


Abstract
We have shown that high epithelial cell density is a major barrier to the distribution of protein-bound drugs in solid tumors, and tumor priming (expansion of interstitial space using an apoptosis-inducing pretreatment) can promote drug delivery. This study evaluated the optimal conditions of paclitaxel tumor priming (time window, particle size) and its effects on the delivery and efficacy of nanomedicines. Paclitaxel tumor priming was applied to mice bearing human xenograft tumors. The kinetics of paclitaxel-induced apoptosis was evaluated to identify the time window of tumor priming. The effects of tumor priming on the tumor delivery and interstitial dispersion of fluorescence-labeled nanoparticles of various sizes, the perfusion of tumor and normal tissues, the delivery of doxorubicin HCl liposomes to tumor and host tissues, and the antitumor activity and host toxicity were studied. Tumor priming by a single i.v. injection of paclitaxel induced apoptosis, expanded the interstitial space, vessel diameter and blood-perfused area, and promoted the delivery and interstitial dispersion of nanoparticles (100- and 200-nm diameter, administered 48 h after paclitaxel) in a tumor-selective manner. Tumor priming also enhanced the tumor delivery and antitumor activity of doxorubicin HCl liposomes (85 nm) without affecting the delivery to noncancerous host tissues or enhancing host toxicity. Tumor priming represents a potentially useful means to promote tumor-selective delivery and efficacy of nanomedicines. The current study will have significant impact on enhancing delivery and efficacy of nanomedicines and dosing regimen optimization of combination chemotherapy in the clinical setting.
Inadequate delivery into solid tumors is a well recognized problem for a wide variety of agents, including small molecules with extensive protein-binding, macromolecules such as monoclonal antibodies, cytokines, sense and antisense oligonucleotides, and particulates such as viral and nonviral gene vectors and liposomes (Jang et al., 2003). Delivery of a systemically administered agent to cells in solid tumors involves three processes: distribution through the vascular compartment, transport across the microvascular wall or extravasation, and dispersion within the tumor interstitium (Jang et al., 2003). Research directed at the barriers for these processes has led to several experimental approaches, including administration of agents that produce vasoconstriction (angiotensin II) or normalize vessels (inhibitor of vascular endothelial growth factor receptor), enzymes that degrade tumor matrix materials (collagenase, hyaluronidase), or hyperthermia (Suzuki et al., 1981; Kong et al., 2000; Eikenes et al., 2004; Tong et al., 2004; Chang et al., 2005; Eikenes et al., 2005). These pharmacological and physical interventions enhance the tumor blood flow and/or gradient between microvascular pressure (MVP) and interstitial fluid pressure (IFP), thereby enhancing the extravasation and intratumoral dispersion of small molecules, proteins, and liposomes. Among these approaches, only angiotensin II has been evaluated in patients, starting in the late 1970s and exclusively in Japan; this agent selectively increased tumor blood flow and improved the activity of chemotherapy in gastric carcinoma (Suzuki et al., 1981; Hoshi and Sato, 1995; Kong et al., 2000; Jang et al., 2003; Eikenes et al., 2004, 2005; Tong et al., 2004; Chang et al., 2005).
We have shown high tumor cell density is a major barrier for protein-bound drugs to penetrate into the inner layers of solid tumors. We further showed that pretreatment with a drug efficient in inducing apoptosis (paclitaxel or doxorubicin) reduces the tumor cell density and expands the interstitial space and thereby promotes the penetration of protein-bound drugs into three-dimensional tumor histocultures in vitro and in vivo (Jang et al., 2001, 2003). The present study evaluated the potential utility of tumor priming in the delivery and efficacy of nanomedicines, using the same tumor model as in the earlier studies, i.e., s.c. human pharynx FaDu xenograft solid tumors. The tumor priming treatment was a single i.v. injection of paclitaxel (40 mg/kg) that is sufficient to induce ≥10% apoptosis in tumors (Jang et al., 2001). The first objective was to establish the optimal tumor priming conditions (time window, particle size), the effects of tumor priming on tumor microvasculature and spatial distribution of nanoparticles, and the tissue selectivity of tumor priming. This was accomplished using latex beads loaded with a non-leaching fluorescent dye (100∼500-nm diameter). The second objective was to evaluate the effects of tumor priming on the delivery and efficacy of nanomedicines. Previous studies with colloidal gold-labeled or fluorescent liposomes show that after extravasation from tumor microvessels through interendothelial cell junctions, liposomes are localized in perivascular space with minimal penetration into tumor matrix (Yuan et al., 1994); the latter is considered a major reason of the lower efficacy in larger tumors (Harrington et al., 2001). Hence, we used doxorubicin-loaded liposomes (doxorubicin HCl liposome injection, Doxil; ∼85-nm diameter) in the present proof-of-concept study (Gabizon et al., 2003).
Materials and Methods
Chemicals and Reagents. Paclitaxel was purchased from Handetech (Houston, TX), and polyoxyethylated castor oil (Cremophor EL), was purchased from Sigma-Aldrich (St. Louis, MO). Paclitaxel was solubilized in polyoxyethylated castor oil:ethanol (1:1) at a concentration of 12 mg/ml and diluted with physiological saline to 4 mg/ml immediately before injection. Fluorescence-labeled latex beads (100-, 200-, and 500-nm diameter, carboxylate-modified, excitation and emission wavelengths of 580 and 605 nm) and 3,3′-diheptyloxacarbocyanine iodide were purchased from Invitrogen (Carlsbad, CA). Doxorubicin HCl liposomes are composed of cholesterol, fully hydrogenated soy phosphatidylcholine, and N-carbamoylmethoxy-polyethylene glycol 2000–1,2-distearoyl-sn-glycerol-3-phosphoethanolamine sodium salt, in a molar percentage ratio of 38:56:5, and was from Alza (Palo Alto, CA). Doxorubicin hydrochloride and epirubicin hydrochloride were gifts from Pfizer (Groton, CT) and the National Cancer Institute (Bethesda, MD), respectively. The O.C.T. embedding matrix was from Miles Inc. (Elkhard, IN). Cell culture supplies were from commercial vendors including Hoechst-Roussel Inc. (Somerville, NJ), Solo Pak Laboratories (Franklin Park, IL), and Invitrogen, as described previously (Jang et al., 2001).
Animal Protocols. Male nu/nu athymic mice (18–21 g; National Cancer Institute) were cared for per institutional guidelines and had free access to food and water. FaDu cells (American Type Culture Collection, Manassas, VA) were maintained in minimum essential medium supplemented with 9% heat-inactivated fetal bovine serum, 2 mM l-glutamine, 0.1 mM nonessential amino acids, 90 μg/ml gentamicin, and 90 μg/ml cefotaxime. Cells with greater than 90% viability, as determined by trypan blue exclusion, were harvested from subconfluent cultures using trypsin. Tumor cells (2 × 106 cells in 0.15 ml of culture medium) were implanted s.c. to the right side of the upper back of each mouse. Experiments were initiated when tumors reached approximately 10 mm in diameter, in ∼2 weeks. Mice were randomized such that the mean tumor sizes (measured as 0.5 × width2 × length) in different groups were about equal (∼600 mm3). Treatments were injected i.v. through a tail vein. The tumor priming treatment was a single i.v. injection of paclitaxel (40 mg/kg).
For the study on the kinetics of tumor priming, a mouse was treated with paclitaxel, and, at predetermined time points, tumors were excised, fixed in 10% formalin, and embedded in paraffin. Histological sections (7 μm thick) were prepared, and the number of apoptotic cells and viable cells in non-necrotic regions were counted under a microscope at 400× magnification, as described previously (Gan et al., 1996, 1998). Five tumors were used per time point. For each tumor, we evaluated at least five randomly selected regions and counted at least 3000 cells per tumor. Based on our previous finding that apoptosis and/or reduction of tumor cell density promotes the delivery of macromolecules, the peak times for these two parameters were selected as the optimal time window of tumor priming for the subsequent studies.
For the study on the effects of tumor priming on nanoparticle delivery to tumors, each mouse was given an i.v. injection of fluorescence-labeled latex beads (100-, 200-, and 500-nm diameter) 48 h after the paclitaxel tumor priming treatment (or vehicle for the control group). Another 24 h later, tumors were harvested, rinsed, blot-dried, and flash-frozen in liquid nitrogen. These procedures were completed in less than 5 min. Frozen tumors were mounted on cryostat chucks using O.C.T. embedding matrix and cut into 10-μm-thick sections at –30°C using a cryotome. Sections obtained at multiple depths were thaw-mounted onto glass slides, and fluorescence microscopic photographs were captured for computer-assisted image analysis. Eight tumors per group were studied. We analyzed at least three randomly selected sections per tumor and 20 to 25 images (100× magnification) covering at least 90% of the tumor cross-section (excluding the necrotic regions and tumor tissues immediately adjacent to normal tissues).
To evaluate the effects of tumor priming on tumor perfusion and spatial distribution of nanoparticles, a mouse was given i.v. injections of paclitaxel (or vehicle for the control group) and latex beads (100-nm diameter) at 48 h after paclitaxel, followed by an i.v. injection of a perfusion marker, 3,3′-diheptyloxacarbocyanine iodide (1 mg/kg in 75:25 dimethyl sulfoxide:phosphate-buffered saline), at 24 h after the beads. The perfusion marker is a green fluorescent marker that stains the endothelial cells and outlines patent blood vessels (Trotter et al., 1989). The mouse was euthanized 2 min after the injection of the perfusion marker, and tumors were excised and processed as described above. The fields showing the highest signals (hot spot method; Vermeulen et al., 1996) were selected for analysis of the density, length, and diameter of the patent, perfused vessels, and for analysis of the fraction of perfused area. The threshold fluorescence intensity (expressed in gray value, range 0∼255 units) was determined using tumors from animals that did not receive the perfusion marker. We analyzed four tumors per group (at least three sections per tumor, five images per section).
To evaluate the tumor selectivity of the paclitaxel priming treatment on apoptosis induction and perfusion, normal tissues including liver, kidney, spleen, and heart were excised and processed as described for tumors. To evaluate the effects of tumor priming on the tumor delivery and efficacy of doxorubicin HCl liposomes, animals were divided into the following groups. The delivery study used three groups, i.e., tumor priming group that received the paclitaxel tumor priming treatment followed by doxorubicin HCl liposomes (20 mg/kg) administered 48 h later (referred to as paclitaxel → doxorubicin HCl liposomes) and two control groups without the benefits of tumor priming (single-agent doxorubicin HCl liposomes, paclitaxel plus doxorubicin HCl liposome combination in the reversed sequence, or doxorubicin HCl liposomes → paclitaxel). The efficacy study used two additional control groups, i.e., vehicle (polyoxyethylated castor oil-ethanol and saline) and single-agent paclitaxel.
Computer-Assisted Image Analysis. The accumulation and dispersion of fluorescent latex beads in a tumor were quantified using Optimas image analysis software (Media Cybernetics, Inc., Silver Spring, MD). The threshold fluorescence intensity was determined using tumors from animals that did not receive the beads. The locations of the fluorescent beads were indicated by pixels with intensity exceeding the threshold value. The percentage of area in tumors occupied by beads was calculated as (total number of bead-derived pixels) divided by (total number of pixels per microscopic field), which provided a measurement of the dispersion area. The average bead concentration in tumors was the average fluorescence intensity, which was calculated as (mean intensity of bead-derived pixels multiplied by total number of bead-derived pixels) divided by (total number of pixels per microscopic field).
Tissue Distribution of Doxil-Derived Doxorubicin. A mouse was given an i.v. injection of doxorubicin HCl liposomes. At predetermined time points, animals were anesthetized, blood samples were obtained via heart puncture, and, immediately afterward, tissues were harvested, rinsed with distilled water, blot-dried, and flash-frozen in liquid nitrogen. These procedures were completed in less than 10 min. We studied tumors and several noncancerous tissues (liver, spleen, kidney, heart). Three to five mice were used per data point. Biological samples were extracted using the previously published high-performance liquid chromatographic method (Chin et al., 2002) with the following modifications. Epirubicin hydrochloride (12.5 μg/100 μl) was used as the internal standard. Plasma or thawed tissues (prehomogenized in normal saline) were mixed with Triton X-100 (3% in phosphate-buffered saline, pH 7.4, which disrupted the lipid bilayer and released the encapsulated drug from liposomes) and zinc sulfate (70% in distilled water, which facilitated the extraction of doxorubicin from the biological matrix), and extracted twice with acetone. The acetone extracts were blown to dry, reconstituted, and analyzed by high-performance liquid chromatography. The high-performance liquid chromatography elution times were 5.5 min for doxorubicin and 7 min for epirubicin. The lower detection limit was 1 ng/ml.
Effects of Tumor Priming on Therapeutic Efficacy. Tumor size, body weight, and overall survival time were monitored. The population mean values at each time point were calculated using the measurements for animals that were alive and the values at time of death for animals that had died from treatment toxicity or became moribund (defined as having tumors of larger than 15-mm diameter). Six to nine mice were used per group.
Antitumor activity was evaluated using the same criteria as in human patients, i.e., Response Evaluation Criteria in Solid Tumors (RECIST) (Tsuchida and Therasse, 2001). Partial response refers to at least 50% reduction of tumor volume, maintained for at least two consecutive measurements. Increase in tumor volume by >50% from the lowest measured value is considered progressive disease. Stable disease indicates neither sufficient increase to qualify for progressive disease nor sufficient shrinkage to qualify for response. The onset of response was calculated as the date of the first measurement when response was attained, and the duration of response was from the onset of response to the first date the response was lost. The onset of stable disease was the date of first treatment, and the duration was from onset to the first date when progressive disease was observed.
Pharmacokinetic and Statistical Analyses. The doxorubicin concentration-time profiles were analyzed using standard noncompartmental methods with WinNonlin 4.0 software (Pharsight, Mountain View, CA). The area under the concentration-time curve for each organ was calculated based on a single set of concentration-time data that represented average values of several animals per data point and, hence, did not provide S.D.s. We used the bootstrap method to obtain the confidence intervals of the parameter values (Davison and Hinkley, 1997). In brief, the original data set was used to simulate additional concentration-time curves (1000 times for each group). The corresponding pharmacokinetic parameters for each curve were obtained and the difference in a parameter for the treatment groups (Δparameter) was calculated. A 95% confidence interval for Δparameter spanning over the value of zero indicates no significant difference and vice versa. The p value was calculated based on the percentage of Δparameter that resided on either side of zero.
Plots of frequency versus range of data showed a bell-shaped distribution, indicating normal data distribution. Two-way analysis of variance (ANOVA) and repeated measures ANOVA were used, respectively, to compare the changes of doxorubicin concentrations in tissues and the changes in tumor volume and body weight over time among treatment groups. Two-sided Student's t test was used to compare the percentage of area occupied by fluorescent beads, the average luminescence intensity of beads in tumors, the diameter, length, and density of patent vessels, and the fraction of perfused area between treatment groups. Kaplan-Meier plots were used to calculate the survival probabilities and the median survival time (defined as duration from the day of tumor cell implantation to the day of death or moribundity). Differences in the survival probabilities among treatment groups were analyzed using the Wilcoxon signed rank test. Statistical analysis was performed using SPSS (SPSS Inc., Chicago, IL), SAS software (SAS Institute Inc. Cary, NC), or R program (The Free Software Foundation, Boston, MA). A p value of less than 0.05 was considered statistically significant.
Results
Kinetics of Paclitaxel Tumor Priming. The paclitaxel tumor priming treatment increased the number of apoptotic tumor cells and decreased the tumor cell density initially. Both parameters showed maximal changes at 48 and 72 h, followed by recovery at 96 h (Fig. 1). These results identified 24 to 96 h as the boundary time window when the apoptotic index was equal or greater than 10%. In comparison, no significant apoptosis was observed in normal tissues. Subsequent tumor priming studies used the 48- to 72-h time span, i.e., the latex beads were administered after 48 h of paclitaxel treatment, and the samples were harvested 24 h later.
Tumor Priming Promoted Particulate Delivery to Tumors. Paclitaxel tumor priming significantly increased the delivered amount and the interstitial dispersion of fluorescence-labeled latex beads, administered 48 h after paclitaxel, in tumors (Fig. 2; Table 1). These benefits were observed for the smaller beads (100- and 200-nm diameter) but not the larger beads (500-nm).
Effects of tumor priming on nanoparticle delivery and dispersion in tumors
The micrographs shown in Fig. 2 were evaluated using computer-assisted image analysis for the accumulation and dispersion of the beads. The percentage of area in tumors occupied by particles was calculated as (total number of bead-derived pixels) divided by (total number of pixels per microscopic field), which provided a measurement of the dispersion area. The average bead concentration in tumors was the average fluorescence intensity, which was calculated as (mean intensity of bead-derived pixels multiplied by total number of bead-derived pixels) divided by (total number of pixels per microscopic field). At least three randomly selected sections per tumor and 20 to 25 images (100× magnification) covering at least 90% of the tumor cross-section (excluding the necrotic regions and tumor tissues immediately adjacent to normal tissues) were analyzed. Eight tumors per data point. Mean ± S.D. Differences between control and tumor priming groups were analyzed using two-sided Student's t test.
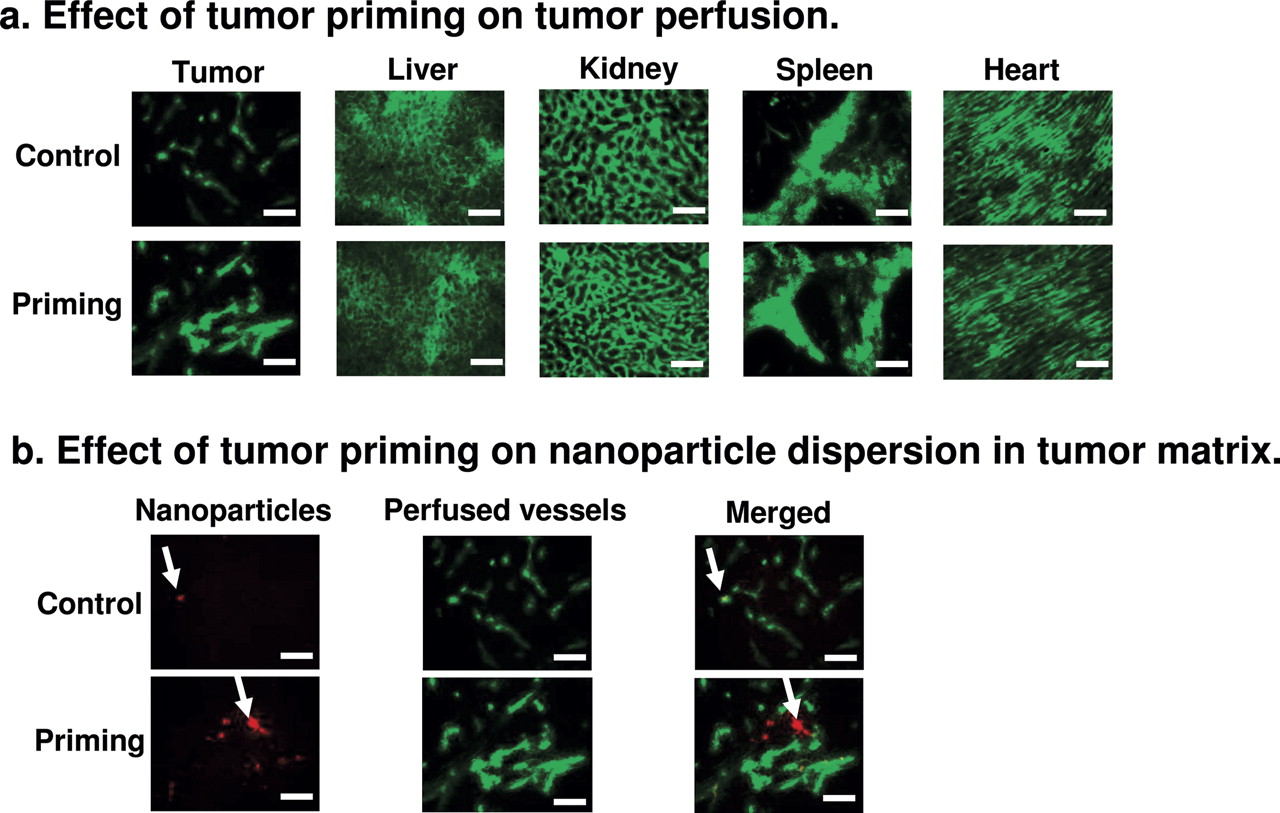
Effects of Tumor Priming on Perfusion of Tumor and Normal Tissues. Potential mechanisms for increasing nanoparticle delivery and dispersion in tumors include increase in perfusion, extravasation, and/or interstitial transport. Changes in perfusion were studied using the perfusion marker 3,3′-diheptyloxacarbocyanine iodide as described under Materials and Methods. The tumor priming treatment significantly increased the diameter of patent vessels that resulted in the trend of a larger fraction of perfused area but did not affect the vessel length or density (Fig. 3; Table 2). These vascular changes were tumor-specific and not observed in normal tissues.
Effects of tumor priming on tumor perfusion
The micrographs shown in Fig. 3 were evaluated using computer-assisted image analysis for the diameter, length, number per field, and area fraction per field of functional tumor vessels. Mean ± S.D. Differences of tumor perfusion between control and tumor priming groups were analyzed using two-sided Student's t test.
We quantified the contribution of priming-promoted perfusion to the priming-promoted nanoparticle delivery in tumor, in two ways (from data shown in Figs. 2 and 3). First, a comparison of the area fractions occupied by the beads and the blood perfusion marker showed that tumor priming increased both area fractions to about the same extent (1.7 and 1.6 times, respectively). We also calculated the ratios of (bead concentration as indicated by fluorescence intensity) to (perfused area fraction), which were nearly identical for tumor priming and control groups (0.10 versus 0.09 for 100-nm particles and 0.11 versus 0.10 for 200-nm particles). These data suggest that ∼90% of the increase in nanoparticle delivery by tumor priming was due to the increase in tumor perfusion.
We studied tumor priming-induced changes in the interstitial transport of nanoparticles in tumor matrix by comparing the spatial distribution of the red fluorescent beads (100-nm diameter, administered 48 h after paclitaxel) with the distribution of the green fluorescent perfusion marker. The marker was administered 24 h after the beads, which is the duration for the majority of an i.v. administered dose of nano-sized particles to transfer from blood to tissues (Ogawara et al., 2002). Without tumor priming, the beads colocalized with the blood-perfused areas or vessels and did not penetrate into the tumor interstitium. With tumor priming, the beads were located away from the perfused areas or vessels and dispersed in the tumor interstitium. These data indicate tumor priming promoted the interstitial transport of nanoparticles.

Kinetics of paclitaxel tumor priming. A mouse was given an i.v. injection of the tumor priming agent (40 mg/kg paclitaxel in polyoxyethylated castor oil and ethanol) or the polyoxyethylated castor oil/ethanol vehicle (control). Tumors and normal tissues were excised and evaluated morphologically. a, kinetics of paclitaxel-induced apoptosis in tumors. Changes in apoptotic index and tumor cell density with time were measured. At least five randomly selected regions and at least 3000 cells per tumor and five tumors per time point were evaluated. Arrows, examples of apoptotic cells. b, no apoptosis in normal tissues (72-h samples). H&E staining (hematoxylin staining for nuclei appeared black in the black-and-white micrograph). Bar, 50 μm. Mean + S.D. Differences between control and tumor priming groups were analyzed using two-sided Student's t test. † and ***, p < 0.001 compared with time 0 samples.

Effects of tumor priming on nanoparticle delivery and dispersion in tumors. A mouse was given an i.v. injection of either the tumor priming agent (40 mg/kg paclitaxel in polyoxyethylated castor oil and ethanol) or the polyoxyethylated castor oil/ethanol vehicle (control), followed by an injection of the red fluorescent latex beads (100-, 200-, and 500-nm diameter, appeared white in the black-and-white micrograph) 48 h later. Tumors were excised 24 h after the injection of the beads and examined. Bar, 100 μm. Note the enhanced delivery of 100- and 200-nm latex beads by tumor priming.
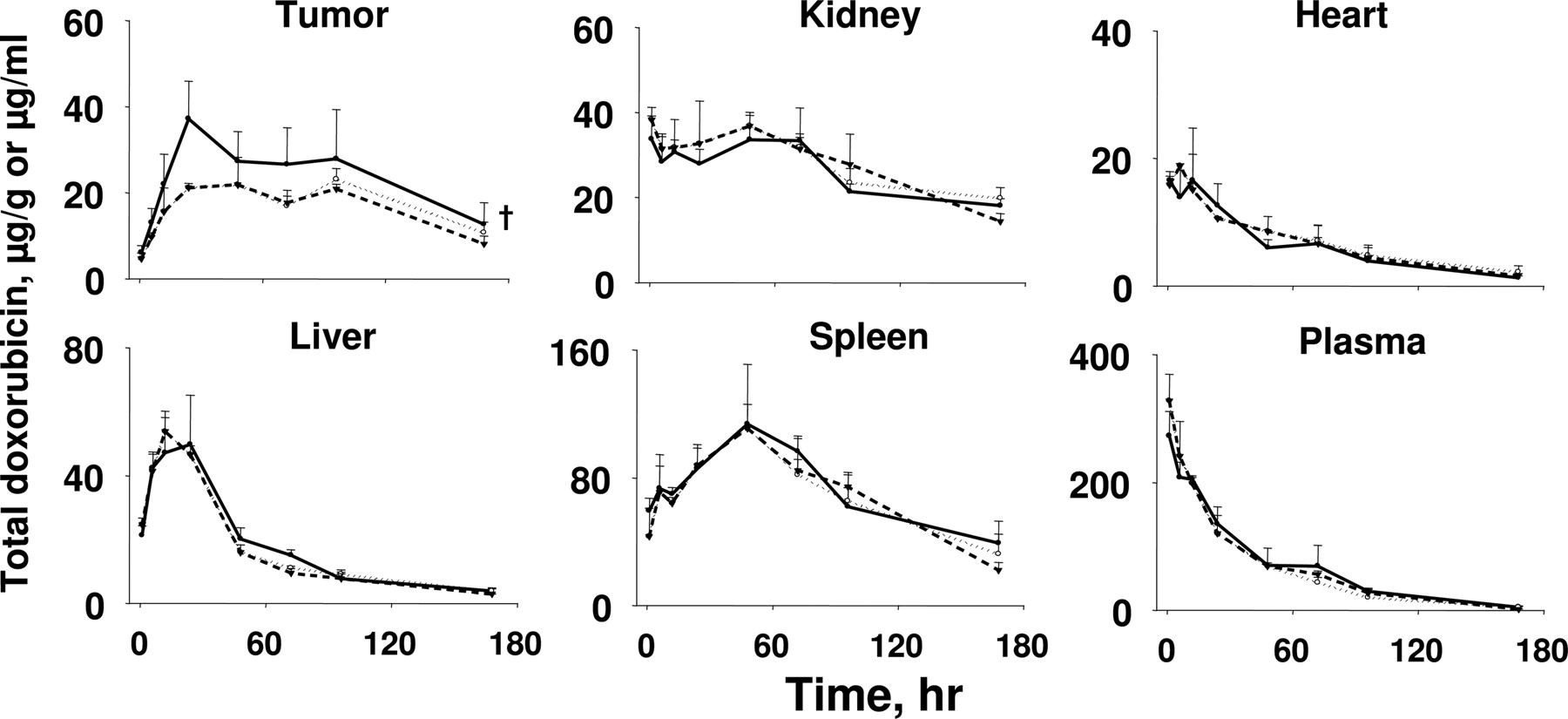
Tumor Priming Selectively Enhanced the Delivery of Drug-Loaded Liposomes to Tumors. We evaluated the delivery of doxorubicin-loaded liposomes to tumors and noncancerous tissues, including organs that are responsible for drug elimination (liver, kidney), enriched in reticuloendothelial system (spleen, liver), and target of doxorubicin toxicity (heart). Figure 4 shows the changes of concentrations of total doxorubicin (sum of free and liposome-entrapped drug) with time. Table 3 shows the areas under the concentration-time curve in tumor and normal tissues. The paclitaxel tumor priming treatment did not affect the doxorubicin delivery to normal tissues but significantly enhanced the delivery to tumors. In contrast, no enhanced tumor uptake was observed in the two control groups without the benefits of tumor priming (i.e., receiving only doxorubicin HCl liposomes without paclitaxel, receiving doxorubicin HCl liposomes before paclitaxel).
Effects of tumor priming on doxorubicin delivery to tumor and normal tissues
The tumor priming group received the PAC tumor priming treatment followed by doxorubicin HCl liposomes (20 mg/kg) administered 48 h later (PAC → doxorubicin HCl liposomes). The two control groups were doxorubicin HCl liposomes alone and paclitaxel plus doxorubicin HCl liposomes combination in the reversed sequence (i.e., doxorubicin HCl liposomes given 48 h before paclitaxel or doxorubicin HCl liposomes → PAC). AUC values, expressed in micrograms per gram per hour or micrograms per milliliter per hour, were calculated using the trapezoidal rule. Statistical analysis of differences between the tumor priming group (PAC → doxorubicin HCl liposomes) and the control group (doxorubicin HCl liposomes alone or doxorubicin HCl liposomes → PAC) was conducted using the bootstrap method with 1000 simulated data sets (see Materials and Methods).
Because more than 98% of the circulating doxorubicin is encapsulated in liposomes (Gabizon et al., 2003), the higher doxorubicin concentrations in tumors indicate that tumor priming enhanced the blood-to-tumor transfer of liposomes. A potential mechanism of the greater tumor delivery of doxorubicin HCl liposomes in the tumor priming group is a reduced doxorubicin clearance by paclitaxel (Briasoulis et al., 2004). This was ruled out because pharmacokinetic results, together with statistical analysis using the bootstrap (see Materials and Methods), indicated that tumor priming did not alter the area under the plasma concentration-time curves of total doxorubicin (sum of free and liposome-entrapped drug).
Tumor Priming Enhanced the Therapeutic Efficacy of Drug-Loaded Liposomes. We evaluated whether tumor priming promoted the efficacy of doxorubicin-loaded liposomes without enhancing the host toxicity. The tumor priming group received paclitaxel 48 h before doxorubicin HCl liposomes (paclitaxel → doxorubicin HCl liposome). The four control groups, all without the benefits of tumor priming, were vehicles without paclitaxel or doxorubicin HCl liposomes, paclitaxel alone, doxorubicin HCl liposome alone, and doxorubicin HCl liposomes → paclitaxel. Comparison of the tumor priming group with the last control group receiving the same two-drug combination but in the opposite order (which would fail the time window requirement of tumor priming) enabled the delineation of the effects of tumor priming from other pharmacological interactions among the two agents.
Tumor growth curves and the overall survival of tumor-bearing mice are shown in Fig. 5, a and b, and Table 4. The vehicle control group showed the fastest tumor growth and the shortest overall survival time. The four drug-treated groups showed either slower tumor growth or tumor regression, and all yielded longer survival. The tumor priming group showed the best therapeutic efficacy, as indicated by the earliest onset, longest duration and greatest extent of tumor regression, and the longest survival. The differences between the tumor priming group and the four control groups were statistically significant. The inferior activity of the doxorubicin HCl liposomes → paclitaxel group compared with the tumor priming paclitaxel → doxorubicin HCl liposomes group confirmed the important role of tumor priming in promoting the therapeutic efficacy of drug-loaded nanoparticles.
Tumor priming enhanced antitumor efficacy of doxorubicin HCl liposomes without enhancing toxicity
Partial response means >50% tumor size reduction. Progressive disease means >50% tumor size increase. Stable disease means <50% tumor size reduction or <50% tumor size increase. Mean ± S.D. NA, not applicable.

Effects of tumor priming on tumor perfusion and dispersion of nanoparticles in tumor matrix. A mouse was given an i.v. injection of the tumor priming agent (40 mg/kg paclitaxel in polyoxyethylated castor oil and ethanol) or the polyoxyethylated castor oil/ethanol vehicle (control), followed by an injection of red fluorescent latex beads (100-nm diameter) at 48 h and an injection of the green fluorescent perfusion marker 3,3′-diheptyloxacarbocyanine iodide at 72 h. Two minutes after the injection of the perfusion marker, tumor and normal tissues were excised and evaluated using computer-assisted image analysis. At least five images per section and at least three sections per tumor were analyzed. Four tumors per data point. Bar, 100 μm. a, effect of tumor priming on tumor perfusion. b, effect of tumor priming on nanoparticle dispersion in tumor matrix. Nanoparticles (red fluorescence), vessels (green fluorescence), nanoparticles merged with vessels (yellow). Arrows, location of nanoparticles. Note the colocalization of nanoparticles with vessels in the control group and the greater dispersion of nanoparticles away from vessels in the tumor priming group.
The gross toxicity of treatments was monitored as body weight loss (Fig. 5c). During the first 10 to 20 days, the vehicle control group showed weight gain, the single-agent paclitaxel group showed no changes, and the single-agent doxorubicin HCl liposome group showed substantial weight loss. Among the two groups that received combinations of paclitaxel and doxorubicin HCl liposomes in opposite sequences, the group that received doxorubicin HCl liposomes → paclitaxel (no tumor priming) showed significant greater body weight loss compared with single-agent doxorubicin HCl liposome and showed 44% toxicity-related deaths within 1 week. In comparison, the tumor priming group showed a trend of less severe body weight loss and a more rapid and more complete recovery and did not cause toxicity-related death. The greater toxicity of the doxorubicin HCl liposome → paclitaxel combination is probably a result of the aggregate toxicity of the two drugs due to the high circulating doxorubicin concentration at 48h(∼70 μg/ml in plasma, Fig. 3) when paclitaxel was administered. Aggregate toxicity is not likely for the reversed sequence because paclitaxel has a much shorter elimination half-life compared with doxorubicin HCl liposome (1–2 versus 20–35 h) (Gabizon et al., 2003; Yeh et al., 2005), and over 99% of the paclitaxel dose would have been eliminated in 48 h when doxorubicin HCl liposome was administered. These data indicate tumor priming did not enhance the toxicity of doxorubicin HCl liposome.
Discussion
The present study established the optimal in vivo conditions for paclitaxel tumor priming in animals; the boundary time window for maximal apoptosis and enhancing the delivery of nanoparticles to tumors was 48 to 72 h postpaclitaxel treatment, and the upper limit of the nanoparticle diameter was between 200 and 500 nm. This time window may explain the observation that the ability of paclitaxel pretreatment (50 mg/kg) to promote the uptake of a cytokine huKS-IL2 in murine colon carcinoma was dependent on the time interval; enhancement was observed when paclitaxel was administered 24 h but not 1 h before the cytokine (Holden et al., 2001). The results also established the tumor selectivity of paclitaxel tumor priming, in agreement with the concept that tumor cells are more prone to paclitaxel-induced apoptosis compared with normal tissues (Rowinsky et al., 1993). The study further provided the proof-of-concept that paclitaxel tumor priming promoted the delivery and efficacy of nanomedicines.
Greater total nanoparticle delivery to tumors can be due to increasing the transport through the vasculature (e.g., improved perfusion) and/or increase in extravasation (Jang et al., 2003). Our results showed that paclitaxel tumor priming reduced the tumor cell density, expanded the microvessel diameter, and promoted tumor perfusion; the latter accounted for ∼90% of the increase in total delivery. The effects on extravasation are less clear. Although increasing the gradient between MVP and IFP can promote the extravasation of albumin, antibodies, and liposomes (Eikenes et al., 2004, 2005; Tong et al., 2004), this is not a likely mechanism because paclitaxel simultaneously reduces MVP and IFP and does not alter the pressure gradient across a vessel (Griffon-Etienne et al., 1999). In addition, our original observation that tumor priming promoted the penetration of protein-bound drugs in tumor fragments was made under in vitro conditions (Jang et al., 2001), which rules out enhancing extravasation as a requirement for the priming-promoted delivery. Whether paclitaxel increases the vessel pore size, as observed for hyperthermia on liposome extravasation (Kong et al., 2000), deserves further investigations.

Effects of tumor priming on delivery of doxorubicin-loaded liposomes to tumor and normal tissues. The tumor priming group received the paclitaxel (PAC) tumor priming treatment followed by doxorubicin HCl liposome (20 mg/kg) administered 48 h later (PAC → doxorubicin HCl liposomes, •, solid line). The two control groups were doxorubicin HCl liposomes alone (▾, dashed line) (a) and paclitaxel plus doxorubicin HCl liposome combination in the reversed sequence (i.e., doxorubicin HCl liposomes given 48 h before paclitaxel or doxorubicin HCl liposomes → PAC, ○, dotted line) (b). Mean + S.D. of three to five mice per data point. The graphs show the changes in total doxorubicin concentrations (sum of free and liposome-entrapped drug) with time. The units are micrograms per gram for tissues and micrograms per milliliter for plasma. Note the different scales for different tissues. Two-way ANOVA was used to compare doxorubicin concentration-time curves in tissues among treatment groups. † and *, p < 0.05.
Tumor priming promoted the interstitial transport of nanoparticles, probably due to the perturbation of tumor structure, i.e., a reduction in tumor cell density leading to greater porosity and lower tortuosity. A recent study demonstrated that tumor structure instead of IFP is the major barrier for penetration of antibodies into solid tumors (Flessner et al., 2005).
Results of the present study indicate paclitaxel tumor priming satisfied several requirements for improving the delivery and efficacy of nanomedicines: increasing the amount delivered to tumors through increasing tumor perfusion and/or extravasation, promoting the interstitial transport to individual tumor cells, promoting the delivery in a tumor-selective manner, enhancing the activity of nanomedicines without enhancing the toxicity, and can be readily applied in clinical settings. In contrast, none of the previously attempted pharmacological and physical interventions has satisfied all five requirements. With respect to promoting the extravasation and delivery of nanoparticles, angiotensin II improved the uptake of small- and moderate-sized molecules (up to ∼12 kDa) but not larger immunotoxins (210 kDa) (Suzuki et al., 1981; Abe et al., 1988; Elizondo and Sung, 1996). Enzyme treatments (collagenase, hyaluronidase) enhanced the tumor center/periphery concentration ratios of antibodies and liposomes (up to 85-nm diameter) (Eikenes et al., 2004, 2005). Applying hyperthermia locally to s.c. tumors improved the extravasation and delivery of i.v. administered liposomes and gene vectors (up to 400-nm diameter) (Kong et al., 2000; Chang et al., 2005). Normalization of tumor vasculature by an inhibitor of vascular endothelial growth factor receptor II (DC101) promoted the extravasation of albumin (67 kDa, ∼7-nm diameter) (Tong et al., 2004). With respect to interstitial transport, angiotensin II did not promote the dispersion of immunotoxin, whereas DC101 promoted the dispersion of albumin (Elizondo and Sung, 1996; Tong et al., 2004). The effects of enzymes and hyperthermia have not been studied. With respect to selectivity, only angiotensin II could promote the delivery of a protein (∼12 kDa) in a tumor-selective manner (Abe et al., 1988). With respect to efficacy in preclinical models, only angiotensin II could improve the antitumor activity of a small molecule (mitomycin C, 334 Da) (Suzuki et al., 1981). With respect to clinical utility, only angiotensin II has been evaluated in patients (Hoshi and Sato, 1995), but this agent is not widely used even though clinical evaluation was initiated nearly 30 years ago, probably because it induces systemic hypertension that requires careful monitoring (Hoshi and Sato, 1995). Because collagenase and hyaluronidase may promote metastasis, and hyperthermia cannot be readily applied to deep-seated tumors and undetectable micrometastases (Brinckerhoff et al., 2000; Kong et al., 2000; Chang et al., 2005; Kovar et al., 2006), these approaches have limited utility. The potential utility of DC101 is unclear because there are no preclinical data to indicate whether it promotes tumor-selective delivery or efficacy of nanomedicines.
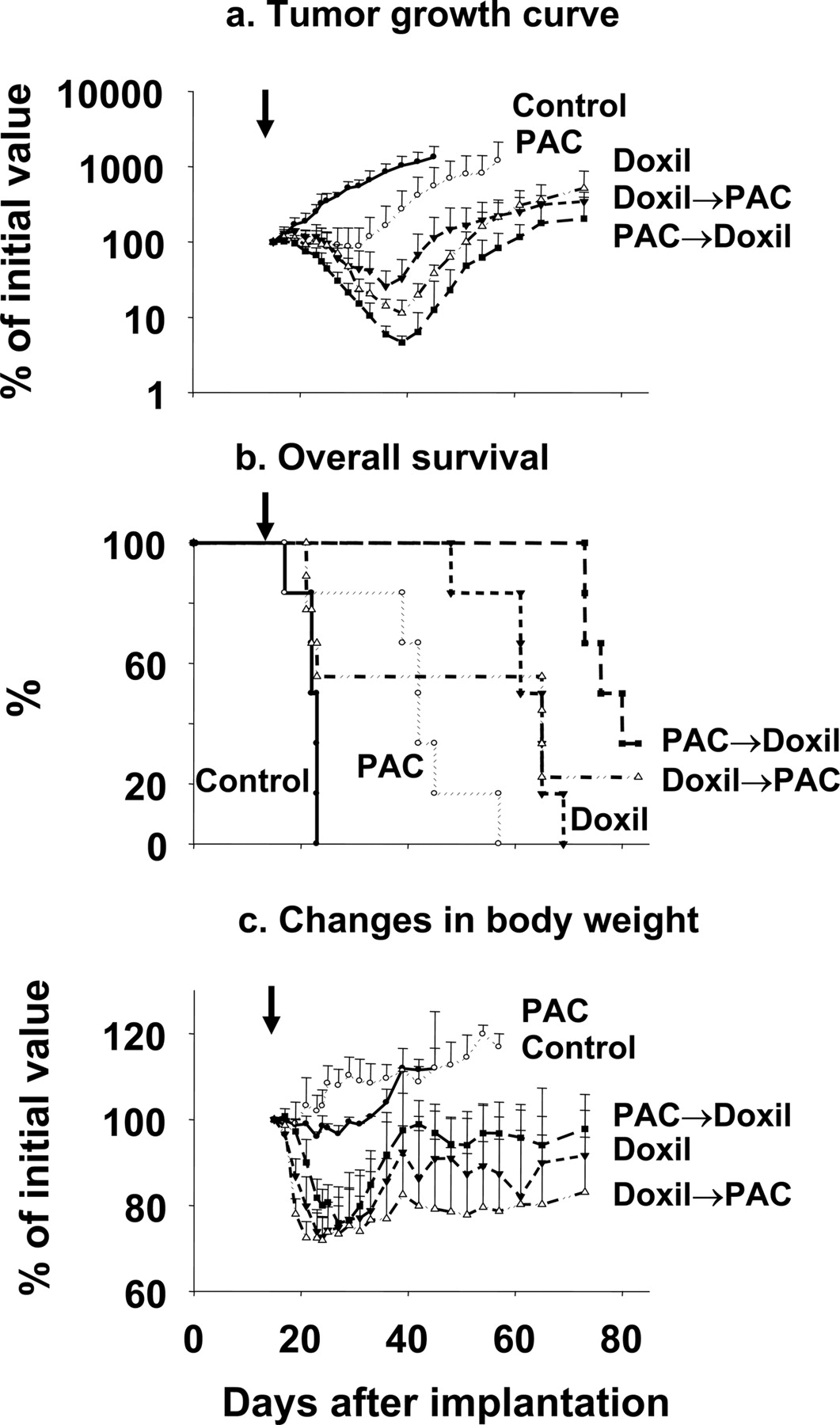
Tumor priming improved the efficacy of doxorubicin-loaded liposomes. The tumor priming group received the PAC tumor priming treatment followed by doxorubicin HCl liposomes (20 mg/kg) administered 48 h later (PAC → doxorubicin HCl liposomes, ▪). a, four control groups were blank vehicle (•), paclitaxel alone (PAC, ○), doxorubicin HCl liposomes alone (▾), and doxorubicin HCl liposome → PAC (▴). Mean + S.D. (n = 6–9 per group). a, tumor growth curve. b, overall survival. Kaplan-Meier plot. c, changes in body weight. Arrows, initiation of treatment.
The therapeutic utility of tumor priming for nanomedicine delivery depends on successful translation of the current findings to humans. The two requirements of effective paclitaxel tumor priming are its dose and the time window, i.e., sufficient to produce and maintain ∼10% apoptosis. The tumor priming dose of paclitaxel in mice, calculated using a body surface area of 0.008 m2 for a 20-g mouse (Davies and Morris, 1993), is approximately 100 mg/m2 in humans, which is lower than the standard dose of 135 to 225 mg/m2 used in patients (Rowinsky et al., 1993) and therefore clinically achievable. With respect to the time window, the major determinant is the kinetics of apoptosis. Paclitaxel induced apoptosis at similar rates in murine and human tumors under in vitro and in vivo conditions. For example, a single dose of paclitaxel (40–60 mg/kg) induced appreciable level of apoptosis beginning at 9 to 12 h and ending at 96 to 120 h, with the peak levels reached between 18 and 72 h in transplantable murine carcinoma (mammary, ovarian) and human xenograft tumors in mice (soft tissue sarcoma, pharynx carcinoma) and in histocultures of tumors obtained from cancer patients (ovarian, head and neck, breast, prostate, and bladder) (Milas et al., 1995; Gan et al., 1996, 1998; Au et al., 1997; Chen et al., 1998; Millenbaugh et al., 1998; Griffon-Etienne et al., 1999). The slow onset and protracted appearance of paclitaxel-induced apoptosis is due to the slow manifestation of apoptosis and the significant intracellular drug retention (Au et al., 1998; Jang et al., 2001). Based on the similarity in the kinetics of paclitaxel-induced apoptosis in animal and human tumors, we propose that the boundary time window of 24 to 96 h postpaclitaxel identified in the current study is a reasonable starting point for tumor priming in humans.
With respect to the size of nanoparticles that may benefit from tumor priming, the present study identified 200 to 500 nm as the upper limit. This size range is in agreement with the pore size of tumor vessels in xenograft tumors, which ranges from 100 to 780 nm in diameter depending on the anatomic location of the tumor (e.g., smaller in cranial tumors compared with s.c. tumors) and the tumor growth rate (e.g., smaller in regressing tumors) (Jang et al., 2003). The pore size of tumor vessels in humans has not been established, and further studies to define the optimal particle size range in humans are warranted. In view of the clinical efficacy of two nano-sized agents, doxorubicin HCl liposomes and Abraxane (albumin-conjugated paclitaxel), that have the respective diameters of 85 and 130 nm (Gabizon et al., 2003; Desai et al., 2006), it is reasonable to infer that in humans, particles of comparable sizes would benefit from tumor priming.
The current findings may have implications in clinic. Paclitaxel can elicit multiple interactions with other drugs. It affects the metabolism of agents such as doxorubicin and epirubicin and perturbs cell cycle progression and thereby affects the pharmacodynamics of cell cycle phase-specific agents such as gemcitabine and methotrexate (Baker and Dorr, 2001; Minotti et al., 2001). The present study showed that paclitaxel also affected the delivery of subsequently administered agents. Furthermore, although an effective paclitaxel tumor priming schedule enhanced the activity without promoting the toxicity of doxorubicin HCl liposomes, a reverse sequence of the combination that disabled tumor priming produced greater toxicity and lethality without producing therapeutic benefits. These complex time- and dose-dependent tumor priming effects of paclitaxel and the multidimensional drug-drug interactions highlight the need of careful considerations in using paclitaxel in combination therapy. For the combination of paclitaxel and doxorubicin HCl liposomes, the several schedules under clinical evaluation involve using concurrent administration (Campos et al., 2003; Briasoulis et al., 2004). Our current finding that a paclitaxel pretreatment improved the tumor-selective delivery and efficacy of doxorubicin HCl liposomes would support a schedule of administering paclitaxel before doxorubicin HCl liposomes to take advantage of the tumor priming property of paclitaxel. Furthermore, because chemotherapy is typically given in multiple cycles, tumor priming would be particularly useful in situations when tumor cells have recovered and begun to repopulate the tumor matrix. In summary, paclitaxel tumor priming can be readily practiced in clinic and represents a potentially useful method for promoting the delivery and efficacy of nanomedicines in patients.
Footnotes
-
This study was supported in part by the National Cancer Institute, Department of Health and Human Services (Grant R21CA111770).
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.107.121632.
-
ABBREVIATIONS: MVP, microvascular pressure; IFP, interstitial fluid pressure; ANOVA, analysis of variance; PAC, paclitaxel.
- Received February 18, 2007.
- Accepted April 6, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}