


Abstract
20-Hydroxyeicosatetraenoic acid (20-HETE) is formed by the ω-hydroxylation of arachidonic acid by cytochrome P450 4A and 4F enzymes, and it induces angiogenic responses in vivo. To test the hypothesis that 20-HETE increases endothelial cell (EC) proliferation via vascular endothelial growth factor (VEGF), we studied the effects of WIT003 [20-hydroxyeicosa-5(Z),14(Z)-dienoic acid], a 20-HETE analog on human macrovascular or microvascular EC. WIT003, as well as pure 20-HETE, stimulated EC proliferation by ∼40%. These proliferative effects were accompanied by increased VEGF expression and release that were observed as early as 4 h after 20-HETE agonist addition. This was accompanied by increased phosphorylation of the VEGF receptor 2. The proliferative effects of 20-HETE were markedly inhibited by a VEGF-neutralizing antibody. Polyethylene glycol-superoxide dismutase (PEG-SOD) markedly inhibited both the increases in VEGF expression and the proliferative effects of 20-HETE. In contrast, administration of the NAD(P)H oxidase inhibitor apocynin had no effect to the proliferative response to 20-HETE. The 20-HETE agonist markedly increased superoxide formation as reflected by an increase in dihydroethidium staining of EC, and this increase was inhibited by PEG-SOD but not by apocynin. 20-HETE also increased the phosphorylation of p42/p44 mitogen-activated protein kinase (MAPK) in EC, whereas an inhibitor of MAPK [U0126, 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene] suppressed the proliferative and the VEGF changes but not the pro-oxidant effects of 20-HETE. These data suggest that 20-HETE stimulates superoxide formation by pathways other than apocynin-sensitive NAD(P)H oxidase, thereby activating MAPK and then enhancing VEGF synthesis that drives EC proliferation. Thus, 20-HETE may be involved in the regulation of EC functions, such as angiogenesis.
Arachidonic acid is metabolized to 20-HETE by cytochrome P450 4A enzymes of (CYP4A) and 4F families in vascular tissues (Miyata and Roman, 2005). 20-HETE plays a role in the angiogenesis induced by electrical stimulation of skeletal muscle (Amaral et al., 2003). Jiang et al. (2004) have also shown that overexpression of CYP4A1 in vascular smooth muscle cells promotes endothelial sprouting in renal arterial microvessels. Furthermore, our group has recently reported that 20-HETE induces neovascularization in vivo (Chen et al., 2005). These data suggested that 20-HETE may induce angiogenic responses in endothelial cells (EC).
Angiogenesis requires the coordinated action of a variety of growth factors and cell adhesion molecules in endothelial and mural cells (Coultas et al., 2005). In normal tissues, blood vessel growth is regulated through a complex balance between the actions of proangiogenic factors [e.g., vascular endothelial growth factor (VEGF)] and angiogenic inhibitors. VEGF is a crucial factor in the regulation of angiogenesis. VEGF is the most important angiogenic molecule associated with neovascularization and a key regulator of vascular EC sprouting (Carmeliet, 2004). VEGF expression is regulated by a number of external factors. Among the most important is hypoxia-inducible factor-1α (HIF-1α) (Pages and Pouyssegur, 2005). Although VEGF has a number of variants, each with discrete effects, the classic regulators of angiogenesis are the VEGF-A isoforms acting on its VEGF receptor 2 subtype. Two high-affinity receptors for VEGF-A, VEGFR1 (Flt-1) and VEGFR2 (kinase insert domain-containing receptor/Flk-1), cooperate to induce vasculogenesis and angiogenesis in the developing embryo. However, VEGFR2 transduces the major signals for angiogenesis via its strong tyrosine kinase activity (Ferrara, 2005).
We hypothesized that 20-HETE induces EC proliferation, a putative marker of angiogenesis, by activating the VEGF-VEGFR2 pathway. To test this hypothesis, we examined the effects of exogenous 20-HETE or a noncyclooxygenase-metabolizable analog, WIT003 [20-hydroxyeicosa-5(Z),14(Z)-dienoic acid], on human umbilical vein endothelial cells (HUVEC) and dermal microvascular endothelial cells (HDMVEC) in vitro. We found that 20-HETE induced EC proliferation in a dose-dependent fashion by increasing VEGF expression and release. HIF-1α was not increased at the time points in which changes in VEGF had its peak. The EC-proliferative effects of 20-HETE seemed to be mediated by superoxide formation, up-regulation of the MAPK signaling pathway, and activation of the VEGF-VEGFR2 pathway, which then mediates 20-HETE-induced EC proliferation. EC migration was also stimulated by 20-HETE. These data suggest that 20-HETE may contribute to regulation of endothelial cell growth.
Materials and Methods
Cell Cultures and Chemicals. HUVEC and HDMVEC were purchased from Cambrex Bio Science (East Rutherford, NJ). Both types of cells were grown according to the manufacturer's recommendations and maintained at 37°C in a humidified incubator containing 5% CO2. 20-HETE and WIT003, a stable analog of 20-HETE, were both synthesized by J.R.F. PEG-SOD, apocynin, U0126, SU5416, tempol, angiotensin II (ANG II), diphenylene iodonium (DPI), and dihydroethidium (DHE) were all purchased from Sigma-Aldrich (St. Louis, MO).
Cell Proliferation Assays. Proliferation studies were performed with cultures plated at a density that ensured exponential growth for at least 5 days. Proliferation was assessed by counting EC plated at 2 × 104 cells in 35-mm dishes. Cells were allowed to grow overnight before exposure to various treatments. All test agents were dissolved in ethanol (EtOH) unless indicated otherwise, and an equal volume of EtOH was added to the cultures as a vehicle control. The concentration of EtOH in the medium never exceeded 0.1%. After 48 h, EC were counted using a hemocytometer.
Human VEGF ELISA. EC were plated at a density of 2 × 104 cells in 35-mm dishes and allowed to grow overnight. The medium was then replaced, and the cells were exposed to 10 μM WIT003 for 4 or 24 h. Culture medium was then collected. The medium was cleared of debris by centrifugation at 10,000g and 4°C. VEGF in the supernatant was measured with a human VEGF ELISA kit (Calbiochem, La Jolla, CA) according to the manufacturer's recommendations. Cell numbers were also obtained by cell counts in these cultures. The concentration of VEGF in the media was normalized against the number of cells in each culture and expressed as picograms/milliliter/103 cells.
Western Blotting. EC were treated with vehicle, 10 μM WIT003, or various inhibitors and washed twice with ice-cold PBS. Western blot was performed as described previously (Guo et al., 2005). The primary antibodies used were anti-human VEGF, anti-phospho-VEGFR2, anti-HIF-1α (Upstate Biotechnology, Waltham, MA), anti-phospho-p42/p44 MAPK (Upstate Biotechnology), and total p42/p44 MAPK (Santa Cruz Biotechnology, Santa Cruz, CA). All primary antibodies were used at a dilution of 1:500 to 1:1000. Stripped membranes were reprobed with actin (Santa Cruz Biotechnology) that served as a loading control.
Measurement of Superoxide Formation. Superoxide formation in cultured HDMVEC was assayed with the fluorescent dye DHE using both fluorescence microscopy and flow cytometry. For fluorescence microscopy with DHE, equal numbers of HDMVEC were plated in 12-well plates and allowed to grow overnight. Cultures were then incubated with or without 10 μM WIT003 in sterile d-PBS (Mediatech, Herndon, VA) containing 0.1% d-glucose and l-arginine (50 μM) for 30 min. DHE (2 μM) was added, and fluorescence microscopy was performed after a 30-min incubation. Some wells were also pretreated with PEG-SOD or apocynin for 1 h before adding WIT003. All chemicals were made fresh on the day of the experiments. Fluorescent images were obtained with a 20× objective (identical exposure times) using a Leica inverted immunofluorescence microscope (Leica Microsystems, Inc., Deerfield, IL). For flow cytometry with DHE, 1 × 106 HDMVEC were also treated with 0.01, 0.1, 1, or 10 μM WIT003 for 30 min again. Some cells were also treated in the presence of apocynin and PEG-SOD. Fluorescence intensities were monitored and recorded using a FACScan cell sorter (BD Biosciences, Franklin Lakes, NJ). A minimum of 104 events/sample was collected. Data were analyzed using CellQuest Pro Software (BD Biosciences). Similar experiments were also repeated using 10 μM ANG II.
Morphological Assessment. HDMVEC were plated in 12-well culture plates and treated with 10 μM WIT003. Cultures were processed for immunofluorescence microscopy 24 h later. For F-actin immunofluorescence staining, cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature and then permeabilized with 0.1% Triton X-100 for 5 min. Cells were washed three times with washing buffer and incubated with a 1:500 dilution of TRITC-conjugated phalloidin for 30 min at room temperature. To stain for α-tubulin, cells were extracted by immersion in microtubule-stabilizing buffer containing 0.1% Triton X-100 for 1 min at room temperature. The extracted cells were fixed in ice-cold methanol for 15 min and incubated with a mouse monoclonal anti-α-tubulin clone DM1A fluorescein isothiocyanate-conjugated antibody at a dilution of 1:50 for at least 2 h. After washing the wells, the nuclei were counterstained by incubating the cells with 4,6-diamidino-2-phenylindole for 2 min at room temperature. Finally, cells were mounted on a slide with antifade-mounting solution, and cytoskeletal elements were visualized with an inverted Leica fluorescence microscope.
EC Migration Assay. Chemotaxis assay of HDMVEC was performed using a QCM cell migration assay kit (Chemicon International, Temecula, CA) following the manufacturer's recommended protocol. In brief, 300 μl of HDMVEC cell suspension containing 1 × 105 cells was loaded in the upper wells along with serum-free medium, whereas serum-free medium containing 10 μM WIT003 was placed in some of the lower wells and medium containing 2% fetal bovine serum was used to load the remaining lower wells that served as positive controls. The chamber was incubated at 37°C for 24 h. Cells were then stained, and nonmigrating cells on the upper surface of the membrane were removed by wiping with a cotton swab. Chemotaxis was quantified in 25 random fields by counting the cells that migrated to the lower side of the membrane. All experiments were performed blindly with a Leica transmitted light microscope (40×), as those doing the counting were unaware of the treatment groups to minimize bias.
Statistical Analysis. Data were analyzed using analysis of variance followed by Tukey's test or a Student's t test when only two groups were studied. A p < 0.05 was considered to be significant.
Results
20-HETE Induces Proliferation of EC
Because WIT003 is thought to be a proangiogenic factor in vivo and EC proliferation is a surrogate marker of the angiogenic response in vitro, we studied the effects of WIT003 on EC proliferation. WIT003 induced a dose-dependent increase in proliferation of HUVEC and HDMVEC in vitro (Fig. 1A). We chose 10 μM WIT003 as our working concentration because it consistently produced maximal proliferation, increasing EC proliferation at 48 h by ∼40%. To confirm the equivalency of WIT003 with 20-HETE, 20-HETE was also tested under identical proliferation conditions with HUVEC and HDMVEC in a parallel experiment (Fig. 1B). We found that 20-HETE also induced a similar and comparable dose-dependent increase in proliferation of both EC. Because there was no significant difference between the effects of 20-HETE and WIT003, we chose to use WIT003 for most subsequent experiments since it lacks double bonds in the 11,12 and 8,9 positions and cannot be metabolized by cyclooxygenase enzymes to prostanoids. Furthermore, responses to WIT003 and 20-HETE did not vary significantly between HUVEC and HDMVEC. Thus, we chose to perform the remaining experiments using HDMVEC only.

WIT003 and 20-HETE induce EC proliferation. HUVEC and HDMVEC were plated onto 35-mm culture dishes and allowed to grow overnight. Cultures were treated with either 0.1, 1, or 10 μM WIT003 (A) or 20-HETE (B), and cell proliferation was assessed by cell counts 48 h later. Data shown are combined results of HUVEC and HDMVEC and are presented as the mean ± S.D. of at least three experiments (each performed in triplicate). *, p < 0.05 versus control; **, p < 0.05 versus 0.1 μM; ***, p < 0.05 versus 1 μM.
VEGF Mediates WIT003-Induced EC Proliferation
To determine whether VEGF is involved in 20-HETE-induced stimulation of EC proliferation, we measured VEGF levels in medium collected from EC treated with 10 μM WIT003 for 0, 4, and 24 h. We detected a ∼5- and ∼3-fold increase in VEGF in medium collected from the EC cultures at 4 and 24 h after the addition of WIT003, respectively (Fig. 2A). These increases in extracellular VEGF were accompanied by a rise in intracellular VEGF protein level at 4 h after exposure to the 20-HETE analog (Fig. 2B). In addition, WIT003 significantly induced phosphorylation of the VEGF receptor in EC as shown by Western blot using an antibody against phosphorylated VEGFR2 (Fig. 2, B and C). HIF is an important regulator of VEGF expression and activity. However, HIF-1α protein levels remained unchanged from control as the intracellular level of VEGF rose at 4 h after treatment (Fig. 2D).

WIT003 activates a HIF-independent VEGF-VEGFR2 pathway. A, human VEGF ELISA was performed with medium harvested from HDMVEC treated with 10 μM WIT003 for 0 (basal), 4, or 24 h. Data were normalized against the cell numbers in the cultures and are presented as the mean ± S.D. of three experiments (each performed in triplicate). *, p < 0.05 versus time 0 (basal). B, VEGF protein level, VEGFR2 phosphorylation, and total VEGFR level 4 h after the addition of 10 μM WIT003 examined by Western blot. C, densitometry analysis of the Western blot on VEGF protein was shown. D, HIF-1α expression in HDMVEC treated with either EtOH (control) or 10 μM WIT003, assessed by Western blot using an anti-HIF-1α antibody. Representative blots from at least three experiments are shown. Actin was used as loading controls.
To confirm the involvement of the VEGF-VEGFR2 pathway in 20-HETE-stimulated EC proliferation, we pretreated EC with SU5416 (10 μM), an inhibitor of VEGFR2 tyrosine kinase activity, or VEGF-neutralizing antibody (1 μg/ml) and studied whether they alter the proliferative effects of WIT003. SU5416 abolished the proliferative effects of WIT003 (Fig. 3A). Although not completely, anti-VEGF antibody significantly and markedly inhibited the proliferative effects of WIT003 (Fig. 3B).
ROS Mediate the Proliferative Effects of 20-HETE
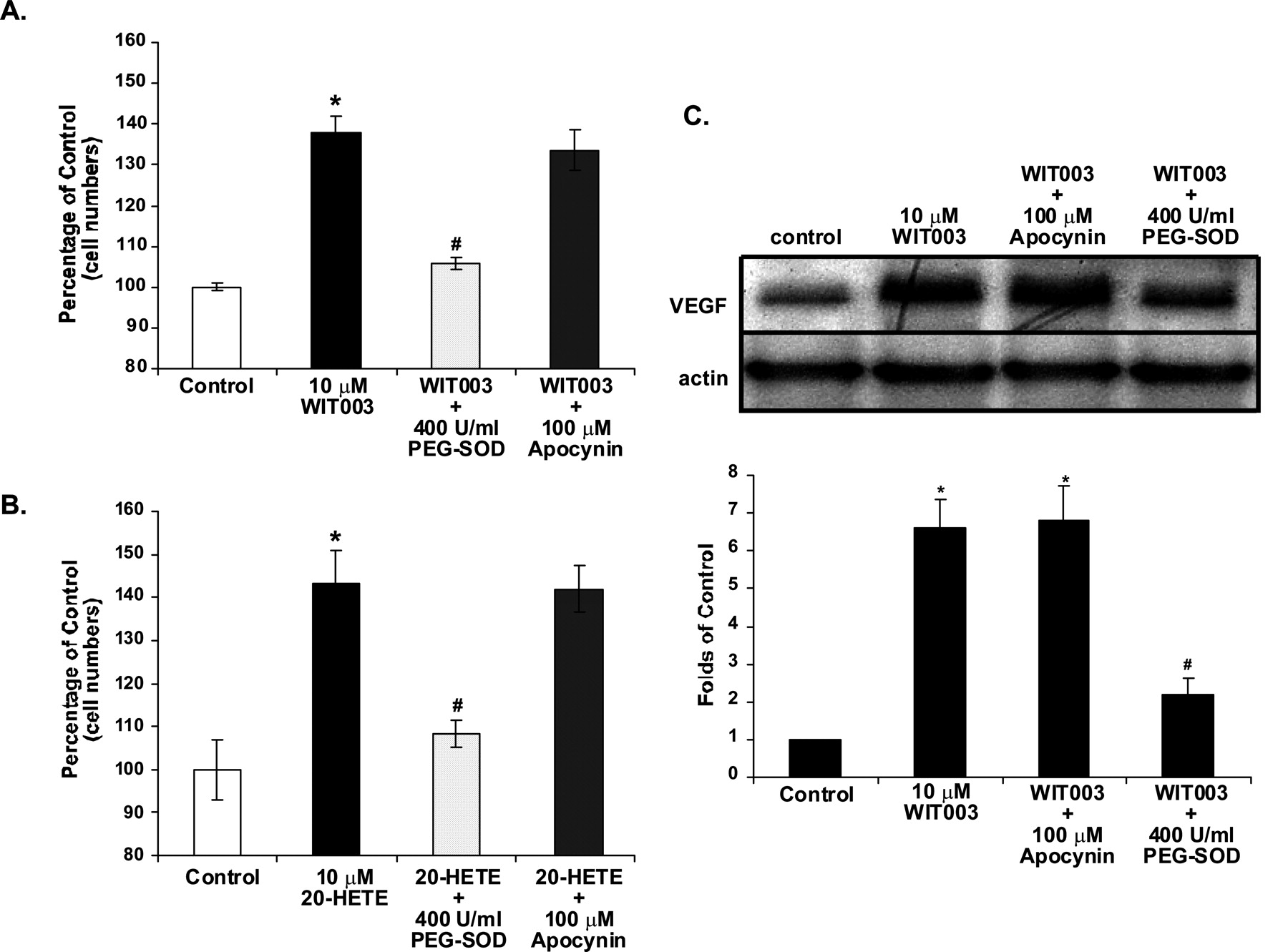
To determine whether oxidative stress plays a role in the proliferative effects of 20-HETE in EC, we coincubated EC with 400 U/ml PEG-SOD in the presence of either 10 μM WIT003 or 20-HETE for 48 h. PEG-SOD almost completely abolished the effects of 20-HETE on EC proliferation (Fig. 4, A and B), suggesting that the proliferative effects of 20-HETE on EC are secondary to stimulation of superoxide formation. Because increased ROS is often produced by stimulation of NADPH oxidase, we also treated EC with apocynin (100 μM), an inhibitor of NADPH oxidase, and studied the effects of WIT003 or 20-HETE on cell proliferation. Apocynin had little or no effect on the proliferative effects of 20-HETE (p = 0.1) (Fig. 4, A and B). The effects of PEG-SOD and apocynin were further confirmed using tempol, a well known superoxide scavenger (50 μM), or DPI, a general inhibitor of flavin-dependent enzymes (10 μM), respectively. Tempol inhibited the proliferative responses to 20-HETE and WIT003, but DPI did not (n = 2 in triplicate; data not shown). Furthermore, the increased intracellular VEGF level observed 24 h after adding WIT003 was significantly abolished by PEG-SOD but not by apocynin (Fig. 4C).

Effects of VEGFR inhibitor and VEGF-neutralizing antibody on 20-HETE-induced EC proliferation. Cell proliferation was studied in HDMVEC treated with 10 μM WIT003 in the presence of either 10 μM SU5416 (A) or 1 μg/ml VEGF-neutralizing antibody (B). Data shown are presented as the mean ± S.D. of at least three experiments (each performed in triplicate). *, p < 0.05, WIT003 versus all groups. #, p < 0.05, WIT003 + anti-VEGF versus control. These data suggest that the proliferative effects of 20-HETE are secondary to the activation of VEGF.
DHE Imaging for Measurements of Superoxide Formation
To confirm that 20-HETE increases superoxide formation in EC, we exposed cultured EC to WIT003 in the presence and absence of PEG-SOD or apocynin and assayed superoxide formation by measuring the DHE fluorescence. Cells treated with WIT003 had markedly increased fluorescence compared with control, and this was inhibited by PEG-SOD but not by apocynin (Fig. 5A). Flow cytometry was performed to quantitate the increases in DHE fluorescence intensity in EC treated with WIT003 (Fig. 5B). The increases in fluorescence (red) were almost completely inhibited by PEG-SOD (400 U/ml) but not by apocynin (Fig. 5C).
Dose-Response Experiments. WIT003 induced dose-dependent superoxide formation, as indicated by increased DHE fluorescence. Under the experimental conditions used, increases in superoxide formation were observed at WIT003 concentrations as low as 0.01 μM. A plateau was observed after 1 μM. (Fig. 5D).
Apocynin Inhibits ANG II-Induced ROS Formation in EC
To ensure that the concentration of apocynin we were using (100 μM) was effective as NAD(P)H oxidase inhibitor of the ECs, we studied the effects of apocynin on ANG II-induced superoxide formation. Using DHE fluorescent microscopy and flow cytometry, we show that 100 μM apocynin significantly inhibited the superoxide formation induced by ANG II (Fig. 6, A and B). This demonstrates that apocynin at the concentrations used did inhibit NAD(P)H oxidase activation in the ECs, thus ruling out NAD(P)H oxidase-dependent production of superoxide as the source of the 20-HETE-induced changes in superoxide formation.
WIT003 Induces Morphological Changes in EC
In the flow-cytometry experiments, we observed that the cells treated with WIT003 appeared to be smaller than the controls, suggesting morphological changes. Indeed, immunocytochemistry for F-actin and α-tubulin showed that incubation with WIT003 led to marked cytoskeletal changes in EC, which became more spindle-shaped (Fig. 7, A, B, C, and D). These changes were observed 24 h after adding WIT003. Cytoskeleton changes were not visible with short incubation times.
WIT003 Stimulates EC Proliferation through Activation of the MAPK Signal Transduction Pathway
Phosphorylation and activation of MAPK pathway play a key role in the regulation of cell growth and proliferation. Treatment of HDMVEC with WIT003 (10 μM) significantly increased the phosphorylation of p42/p44 MAPK (Fig. 8A) without affecting total p42/p44 MAPK levels. At the concentration we used, U0126 completely abolished the activation of phospho-p42/p44 MAPK induced by WIT003 (Fig. 8B). Parallel cell proliferation studies carried out in EC treated with WIT003 and the MAPK inhibitor U0126 (10 μM) showed that the proliferative effects of 20-HETE were significantly inhibited by U0126 (Fig. 8C). We also tested the inhibitory effects of U0126 on 20-HETE-induced superoxide formation and VEGF stimulation and found that 10 μM U0126 did not inhibit the effects of 20-HETE on superoxide formation (Fig. 8D), whereas it significantly inhibited the increase in intracellular VEGF induced 24 h after administration of WIT003 (Fig. 8E).

Effects of antioxidants on WIT003- and 20-HETE-induced proliferation of EC and VEGF up-regulation. A and B, HDMVEC were exposed to either 10 μM WIT003 (A) or 20-HETE (B) in the presence of 100 μM apocynin or 400 U/ml PEG-SOD (1-h pretreatment). Cell proliferation was assessed by cell count 48 h later. Results shown are presented as the mean ± S.D. of three experiments (each performed in triplicate). *, p < 0.05 versus control; #, p < 0.05 versus WIT003 or 20-HETE. C, HDMVEC were exposed to 10 μM WIT003 alone or after 1-h pretreatment with 100 μM apocynin or 400 U/ml PEG-SOD. Western blot was performed using a polyclonal VEGF antibody, with actin serving as a loading control. Densitometry analysis was shown as the bar graph. Representative images from three individual experiments are shown. *, p < 0.05 versus control; #, p < 0.05 versus WIT003. The data suggest that the proliferative effects of 20-HETE on EC are secondary to stimulation of superoxide formation by pathways other than NADPH oxidase.
20-HETE Induces EC Migration
Another step in the angiogenic response is EC migration. To determine the effects of 20-HETE on HDMVEC migration, a chemotaxis assay was performed with WIT003 using a QCM cell migration assay kit (Chemicon International) based on the manufacturer's suggested protocol. 20-HETE produced a significant 3-fold increase in EC migration (137 ± 17 with WIT003 versus 48 ± 8 cells/25 fields in the controls). However, 20-HETE is a relatively weak chemotaxic agent compared with 2% serum where the number of migrating cells was ∼1000 cells/25 fields (data not shown).
Discussion
The CYP4A-20-HETE system is present in the vasculature, and 20-HETE is present in plasma (Kroetz and Xu, 2005; Miyata and Roman, 2005). The CYP4A-20-HETE system may play an important role in the regulation of vascular tone. Several reports have demonstrated that 20-HETE is proangiogenic (Amaral et al., 2003; Jiang et al., 2004; Chen et al., 2005). Angiogenesis predominantly involves EC (Carmeliet, 2004), suggesting that 20-HETE interacts with EC. To delineate the mechanisms by which 20-HETE elicits angiogenic responses, we investigated the effects of 20-HETE on EC proliferation, a surrogate marker of the angiogenic response. We primarily used a stable 20-HETE analog, WIT003, because comparison with pure 20-HETE did not show significantly different responses. We found that 20-HETE induced a dose-related proliferative response in both microvascular and macrovascular EC. Responses did not vary greatly between EC subtypes. A fundamental component of the angiogenic process is VEGF, and we tested whether the 20-HETE analog stimulates EC proliferation through increasing the production and release of VEGF. We found that a single addition of WIT003 to the cells resulted in rapid increases (less than 4 h) in both VEGF release and intracellular VEGF protein levels. VEGF concentrations in the medium were lower at 24 h after exposure to the 20-HETE analog than in samples collected at 4 h. This may be due to loss of VEGF protein stability, because intracellular protein levels were consistently increased at this time point. 20-HETE also resulted in a concomitant increase in VEGFR2 phosphorylation. SU5416, which inhibits the tyrosine kinase activity of activated VEGFR2, and a neutralizing antibody against human VEGF both inhibited the proliferative effects of 20-HETE, suggesting that they are mediated by activation of the VEGF-VEGFR2 pathway. We cannot exclude that other pathways besides VEGF are also involved, because inhibition by the VEGF antibody was not complete. However, VEGF is clearly the main mediator of the proliferative effects of 20-HETE.

DHE imaging and flow cytometry of superoxide formation in 20-HETE-treated HDMVEC. A, cells were plated in 12-well plates and treated with EtOH (control), 10 μM WIT003 alone, WIT003 or 400 U/ml PEG-SOD (1-h pretreatment), WIT003, and 100 μM apocynin (also 1-h pretreatment) for 30 min in d-PBS. DHE (2 μM) was then added, and fluorescence microscopy was performed 20 min later. Images were obtained using an inverted Leica immunofluorescence microscope with a 20× objective. All fluorescence microscopic images were taken using identical exposure times. Representative images from five experiments are shown. B, 1 × 106 HDMVEC were treated with either EtOH (control) or 10 μM WIT003 for 30 min in d-PBS. Flow cytometry was performed 20 min after adding 2 μM DHE. Representative histograms from four experiments are shown. C, flow cytometry of DHE fluorescence intensity in HDMVEC exposed to either EtOH alone, 10 μM WIT003 alone, WIT003 plus 400 U/ml PEG-SOD, or WIT003 plus 100 μM apocynin was performed. Data shown are presented as the mean ± S.D. of three experiments (each performed in triplicate). *, p < 0.05 versus control. D, dose-response study with DHE flow cytometry in HDMVEC treated with various concentrations of WIT003 (n = 2) is shown. *, p < 0.05 versus control; **, p < 0.05 versus 0.01 μM; ***, p < 0.05 versus 0.1 μM.
VEGF expression is regulated by a number of external factors. Hypoxia-induced VEGF production serves as a driving force for the development of neovessels that is mediated by changes in HIF-1α (Pages and Pouyssegur, 2005). However, hypoxia was not present in our experiments. At the time points when increases in VEGF were maximal (4 h), we did not observe changes in immunoreactive HIF-1α, suggesting that 20-HETE may have stimulated VEGF production at this particular time point via nonhypoxic regulatory pathways.
Because accumulating evidence links oxidative stress to angiogenesis and VEGF regulation (Ushio-Fukai, 2006), we studied whether the proliferative effects of 20-HETE would be affected by treating the cells with 20-HETE in the presence of antioxidants, such as tempol or PEG-SOD. The effects of 20-HETE on proliferation and VEGF were abolished by both agents, suggesting that these 20-HETE-induced changes are dependent on superoxide formation. We confirmed using the DHE fluorescence assay that 20-HETE increases superoxide formation by EC. The changes in DHE fluorescence induced by WIT003 were due to the formation of superoxide, because they were markedly inhibited by PEG-SOD and tempol. Overall, these results indicate that the 20-HETE analog WIT003 is a potent inducer of superoxide.

Apocynin inhibits ANG II-induced superoxide formation. A, HDMVEC were plated in 12-well plates and treated with EtOH (control), 10 μM ANG II alone, or ANG II together with 100 μM apocynin (1-h pretreatment) for 30 min in d-PBS. DHE (2 μM) was then added, and fluorescence microscopy was performed 20 min later. Images were obtained as described in Fig. 5A. B, flow cytometry of DHE fluorescence intensity in HDMVEC exposed to either EtOH alone, 10 μM ANG II alone, 100 μM apocynin alone (1-h pretreatment), or ANG II together with 100 μM apocynin. Data shown are presented as the mean ± S.D. of two experiments. *, p < 0.05 versus control; #, p < 0.05 versus ANG II treated. These data confirm that ANG II-induced superoxide formation in EC is NAD(P)H oxidase-dependent and that 100 μM apocynin effectively inhibits NAD(P)H oxidase activity.
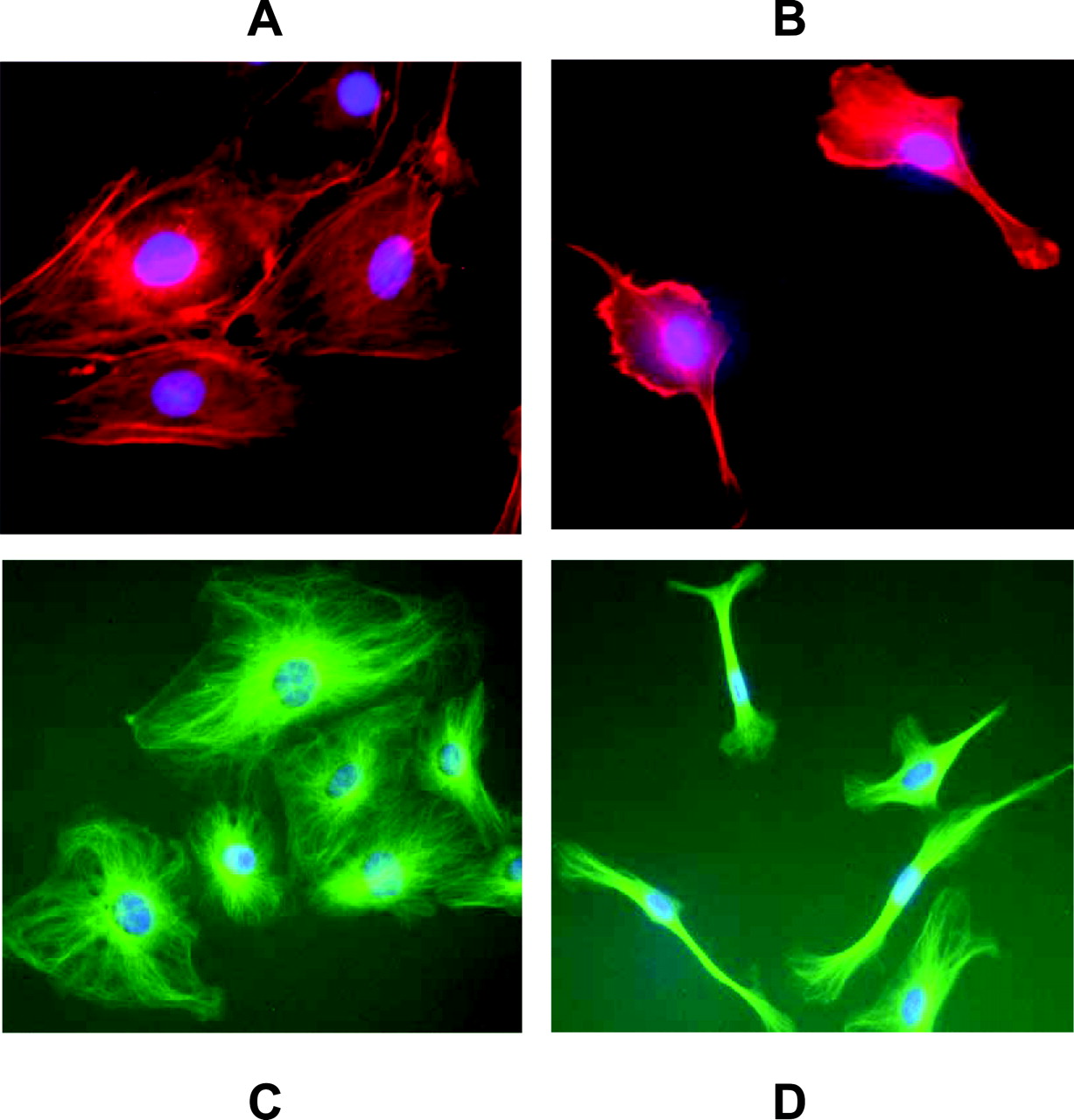
20-HETE significantly alters EC morphology and cytoskeleton structure. HDMVEC were plated in 24-well culture plates and treated with either EtOH (A and C) or 10 μM WIT003 (B and D) for 24 h. Cultures were then stained with TRITC-conjugated phalloidin for F-actin (red) and fluorescein isothiocyanate-conjugated anti-α-tubulin antibody for α-tubulin (green) as described under Materials and Methods. Images were obtained using a 20× objective. 4,6-Diamidino-2-phenylindole was used to visualize the nuclei (blue). Images shown are representative of at least three independent experiments.

Effects of WIT003 on MAPK in EC. A, HDMVEC were exposed to 10 μM WIT003 for various times, and Western blot was carried out using antibodies against phospho-p42/p44 MAPK. Total p42/p44 MAPK was used as a control. B, Western blot analysis of HDMVEC treated with either EtOH, WIT003, or WIT003 together with 10 μM U0126 using an antibody against phospho- and total p42/p44 MAPK. C, cultures were pretreated with 10 μM U0126, an inhibitor of the MAPK pathway, for 1 h before adding WIT003. Cell proliferation was assessed by cell count 48 h later. Results shown are presented as the mean ± S.D. of three experiments (each performed in triplicate). *, p < 0.05 versus control; #, p < 0.05 versus WIT003. D, DHE fluorescence microscopy shows that 20-HETE-induced ROS formation was not affected by U0126. E, Western blot using an anti-VEGF polyclonal antibody in EC pretreated with U0126 before adding WIT003 for 24 h showing inhibition of the 20-HETE analog-induced increases in VEGF. Densitometry analysis was shown in bar graph. *, p < 0.05 versus control; #, p < 0.05 versus WIT003. Representative Western blots from three experiments are shown. These data suggest that the 20-HETE analog-induced EC proliferation requires activation of p42/p44 MAPK.
These changes in DHE fluorescence were further confirmed using a flow-cytometric assay. The dose response with DHE flow cytometry showed that concentrations of the 20-HETE mimetic WIT003 as low as 10 nM induced rapid and significant increases in DHE fluorescence. These increases were abolished by coincubation with PEG-SOD, paralleling the effects of these antioxidants on 20-HETE-induced proliferation and DHE fluorescence microscopy.
The changes in superoxide formation induced by 20-HETE in EC were observed within minutes. VEGF levels and release both increased markedly within 4 h after stimulation with 20-HETE, suggesting that stimulation of VEGF is secondary to the increase in superoxide formation induced by 20-HETE. Superoxide produced via the NAD(P)H oxidase pathway has been reported to be essential for stimulation of angiogenesis responses by EC (Ushio-Fukai, 2006). We tested whether inhibiting NAD(P)H assembly with apocynin would inhibit 20-HETE-induced EC proliferation and VEGF responses. Apocynin has been used extensively as an inhibitor of NAD(P)H oxidase, because it attenuates the catalytic activity of the various forms of NAD(P)H oxidase by blocking translocation of p47phox and p67phox from the cytosol to the membrane. Contrary to our expectations, apocynin treatment had no effect on the ability of 20-HETE to increase VEGF expression and EC proliferation. DPI, which inhibits all flavin-dependent enzymes (Cifuentes and Pagano, 2006), also had little effect on 20-HETE-induced proliferation (data not shown). To confirm these data, we tested whether treatment with apocynin would affect the strong induction of DHE fluorescence induced by 20-HETE in DHE-loaded EC. 20-HETE-induced increase in DHE fluorescence remained unchanged in EC treated with apocynin. At the concentration used (100 μM), apocynin did inhibit NAD(P)H oxidase because it almost abolished the superoxide production induced by ANG II, known to be NADP(H)-oxidase dependent (Ushio-Fukai, 2006). Thus, these data suggest that the 20-HETE analog WIT003 and ANG II both induce superoxide formation in EC. NADP(H) oxidase mediates the effects of ANG II but not those of WIT003/20-HETE. Because neither apocynin nor DPI had any effect on the pro-oxidant, proliferative, or VEGF-increasing effects of 20-HETE, we concluded that 20-HETE induces superoxide formation in EC via a pathway(s) other than NAD(P)H oxidase, an unexpected finding. Possible candidates include mitochondrial enzymes, lipoxygenase, cyclooxygenase, nitric-oxide synthase, xanthine oxidase, peroxidases, and other hemoproteins (Cifuentes and Pagano, 2006).
Immunofluorescence microscopy for F-actin and α-tubulin showed that incubation with 20-HETE led to marked morphologic changes in EC, which became more spindle-shaped. We do not know whether these changes in the cytoskeleton and cell morphology are driving the changes in VEGF, oxidative stress, and cell proliferation. However, it has been suggested that the cytoskeletal changes may be a central point of cross-talk in growth- and redox-signaling pathways, involving some factors known to regulate vascular cell growth and function (Touyz et al., 2005). VEGF-induced angiogenic responses require cytoskeletal changes in EC (van Nieuw Amerongen et al., 2003). The alterations in EC morphology were observed 24 h after the addition of the 20-HETE analog, suggesting that it is associated with secondary responses.
There is evidence of an association between the growth effects of VEGF in EC and MAPK (Pages and Pouyssegur, 2005). 20-HETE up-regulates MAPK in vascular smooth muscle cells (Muthalif et al., 1998). Many of the effects of growth factors on EC function depend on activation of MAPK (Campochiaro, 2006a,b). For example, ROS-mediated activation of ERK1/2 (p42/p44 MAPK) is an important mechanism that modulates trans-endothelial permeability (Rousseau et al., 2000). We tested whether 20-HETE activates ERK1/2 and found that the addition of WIT003 markedly increased ERK1/2 phosphorylation, indicating activation of this pathway. Further inhibition of ERK1/2 activation with the specific mitogen-activated protein kinase kinase inhibitor U0126 markedly inhibited the proliferation and the VEGF increases induced by 20-HETE. At the U0126 dose used, the ERK1/2 phosphorylation induced by WIT003 was completely abolished by treatment with U0126. However, proliferative activity was greatly reduced (∼80–90%) but not completely abolished. Although this suggests that the main pathway activated by 20-HETE to induce EC proliferation is p42/p44 MAPK, we cannot exclude that 20-HETE may also activate additional pathways; however, their contribution seems to be, if anything, minor. On the other hand, we still observed robust changes in red fluorescence in DHE-loaded cells treated with 20-HETE and U0126, suggesting that activation of MAPK occurs downstream from superoxide signaling. That suggests that the cascade of events triggered by 20-HETE is superoxide → MAPK activation → VEGF → proliferation. We also found that 20-HETE induced EC migration, but this effect was weak, ∼3-fold over basal.
Our data indicate that 20-HETE induces EC proliferation that was associated with a marked increase in superoxide formation via pathways other than apocynin-sensitive NAD(P)H oxidases. The proliferative effects of 20-HETE are transduced via pathways dependent on MAPK activation and are mediated by VEGF. These data partly explain the mechanisms by which 20-HETE induces angiogenesis (Amaral et al., 2003; Jiang et al., 2004; Chen et al., 2005). Because 20-HETE synthase activity is present in vascular cells (Miyata and Roman, 2005), stimuli that lead to the release of arachidonic acid within vascular cells would in turn increase 20-HETE synthesis and release. EC are the main vascular cells involved in angiogenic responses, and clearly, they respond to 20-HETE with proliferation and also migration, both putative angiogenic responses. These data suggest that 20-HETE is a nonhypoxic regulator of VEGF and thus may contribute to regulation of angiogenesis. Furthermore, 20-HETE itself induces oxidative stress. This is consistent with recent reports indicating that overexpression of 20-HETE synthase induces endothelial dysfunction (Wang et al., 2006). Besides being an important regulator of angiogenesis and vascular permeability, VEGF may also act as a cytokine, regulating inflammation, survival, and cell growth in several types of cancer. Because 20-HETE seems to be an upstream regulator of VEGF and a strong inducer of ROS, its role may extend beyond its known effects on vascular tone and renal function.
Acknowledgments
We thank Dr. Gloria Scicli for assistance with the DHE immunofluorescence microscopy. We also thank Drs. Asad Abbas and Vamshi Thandra for help in EC culturing, cytoskeleton immunochemistry, and migration experiments.
Footnotes
-
This work was supported by National Institutes of Health Grants EY014385 (to A.G.S.), GM31278 (to J.R.F.), and HL 036279 (to R.J.R.) and by a grant from the Robert A. Welch Foundation (to J.R.F.). This work was presented in part: Guo M, Roman R, Falck J, Chen P, and Scicli AG (2006) 20-HETE-induced proliferation of endothelial cells is mediated by activation of VEGF receptors, in The 8th Annual Winter Eicosanoid Conference; 2006 March 12–15; Baltimore, MD. New York Medical College, Valhalla, NY.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.115360.
-
ABBREVIATIONS: 20-HETE, 20-hydroxyeicosatetraenoic acid; WIT003, 20-hydroxyeicosa-5(Z), 14(Z)-dienoic acid; HUVEC, human umbilical vascular endothelial cells; HDMVEC, human dermal microvascular endothelial cells; PEG-SOD, polyethylene glycol-superoxide dismutase; DPI, diphenylene iodonium; DHE, dihydroethidium; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor; ANG II, angiotensin II; HIF, hypoxia-inducible factor; ROS, reactive oxygen species; MAPK, mitogen-activated protein kinases; ERK1/2, extracellular signal-regulated kinase; U0126, 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene; ELISA, enzyme-linked immunosorbent assay; PBS, phosphate-buffered saline; TRITC, tetramethylrhodamine B isothiocyanate; SU5416, 3-[(2,4-dimethylpyrrol-5-yl)methylidenyl]-iodolin-2-one.
- Received October 16, 2006.
- Accepted January 5, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}