twice in release buffer (HBSS supplemented with 10.9 mM HEPES, 4.2 mM sodium bicarbonate, 10 mM dextrose, and 0.1% bovine serum albumin, pH 7.4) and then pretreated with vehicle (Veh) or BK (10 μM). Fifteen minutes later, cells were treated with opioid ligands or vehicle (15 min), followed by PGE2/BK (1 μM/10 μM) or vehicle for an additional 15 min. Levels of iCGRP obtained from the supernatant (500 μl) were measured with radioimmunoassay.
Extracellular Signal-Regulated Kinase 1/2 Phosphorylation.
TG cells were harvested and grown in 48-well plates culture for 5 days as described above. Cells were washed twice with HBSS containing 20 mM HEPES, pH 7.4 (wash buffer) and pre-equilibrated in 250 μl of wash buffer per well for 30 min at 37°C (room air). Cells were then incubated with the KOR agonist U50488 (100 nM) in the absence or presence of nor-BNI (3 nM) for 0 to 15 min at 37°C. BK (10 μM) or vehicle was added during the pre-equilibration period 15 min before U50488. Incubations were terminated by aspiration of buffer and addition of 50 μl of lysis buffer supplied with the SureFire phospho-extracellular signal-regulated kinase (pERK) assay kit (PerkinElmer Life and Analytical Sciences). Samples were processed according to the manufacturer's directions. The fluorescence signal from pERK was measured in duplicate using a Fluostar microplate reader (BMG Labtech GmbH, Offenburg, Germany) with AlphaScreen settings.
Behavioral Assay.
Opioid agonist-mediated changes in paw withdrawal latency (PWL) to a thermal stimulus were measured with a plantar test apparatus (Hargreaves et al., 1988) as described previously (Rowan et al., 2009). In brief, rats were placed in plastic boxes (10 × 20 cm) with a glass floor. After a 30-min habituation period, the plantar surface of the hindpaw was exposed to a beam of radiant heat through the glass floor. The rate of increase in temperature of the glass floor was adjusted so that baseline PWL values were approximately 10 ± 2 s; cutoff time was 25 s to prevent tissue damage. PWL measurements were taken in duplicate at least 30 s apart at 5-min intervals, and the average was used for statistical analysis. After baseline PWL was determined, animals were pretreated with BK (25 μg) or Veh via intraplantar injection 15 min before coinjection with U50488 (with indicated doses) and PGE2 (0.3 μg). Where indicated, antagonists were injected intraplantarly with BK or Veh pretreatment. Data are expressed as the change (s) from individual PWL baseline values. All drugs, prepared in phosphate-buffered saline, were administered via intraplantar injection at a final volume of 50 μl. Observers were blinded to the treatment allocation.
Data Analysis.
For TG cell culture experiments, concentration-response data were fit to a logistic equation (equation 1) using nonlinear regression analysis to provide estimates of maximal response (Rmax), potency (EC50), and slope factor (n).
 where R is the measured response at a given agonist concentration [A], Ro is the response in the absence of agonist, Ri is the response after maximal inhibition by the agonist, EC50 is the concentration of agonist that produces half-maximal response, and n is the slope index. Rmax (the maximal inhibition produced by the agonist) was calculated as Ro − Ri. Experiments were repeated at least three times. Statistical differences in concentration-response curve parameters between groups were analyzed with Student's paired t test. When only a single concentration was used, statistical significance was assessed using one-way analysis of variance (ANOVA) followed by Bonferroni's post hoc or Student's t test (paired) using Prism software (GraphPad Software, Inc., San Diego, CA). p < 0.05 was considered statistically significant.
where R is the measured response at a given agonist concentration [A], Ro is the response in the absence of agonist, Ri is the response after maximal inhibition by the agonist, EC50 is the concentration of agonist that produces half-maximal response, and n is the slope index. Rmax (the maximal inhibition produced by the agonist) was calculated as Ro − Ri. Experiments were repeated at least three times. Statistical differences in concentration-response curve parameters between groups were analyzed with Student's paired t test. When only a single concentration was used, statistical significance was assessed using one-way analysis of variance (ANOVA) followed by Bonferroni's post hoc or Student's t test (paired) using Prism software (GraphPad Software, Inc., San Diego, CA). p < 0.05 was considered statistically significant.
For behavioral experiments, time-course data were analyzed with repeated measures two-way analysis of variance, followed by Bonferroni's post hoc test using Prism. Area under the time-response curve was calculated and analyzed with one-way ANOVA followed by Bonferroni's post hoc test using Prism. Statistical inference was made when p < 0.05. Data are presented as mean ± S.E.M.
Results
Characterization of KOR Responses in Primary Cultures Derived from TG.
In primary cultures derived from adult rat TG incubation with the selective KOR agonist U50488 did not alter PGE2-stimulated adenylyl cyclase activity. After 15 min of pretreatment with the inflammatory mediator BK, U50488 inhibited adenylyl cyclase activity by approximately 60% in a pertussis toxin-sensitive manner (Fig. 1A). Figure 1B shows concentration-response curves for U50488 in which the EC50 was 2.3 nM (pEC50 8.63 ± 0.53) with a maximal inhibition of 51 ± 2%. The response to U50488 was blocked by the selective KOR antagonist nor-BNI, but not by the DOR antagonist naltrindole. Likewise, U50488 did not alter stimulated iCGRP release unless TG neurons were first pre-exposed to BK (Fig. 2).

Effect of the KOR agonist U50488 on PGE2-stimulated cAMP accumulation in TG cultures. A, pretreatment with BK is necessary for U50488 to inhibit cAMP accumulation. TG primary cultures were incubated with or without PTx (400 ng/ml, 24 h). Cells were then pretreated with or without BK (10 μM) for 15 min. After pretreatment, cells were incubated with U50488 (100 nM) for 15 min followed by the addition of PGE2 (1 μM) and further incubation for 15 min. Cellular levels of cAMP were determined by RIA. Neither BK nor PTx significantly affected PGE2-stimulated cAMP accumulation (p = 0.72, one-way ANOVA). Basal levels of cAMP (mean pmol ± S.E.M.) were 1.09 ± 0.21 (n = 3) for vehicle conditions, 2.28 ± 0.29 (n = 7) for BK-pretreated cells, and 1.22 ± 0.19 (n = 4) for cells treated 24 h with PTx. The data shown represent the mean percentage above basal cAMP accumulation ± S.E.M. of three to seven experiments. *, p < 0.05, Veh versus U50488, one-way ANOVA with Bonferroni's post test. B, concentration-response curve to U50488 for inhibition of PGE2-stimulated cAMP accumulation in TG cultures pretreated with BK (10 μM, 15 min). The U50488 response is blocked by the KOR antagonist nor-BNI (3 nM), but not by the DOR antagonist naltrindole (20 nM). Antagonists were added during the BK pretreatment period. The data shown represent the mean percentage of PGE2-stimulated cAMP accumulation ± S.E.M. of four experiments.

Effect of the KOR agonist U50488 on BK/PGE2-evoked iCGRP release in TG cultures. Pretreatment with BK is necessary for U50488 to inhibit iCGRP release. Cells were pretreated with or without BK (10 μM) for 15 min, washed, incubated for 15 with or without U50488 (100 nM), and then further incubated with PGE2 (1 μM) + BK (10 μM) for 15 min. Samples of the media were assayed for iCGRP with RIA. Data are expressed as the percentage over basal iCGRP release and represent the mean ± S.E.M. of three to five experiments. Horizontal line at 100% represents baseline release. *, p < 0.05, Veh versus BK pretreatment conditions. **, p < 0.01, Veh versus U50488 conditions, one-way ANOVA with Bonferroni's post test.
Unlike the results from the adenylyl cyclase and the iCGRP experiments, U50488-mediated stimulation of ERK activation (measured as production of pERK) did not depend on prior treatment with BK. Figure 3 shows the time course of pERK production over 15 min in response to U50488. Neither the time course nor the maximal response (≈130% basal) were altered by BK. Stimulation of pERK production by U50488 was completely blocked by nor-BNI (3 nM).

U50488 stimulation of ERK activity in TG cultures does not require BK pretreatment. TG cultures were pretreated with or without BK (10 μm) for 15 min. Cells were then treated with U50488 (100 nM) for the times indicated, and the level of pERK was measured using the pERK Surefire assay kit from PerkinElmer Life and Analytical Sciences, according to the manufacturer's protocol. Nor-BNI was present 15 min before the addition of U50488. Data are expressed as the percentage increase in pERK levels over basal (no ligand) activity and represent the mean ± S.E.M. of four experiments. *, p < 0.05, nor-BNI versus Veh; ***, p < 0.0001, U50488 versus basal, two-way ANOVA with Bonferonni post test.
In addition to opioid receptors, cells of the trigeminal ganglion express other Gi protein-coupled receptors, such as 5-HT1B/1D receptors (Ahn and Basbaum, 2006) and receptors for neuropeptide Y (NPY) (Gibbs et al., 2007). Figure 4 shows that inhibition of PGE2-stimulated adenylyl cyclase activity occurred without BK pretreatment in response to maximal concentrations of the 5-HT1 agonist 5-carboxamidotryptamine (5-CT) or by the NPY1 receptor agonist Leu-Pro NPY (L,P-NPY). The response to either agonist was not altered by BK and was sensitive to inhibition mediated by treatment with pertussis toxin.

Inhibition of PGE2-stimulated cAMP accumulation by the 5-HT1 receptor agonist 5-CT or the NPY1 receptor agonist L,P-NPY in TG cultures does not require BK pretreatment. TG primary cultures were incubated with or without PTx (400 ng/ml, 24 h). Cells were then pretreated with or without BK (10 μM) for 15 min. After pretreatment, cells were incubated with 5-CT (100 nM) or L,P-NPY (1 μM) for 15 min followed by the addition of PGE2 (1 μM) and further incubation for 15 min. Cellular levels of cAMP were determined by RIA. Basal levels of cAMP (mean pmol ± S.E.M.) were 1.07 ± 0.13 (n = 11) for vehicle conditions, 1.3 ± 0.13 (n = 7) for BK-treated cells, and 1.14 ± 0.23 (n = 4) for cells treated 24 h with PTx. The data shown represent the mean percentage above basal cAMP accumulation ± S.E.M. of three to seven experiments. BK pretreatment did not significantly alter the inhibition response of either 5-CT (p = 0.94) or L,P-NPY (p = 0.72) compared with vehicle, one-way ANOVA with Bonferroni's post test. ***, p < 0.001 Veh versus 5-CT or Veh versus L,P-NPY.
Mechanism of BK-Mediated Competency of KOR Function in TG Primary Cultures.
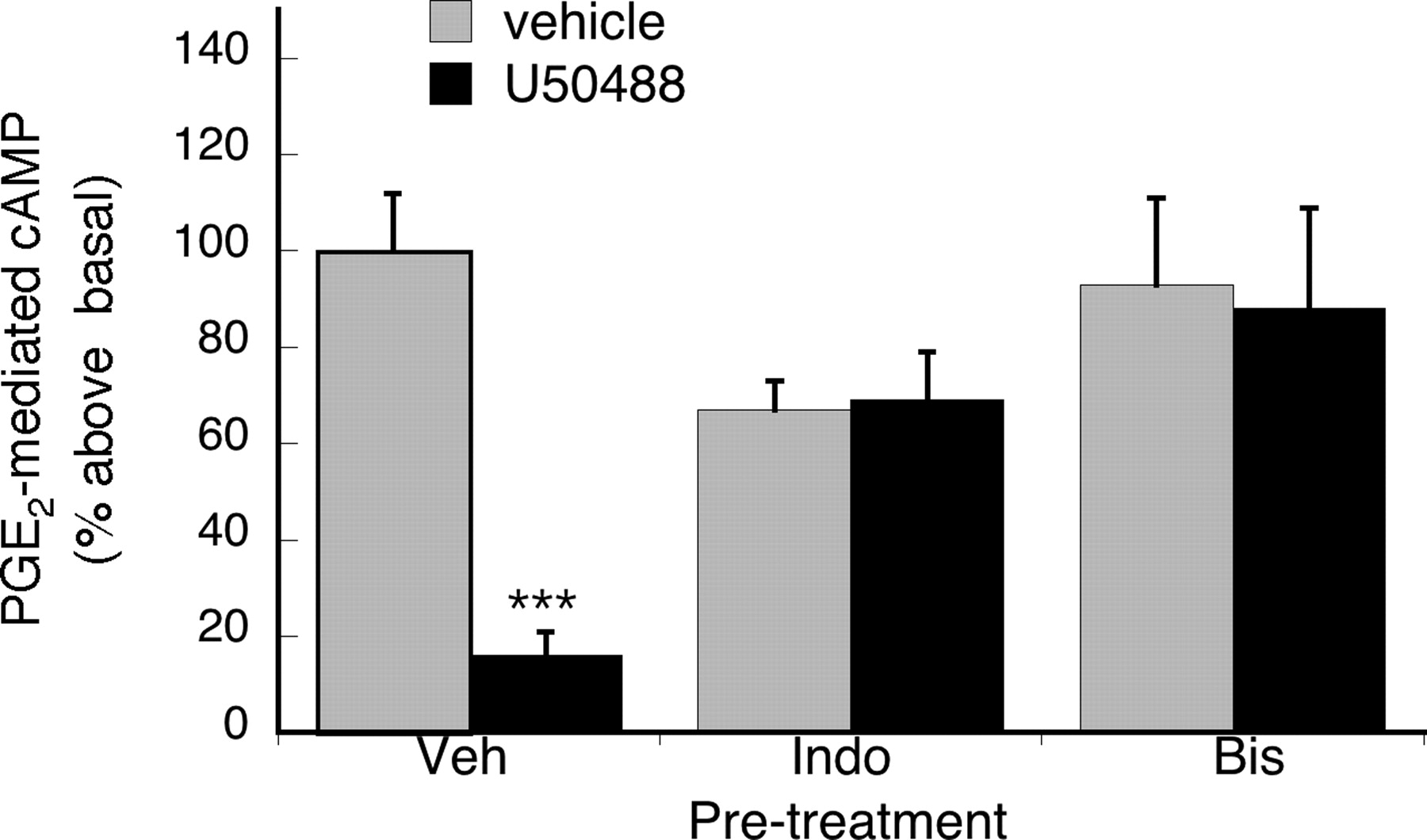
Figure 5 shows that the effect of BK to promote functional competency of KOR to inhibit PGE2-stimulated adenylyl cyclase activity was blocked by the inhibitor of PKC, Bis, and the cyclooxygenase inhibitor indomethacin, suggesting that PKC and cyclooxygenase are involved in mediating the action of BK on KOR signaling. Furthermore, the BK effect was also blocked by an antagonist of the arginine-glycine-asparagine (RGD)-binding integrins, soluble Gly-Arg-Gly-Asp-Ser-Pro (GRGDSP) peptide, but not by the inactive, reverse sequence peptide, Gly-Asp-Gly-Arg-Ser-Pro (GDGRSP) (Fig. 6).

Effect of inhibition of PKC or cyclooxygenase on U50488-mediated inhibition of PGE2-evoked cAMP accumulation. TG cultures were treated with vehicle or either the cyclooxygenase inhibitor Indo (2 μM), or the PKC inhibitor Bis (1 μM), for 15 min before pretreatment with BK (10 μM) for 15 min. After pretreatment, TG cells were incubated with or without U50488 (100 nM) for 15 min followed by addition of PGE2 (1 μM) and further incubation for 15 min. Cellular cAMP levels were determined by RIA. Data shown are the mean ± S.E.M. of four experiments. Data are expressed as the mean percentage above basal cAMP levels ± S.E.M. Neither Indo nor Bis altered PGE2-stimulated cAMP levels (p > 0.05 versus vehicle, one-way ANOVA). ***, p < 0.001, Veh versus U50488, one-way ANOVA with Bonferroni's post test.

BK effects on KOR-mediated inhibition of PGE2-stimulated cAMP accumulation is dependent on RDG-binding integrin activation. Soluble RGD peptides (GRGDSP), but not the inactive, reverse sequence peptides (GDGRSP) block BK pretreatment effects on U50488-mediated inhibition of PGE2-stimulated cAMP accumulation. TG cultures were incubated with vehicle, soluble GRGDSP peptides (100 μM) or the inactive soluble GDGRSP (100 μM) for 30 min, followed by addition of BK (10 μM) and further incubation for 15 min. After BK pretreatment, cells were incubated with vehicle or U50488 (100 nM) for 15 min, followed by addition of PGE2 (1 μM) and further incubation for 15 min. Cellular cAMP levels were determined by RIA. Data shown are the mean ± S.E.M. of four experiments. Data are expressed as the mean percentage above basal cAMP levels ± S.E.M. PGE2-stimulated cAMP levels were not significantly altered by either RGD or DGR treatment, p > 0.05 versus vehicle, one way ANOVA. ***p < 0.001, Veh versus U50488, one-way ANOVA with Bonferroni's post test.
KOR-Mediated Inhibition of PGE2-Induced Thermal Allodynia.
Administration of PGE2 to the hindpaw of rats by intraplantar injection reduces PWL in response to a thermal stimulus (thermal allodynia) for at least 20 min (Rowan et al., 2009). Intraplantar injection of BK produces a transient thermal allodynia that lasts less than 15 min (Rowan et al., 2009). Coadministration of U50488 to the hindpaw, at doses up to 10 μg, with PGE2 did not alter PWL (Fig. 7). However, if U50488 was coadministered at a dose as low as 0.1 μg 15 min after administration of BK the thermal allodynia was significantly reversed (Fig. 7). It is noteworthy that after BK administration the dose-response curve for U50488 for antiallodynia was an inverted-U shape. The largest antiallodynic effect of U50488 (as assessed by the area under the time-response curve) occurred for the 0.1-μg dose, whereas the response to 10 μg of U50488 was less (Supplemental Fig. 1). The effect of U50488 (1 μg) in BK-pretreated hindpaws was blocked by the KOR antagonists nor-BNI and 5′-GNTI (Fig. 8A). The antagonists did not alter PGE2-induced thermal allodynia when administered to BK-pretreated hindpaws (data not shown; p > 0.05, two-way ANOVA with Bonferroni's post test). The effect of U50488 was restricted to the hindpaw ipsilateral to the BK injection (Fig. 8B), indicating the effect of U50488 was mediated by peripheral KOR.

The antiallodynic effect of U50488 on PGE2-mediated thermal allodynia requires pretreatment with BK. Separate groups of animals were injected intraplantarly with BK (25 μg) or vehicle 15 min before coinjection with U50488 (doses indicated) and PGE2 (0.3 μg). PWL was measured in duplicate every 5 min for 20 min after the last injection. Data are expressed as change from individual baselines and represent mean ± S.E.M. of six animals per group. Baseline PWLs were 10.06 ± 0.21. **, p < 0.01; ***, p < 0.001, U50488 versus corresponding Veh time point, two-way repeated ANOVA with Bonferroni's post test.

The antiallodynic effect of U50488 on PGE2-mediated thermal allodynia is blocked by KOR antagonists and is peripherally mediated. A, separate groups of animals were injected intraplantarly with BK (25 μg) with or without the KOR-selective antagonists nor-BNI (10 μg) or 5′-GNTI (2 μg) 15 min before coinjection with U50488 (1 μg) and PGE2 (0.3 μg). PWL was measured in duplicate every 5 min for 20 min after the last injection. Data are expressed as change from individual baselines and represent mean ± S.E.M. of six animals per group. Baseline PWLs were 9.74 ± 0.26 s. **, p < 0.01; ***, p < 0.001 versus Veh, two-way repeated ANOVA with Bonferroni's post test. B, administration of U50488 to the contralateral hindpaw did not alter PGE2-induced thermal allodynia in the hindpaw ipsalateral to the BK injection. Fifteen minutes after ipsilateral BK injection (25 μg), rats received single injections into both the ipsilateral and contralateral hindpaws. Animals received either coinjection of PGE2 (0.3 μg) with U50488 (10 μg) in the ipsilateral hindpaw (Ipsi), along with injection of vehicle in the contralateral hindpaw (Contra), or coinjection of PGE2 (0.3 μg) with vehicle ipsilaterally, along with injection of U50488 (10 μg) contralaterally. PWLs of the ipsilateral hindpaw were measured at 5-min intervals for 20 min after the last injection. Baseline PWLs were 9.93 ± 0.23 s. Data are expressed as change from individual baselines and represent mean ± S.E.M. of six animals per group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus Contra, two-way repeated ANOVA with Bonferroni's post test.
Discussion
An understanding of the mechanisms by which peripheral opioid receptor system function is regulated may be of value for improving the efficacy of peripherally acting opioid drugs and limiting the adverse effects that restrict doses used to treat pain. We found that MOR and DOR agonists do not inhibit PGE2-stimulated adenylyl cyclase activity nor BK/PGE2-stimulated neuropeptide release from cultures of adult rat sensory neurons unless the cells are pretreated with BK or the protease-activated receptor 2 agonist Ser-Leu-Ile-Gly-Arg-Leu-NH2, both of which activate Gq/11-mediated PLC signaling (Patwardhan et al., 2005, 2006; Berg et al., 2007a). Moreover, DOR agonists do not reduce PGE2-stimulated thermal allodynia when administered to the rat hindpaw at peripherally restricted doses unless BK is first preadministered to the hindpaw (Rowan et al., 2009).
As outlined in Fig. 9, the cellular mechanism by which MOR system functional competence is induced involves an increase in receptor-G protein coupling efficiency (assessed as an increase in GTP[γ35S] binding to Gi/o proteins) in response to a cyclooxygenase-dependent metabolite of arachidonic acid that is downstream from PKC (Berg et al., 2007a). BK induction of functional competence of opioid receptor systems in both in vitro and in vivo studies is blocked by inhibitors of PKC and cyclooxygenase (Patwardhan et al., 2005; Berg et al., 2007a; Rowan et al., 2009). The effect of BK on MOR signaling also is mediated by the RGD-binding class of integrins because RGD-binding integrin inhibitors block the induction of MOR functional competence to inhibit adenylyl cyclase activity (Berg et al., 2007b). Here, we found that the KOR agonist U50488 was ineffective at inhibition of adenylyl cyclase activity and iCGRP release from primary cultures of sensory neurons and did not inhibit PGE2-induced thermal allodynia when administered via intraplantar injection unless cells or tissue were pretreated with BK. In addition, the effect of BK in sensory neurons was blocked by inhibitors of PKC (Bis), cyclooxygenase (indomethacin), and RGD-binding integrins (soluble RGD peptide). These data suggest that all three peripheral opioid receptor systems require stimuli associated with inflammation to become capable of inhibiting adenylyl cyclase activity, neuropeptide release, and thermal allodynia.

Proposed mechanism for the regulation of opioid receptor functional competence by activation of BK signaling, based on Patwardhan et al. (2005, 2006), Berg et al. (2007a,b), Rowan et al. (2009), and the present study. BK, acting via bradykinin B2 receptors, activates PLC to produce inositol trisphosphate (IP3) and diacylglycerol (DAG). The B2-selective antagonist, d-Arg-l-Arg-l-Pro-l-Hyp-Gly-l-(2-thienyl)Ala-l-Ser-d-1,2,3,4-tetrahydro-3-isoquinolinecarbonyl-l-(2α,3β,7aβ)-octahydro-1H-indole-2-carbonyl-l-Arg (HOE 140), but not the B1-selective antagonist, Lys-[Leu8]des-Arg9-BK, prevents BK from eliciting functional competence. Agonists at other Gq-coupled receptors, such as the protease-activated receptor 2, will also induce functional competence. DAG activates PKC. Inhibitors of PKC, such as Bis or 5,6,7,13-tetrahydro-13-methyl-5-oxo-12H-indolo[2,3-a]pyrrolo[3,4-c]carbazole-12-propanenitrile (Go 6976), prevent the induction of functional competence by BK and activators of PKC, such the phorbol ester, phorbol dibutyrate (PdBu), promote functional competence. Activation of PKC in turn increases phospholipase A2-mediated AA release, which is metabolized by a COX. Inhibitors of COX, such as Indo, prevent induction of functional competence by BK and PdBu. Application of exogenous AA induces functional competence, which is not blocked by inhibitors of PKC; however, inhibition of COX blocks functional competence induced by PdBu, suggesting that the phospholipase A2 (PLA2)-AA-COX pathway is downstream from PKC. As indicated by the dashed lines, the COX-generated AA metabolite that promotes functional competence is not known. Inhibitors of the RGD-binding class of integrins also block induction of functional competence by BK, suggesting that RGD-binding integrins are necessary for production of functional competence by BK. It is not known whether integrins are an intermediary in BK signaling or act in parallel with BK to promote functional competence of opioid receptors systems. The effect of BK on the opioid receptor signaling system seems to be at the receptor-G protein level because BK pretreatment enhances opioid agonist-stimulated GTP[γ35S] binding.
The requirement of a stimulus, such as BK, to enable opioid receptor inhibition of adenylyl cyclase seems to be somewhat specific to peripheral opioid receptors and the adenylyl cyclase signaling pathway. Central opioid receptors do not require a pretreatment stimulus to be functional to inhibit adenylyl cyclase or to promote analgesia (Waldhoer et al., 2004; Corbett et al., 2006). Moreover, BK pretreatment was not required for, and did not modify, inhibition of adenylyl cyclase activity produced by other PTx-sensitive Gi-coupled receptor agonists, such as 5-CT and L,P-NPY, which act on 5-HT1 and NPY1 receptors, respectively. This suggests that the target of the BK effect may be at the opioid receptors themselves, leading to increases in receptor-G protein coupling efficiency (Berg et al., 2007a). BK pretreatment was also not required for, and did not modify, the stimulation of the ERK pathway by U50488. However, BK pretreatment was necessary for U50488 to inhibit thermal allodynia, suggesting that the adenylyl cyclase signaling pathway and not the ERK pathway plays a role in mediating the antiallodynic effect of U50488. It is noteworthy that ERK activation in dorsal root ganglion primary afferents that express transient receptor potential channel V1 occurs rapidly (2–5 min) in response to noxious heat stimulation and capsaicin injection into the skin (Dai et al., 2002). This suggests that stimulation of ERK activity in primary sensory neurons is associated with increased pain transmission. KOR ligands with differential activity toward kinase signaling pathways have been identified (Bruchas and Chavkin, 2010), and it is interesting to speculate that KOR ligands that display functional selectivity toward inhibition of adenylyl cyclase activity versus activation of ERK may have a higher degree of analgesic efficacy.
The duration of the antiallodynic effect of U50488 was inversely related to its dose. Analgesic efficacy of U50488 was maintained for at least 15 min when administered at a dose of 0.1 μg (intraplantar). However, at a dose of 1 μg the antiallodynic effect lasted for less than 15 min, and for the dose of 10 μg the duration of the effect was less than 10 min. Likewise, the area under the time-response curve was significantly less for the 10-μg dose than for the 0.1-μg dose of U50488. These results suggest that in addition to its antiallodynic effects U50488 may initiate pronociceptive mechanisms that compete temporally with antinociceptive mechanisms. It is interesting that this profile required BK pretreatment because U50488 did not alter paw withdrawal latency in the absence of BK pretreatment. Many studies have shown that acute administration of opioids can enhance pain sensation (“paradoxical pain”) via a variety of mechanisms (for review see Ossipov et al., 2004), and MOR, DOR, and KOR agonists have been shown to prolong action potentials of dorsal root ganglion neurons maintained in long-term organotypic explants (Crain and Shen, 1990). The reduced duration of the antiallodynic action of U50488 could also result from rapid desensitization of U50488-mediated KOR signaling. Opioid receptor systems, such as many seven-transmembrane-spanning receptor systems, can undergo rapid, agonist-induced desensitization that can involve receptor phosphorylation, internalization, and down-regulation as well as postreceptor effects (Marie et al., 2006; Kelly et al., 2008). MOR signaling to voltage-gated calcium channels in dorsal root ganglion neurons desensitizes rapidly in response to agonist pretreatment of cultures (Nomura et al., 1994; Samoriski and Gross, 2000; Tan et al., 2003; König et al., 2010); however, prolonged (4 day) morphine pretreatment of rats did not reduce morphine-induced receptor-G protein coupling nor the inhibition of adenylyl cyclase activity in dorsal root ganglia innervating inflamed hindpaw (Zöllner et al., 2008). It was found that the lack of MOR system desensitization required the availability of endogenous opioids in the inflamed tissue, which may explain differences when agonist treatment is applied to cells in culture versus the intact animal. Additional experiments are required to determine the mechanism underlying the shortened duration of the antiallodynic effect of high doses of U50488.
Many studies have demonstrated analgesic effects mediated by opioid receptors located in the periphery after inflammation or tissue damage (see Joris et al., 1987; Przewlocki and Przewlocka, 2001; Stein and Zöllner, 2009), although peripheral efficacy of opioids in humans is variable and weak (Gupta et al., 2001; Kalso et al., 2002). In addition, there is a prominent role for endogenous opioids released from circulating leukocytes of the immune system that migrate into sites of inflammation in regulating pain sensitivity (Stein and Zöllner, 2009; Busch-Dienstfertig and Stein, 2010). As a result, there has been strong interest in developing new pharmacological approaches to treating pain that involve either direct activation of opioid receptors on sensory neurons or indirect opioid receptor activation by stimulating release of endogenous opioids. Efforts to directly activate opioid receptors focus on developing drugs with low permeability to penetrate into the CNS via the blood-brain barrier (DeHaven-Hudkins and Dolle, 2004). By limiting access to the CNS, adverse effects mediated by CNS opioid receptors would be expected to be reduced or eliminated. Although drugs that target MOR are currently the gold standard for the treatment of pain, activation of peripheral MOR leads to adverse effects such as constipation and itch (Manara et al., 1986; Yamamoto and Sugimoto, 2010). In various animal models and clinical studies, there is evidence that peripherally restricted DOR and KOR agonists can be effective in reducing inflammatory pain (for reviews see Rivière, 2004; Stein and Zöllner, 2009; Kivell and Prisinzano, 2010). It is expected that the adverse effect profile of DOR or KOR peripherally restricted ligands would be less than that of MOR; consequently, approaches that target peripheral DOR or KOR may provide for a better therapeutic index.
In summary, we found that regulation of KOR-mediated inhibition of adenylyl cyclase activity and neuropeptide release from primary sensory TG neurons by U50488 and KOR-mediated antiallodynia in vivo by U50488 is similar to regulation of MOR and DOR. However, BK pretreatment was not required for U50488-mediated stimulation of ERK activity nor for 5-HT1B/1D or NPY1 receptor agonists to inhibit adenylyl cyclase activity. Further understanding of the mechanisms by which inflammation and/or tissue damage regulates the efficacy of opioid drugs along with the roles and regulation of each of the multiple signaling pathways coupled to peripheral opioid receptors may lead to improved pharmacotherapy for treatment of pain with reduced adverse effects.
Authorship Contributions
Participated in research design: Berg, Patwardhan, Milam, and Clarke.
Conducted experiments: Berg, Rowan, Sanchez, Silva, and Patwardhan.
Performed data analysis: Berg, Rowan, Patwardhan, and Clarke.
Wrote or contributed to the writing of the manuscript: Berg, Hargreaves, and Clarke.
Other: Berg and Clarke acquired funding for the research.
Footnotes
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grants DA024865 (to W.P.C.), DA026619 (to K.A.B.)].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.110.177493.
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- CNS
- central nervous system
- AA
- arachidonic acid
- BK
- bradykinin
- CGRP
- calcitonin gene-related peptide
- iCGRP
- immunoreactive CGRP
- COX
- cyclooxygenase
- MOR
- μ-opioid receptor
- KOR
- κ-opioid receptor
- DOR
- ∂-opioid receptor
- PGE2
- prostaglandin E2
- 5′-GNTI
- 5′-guanidinonaltrindole
- nor-BNI
- nor-binaltorphimine
- PLC
- phospholipase C
- PKC
- protein kinase C
- RIA
- radioimmunoassay
- TG
- trigeminal ganglion
- PWL
- paw withdrawal latency
- ANOVA
- analysis of variance
- U50488
- trans-(1S,2S)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzeneacetamide hydrochloride hydrate
- 5-HT
- 5-hydroxytryptamine
- ERK
- extracellular signal-regulated kinase
- pERK
- phospho-ERK
- Veh
- vehicle
- PdBu
- phorbol dibutyrate
- Bis
- bisindolylmaleimide
- HBSS
- Hanks' balanced salt solution
- NPY
- neuropeptide Y
- 5-CT
- 5-carboxamidotryptamine
- PTx
- pertussis toxin
- HOE 140
- d-Arg-l-Arg-l-Pro-l-Hyp-Gly-l-(2-thienyl)Ala-l-Ser-d-1,2,3,4-tetrahydro-3-isoquinolinecarbonyl-l-(2α,3β,7aβ)-octahydro-1H-indole-2-carbonyl-l-Arg
- Go 6976
- 5,6,7,13-tetrahydro-13-methyl-5-oxo-12H-indolo[2,3-a]pyrrolo[3,4-c]carbazole-12-propanenitrile
- Indo
- indomethacin
- DAG
- diacylglycerol.

- Received November 22, 2010.
- Accepted April 5, 2011.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}