


Abstract
The dextran sulfate (DSS) model of colitis causes intestinal injury sharing many characteristics with inflammatory bowel disease, e.g., leukocyte infiltration, loss of gut epithelial barrier, and cachexia. These symptoms are partly mediated by entrapped leukocytes binding to multiple endothelial adhesion molecules (MAdCAM-1, VCAM-1, ICAM-1, and E-selectin). Pravastatin, an 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitor, has anti-inflammatory potency in certain inflammation models; therefore, in this study, we measured the effects of pravastatin in DSS-induced colitis. The administration of pravastatin (1 mg/kg) relieved DSS-induced cachexia, hematochezia, and intestinal epithelial permeability, with no effect on serum cholesterol. Histopathologically, pravastatin prevented leukocyte infiltration and gut injury. Pravastatin also blocked the mucosal expression of MAdCAM-1. DSS treatment promoted mucosal endothelial nitric-oxide synthase (eNOS) mRNA degradation, an effect that was blocked by pravastatin. Importantly, the protective effects of pravastatin in DSS-induced colitis were not found in eNOS-deficient mice. Our results demonstrate that HMG-CoA reductase inhibitors preserve intestinal integrity in colitis, most likely via increased eNOS expression and activity, independent of cholesterol metabolism.
Inflammatory bowel disease (IBD) (Crohn's colitis and ulcerative colitis) is characterized by tissue edema, increased gut epithelial permeability, and extensive infiltration of the gut by leukocytes. The general morbidity and weight loss in individuals with IBD can be attributed to leukocyte sequestration in the gut in this condition (Perkal and Seashore, 1989; Shanahan, 2002). The current literature suggests that multiple immune, genetic, and environmental factors influence both the initiation and progression of colitis (Farrell and Peppercorn, 2002). Despite the fact that the normal intestinal mucosa maintains a high density of leukocytes compared with most tissues, it is not typically inflamed or edematous. However, during active periods of colitis, the colon is even more extensively colonized by lymphocytes and neutrophils that promote extensive oxidant and protease-dependent injury to the gut. Therefore, it is assumed that the intestine has several specialized mechanisms that normally contain these immune responses and that the impairment of these immune-limiting processes causes the entrapment and activation of leukocytes seen in IBD injury. Among the several endogenous agents that control inflammation, nitric oxide has received a great deal of interest as a factor that can limit forms of inflammation. Endothelial cells release nitric oxide (NO) through both by the “constitutive” (eNOS and NOS3) and inducible nitric oxide synthases (iNOS and NOS1). NO released by microvascular endothelial cells reduces several indices of inflammation in vivo and in vitro. NO is a potent reactive oxygen species scavenger and can block many oxidant-mediated inflammatory responses including leukocyte and platelet endothelial adhesion and, equally important, the activation of numerous inflammation-associated genes (Laroux et al., 2001).
Depending on environmental conditions, the level of oxidant, and NO fluxes, however, NO may also promote inflammation; consequently, the nature of NO in chronic inflammation remains particularly controversial (Garcia-Gonzalez and Pena, 1998; Guslandi, 1998; Guihot et al., 2000). For example, although some reports suggest that iNOS-derived NO exacerbates injury in experimental models of colitis (Yoshida et al., 2000; Hokari et al., 2001; Krieglstein and Granger, 2001), Binion et al. (1998, 2000) have reported that human intestinal endothelial cells in Crohn's are deficient in iNOS, which renders Crohn's intestinal endothelium hyperadhesive for leukocytes. The increased leukocyte adhesivity in Crohn's endothelium was corrected by NO donors, supporting a protective role for NO. These differences might also reflect differences based on the cell type (macrophage, endothelial) in which iNOS is expressed.
On the other hand, models using NO synthase inhibitors and eNOS gene-deficient mice consistently argue that NO, which is derived from the constitutive NOS (eNOS) will block leukocyte-endothelial adhesion and leukocyte-dependent injury (Amin-Hanjani et al., 2001). Therefore, therapies that can augment eNOS or eNOS-derived NO might be beneficial in the treatment of chronic inflammatory phenomena like IBD.
The statins are a new class of anticholesteremic HMG-CoA reductase inhibitors that have several beneficial effects on the cardiovascular system not strictly related to their effects on cholesterol metabolism (Fenton et al., 2000; Amin-Hanjani et al., 2001; Davignon and Mabile, 2001; Puddu et al., 2001). For example, statins reduce leukocyte and platelet adhesion, dramatically lower the progression of atherosclerosis, and also appear to protect against postischemic cardiac injury (Kubes et al., 1991; Kim and Berstad, 1992;Kinlay et al., 1996; Garcia-Gonzalez and Pena, 1998; Kreiglstein et al., 2001; Kreiglstein and Granger, 2001). Among several possible mechanistic effects of statins, these agents apparently stabilize mRNAs for the constitutive nitric oxide synthase, which may increase eNOS protein and NO bioavailability. Therefore, if statins increase the endothelial NO supply within the gut, this statin-derived NO might reduce leukocyte adhesion, extravasation, and tissue injury associated with IBD. For that reason, in this article, we examined the effects of pravastatin, a water-soluble statin HMG-CoA reductase inhibitor, on the course of experimental colitis in mice (using the dextran sulfate model).
Materials and Methods
Animals.
The animals used in this study were C57BL/6 (wild-type eNOS) and eNOS−/− (eNOS “knockout”) mice (B6.129P2-NOS3Tm1Unc). These animals were obtained from The Jackson Laboratory (Bar Harbor, ME). All mice were males at 8 to 10 weeks of age (at the beginning of the trial; weight, 23–25 g). They were kept in an environmental room at 24°C with a controlled 12-h light/dark cycle and given free access to a standard pellet diet and water. The mice were kept in cages containing up to five animals and allowed to acclimate for at least 7 days before initiating these experiments.
Induction of Colitis.
Dextran sulfate sodium (DSS) colitis was induced by adding DSS to the drinking water, as previously described (Umene et al., 1994; Taniguchi et al., 1998; Wu and Ling, 1998; Soriano et al., 2000) (n = 9) and DSS + pravastatin (n = 9) groups were administered 3% (w/v) DSS (molecule mass 44 kDa; TdB Consultancy AB, Uppsala, Sweden) in distilled water ad libitum. Control (n = 9) and pravastatin (n = 9) groups received distilled water without DSS.
Pravastatin Administration.
Pravastatin [1 mg/kg, diluted in phosphate-buffered saline (PBS)] or vehicle (PBS alone) was injected intraperitoneally. After, mice were injected daily with pravastatin (in pravastatin alone and DSS + pravastatin groups) or vehicle (the control and DSS alone group).
Evaluation of Clinical Colitis.
In all animals, daily weight, presence of gross blood, and daily stool consistency were determined as previously described. Disease activity index (DAI) was measured as the combined scores of 1) weight loss, 2) stool consistency, and 3) bleeding divided by 3. Each score was determined as follows: by change in weight (0, ≤1%; 1, 1–5%; 2, 5–10%; 3, 10–15%; 4, >15%), hemoccult positivity (0, negative; 2, positive) or gross bleeding (4), and stool consistency (0, normal; 2, loose stools; 4, diarrhea), as previously described (Kim and Berstad, 1992).
Evaluation of Colonic Epithelial Permeability.
Colonic epithelial permeability was assessed by the penetrance of Evans blue (EB; Sigma-Aldrich, St. Louis, MO) from the lumen into the wall of colon on 10th day after administration of DSS. In all animals, the surgical procedure was performed under anesthesia. The proximal colon was ligated at the cecum, and EB was perfused through the colon for 15 min, followed by a 10-min wash with PBS. Perfused colons were dissected free, and the loops were opened and rinsed with 6 mMN-acetylcysteine dissolved in PBS (to clear the tissues of mucus and any adventitiously adsorbed EB). The loop was weighed and extracted overnight in 1 ml ofN,N-dimethyl-formamide (DMF) at 25°C. The absorbance of the extracted EB/DMF was determined at 620 nm using a Titertek MMC/340 plate reader (MTX Lab Systems, Inc., Vienna, VA).
Histological Analysis.
Distal colon samples were fixed in Zamboni's fixative overnight and embedded in JB-4 (Polysciences, Warrington, PA). Five-micrometer sections were stained with hematoxylin/eosin and scored (in a blinded fashion) by a gastrointestinal pathologist (Dr. S. Bharwani). Histological damage was scored using the criteria, described by Cooper et al. (1993). The crypt was scored on 0 to 4 grade [grade 0, intact crypt; grade 1, loss of the basal one-third of the crypt; grade 2, loss of two-thirds of the crypt; grade 3, loss of entire crypt with the surface epithelium remaining intact; grade 4, loss of the entire crypt and surface epithelium (erosion)], and these changes were quantitated as to the percentage involvement by the disease process: 1, 1 to 25%; 2, 26 to 50%; 3, 51 to 75%; 4, 76 to 100%. Crypt damage score was determined as the sum of the grade of the crypt and percent area score. The inflammation was evaluated subjectively on a 0 to 3 grade, and the extent of involvement estimated as: 1, 0 to 25%; 2, 26 to 50%; 3, 51 to 75%; 4, 76 to 100% of the total surface area. The inflammation score was determined as the sum of the inflammation grade and the percent extent score.
Cholesterol Measurement.
Serum cholesterol in mouse serum samples was determined at the LSU Health Science Center Clinical Hematology laboratory (using the cholesterol oxidase/reductase method; Johnson and Johnson, New Brunswick, NJ).
Expression of Endothelial MAdCAM-1.
Tissue samples from distal colon samples were embedded in Tissue-Tek O.C.T. compound (Sakura Finetek, Torrance, CA) was frozen at −20°C. Ten-micrometer sections were cut by cryostat. Nonspecific staining was blocked by incubating samples in normal donkey serum (10%; Sigma-Aldrich) diluted in antibody diluent (Biogenex, San Ramon, CA) for 30 min at 25°C. Sections were incubated in 1° antibody (rat anti-MAdCAM-1; 10 μg/ml) (1 h, 25°C), washed in PBS (three times; 10 min), and incubated in 2° antibody (diluted 1:200) goat anti-rat conjugated to Cy3 (Jackson ImmunoResearch Laboratories, West Grove, PA). After tissues were incubated with 2° antibody, they were washed in PBS (3×, 10 min) and mounted in 10 μl of Vectashield mounting medium (Vector Laboratories, Burlingame, CA) to minimizing photo-bleaching. MAdCAM-1-positive vessels in the lamina propria were counted in three consecutive colon sections from in five mice (n = 5) for each treatment. Data are expressed as the average number of vessels ± standard error per section.
Analysis of Colonic eNOS mRNA Expression by RT-PCR.
eNOS message expression was determined in colon tissue using a semiquantitative reverse transcription-polymerase chain reaction (RT-PCR) on 10th day after administration of DSS. Total RNA was isolated from distal colon tissue using TRIzol reagent (Invitrogen, Carlsbad, CA). One-microgram of RNA was reverse-transcribed to complementary DNA using MuLV Reverse Transcriptase (Applied Biosystems, Foster City, CA) and amplified using the following primers for mouse eNOS gene: sense primer, 5′-GCAGAAGAGTCCAGCGAACA-3′ and antisense primer, 5′-GGCAGCCAAACACCAAAGTC-3′. Thermal cycle conditions were 94°C for 30 s, 58°C for 30 s, and 72°C for 30 s, for a total of 30 cycles. The final cycle was followed with a 5-min incubation at 72°C. PCR amplification of a housekeeping gene (GAPDH) was performed using the same cDNA reaction. RT-PCR products were viewed by ethidium bromide staining and analyzed by densitometry using an Alpha Innotech gel documentation system (San Leandro, CA). eNOS mRNA expression was illustrated by determining the ratio of band intensity of eNOS and GAPDH and are presented as a percentage of controls.
Statistical Analysis.
Results are expressed as means ± S.E. Significant differences were assessed by the Fisher's PLSD test.P values <0.05 were accepted as statistically significant.
Results
Severity of DSS—Induced Colonic Injury.
The average weight of the mice used in this study was 24.1 ± 0.3 g (day 0). Body weight was monitored for 10 days. Histological parameters were evaluated in day 10 tissue samples. Clinically, a progressive loss of body weight (b.wt.), hematochezia, and diarrhea were noted after the 3rd day following administration of 3.0% DSS. Following induction of colitis, b.wt. was significantly decreased only in the DSS group. A significant decrease in b.wt. of 28.2 ± 1.2% in the DSS group was seen compared with the control group. Pravastatin-treated mice receiving DSS, however, lost only 18.8 ± 2.5% body weight. Although this was significantly different from the control group, it was significantly less than that in the DSS group (Fig.1).

Body weight change of mice in DSS/pravastatin model. Body weight of mice treated with pravastatin was not significantly different from controls. Dextran sulfate significantly reduced body weight (★, p < 0.05); this decrease in body weight was minimized by treatment with pravastatin (★,p < 0.05 from DSS; #, p < 0.05 from control). One-way ANOVA with Fisher's PLSD test.
Several other indices of colitis were observed at different times following DSS. Occult blood was commonly observed after 3 days on DSS, diarrhea appeared at day 7, and hematochezia observed on day 8 in DSS-treated mice. A progressive increase in the overall DAI was observed in the DSS-treated mice. An increase of the DAI to 4.0 ± 0 in the DSS group was markedly greater than that observed in the DSS + pravastatin group (3.1 ± 0.3 point) on the 10th day of DSS administration (Fig. 2). We set the parameters of this study to include animals with body weight within two standard deviations of the group mean (by this standard, one animal was excluded from the DSS group).
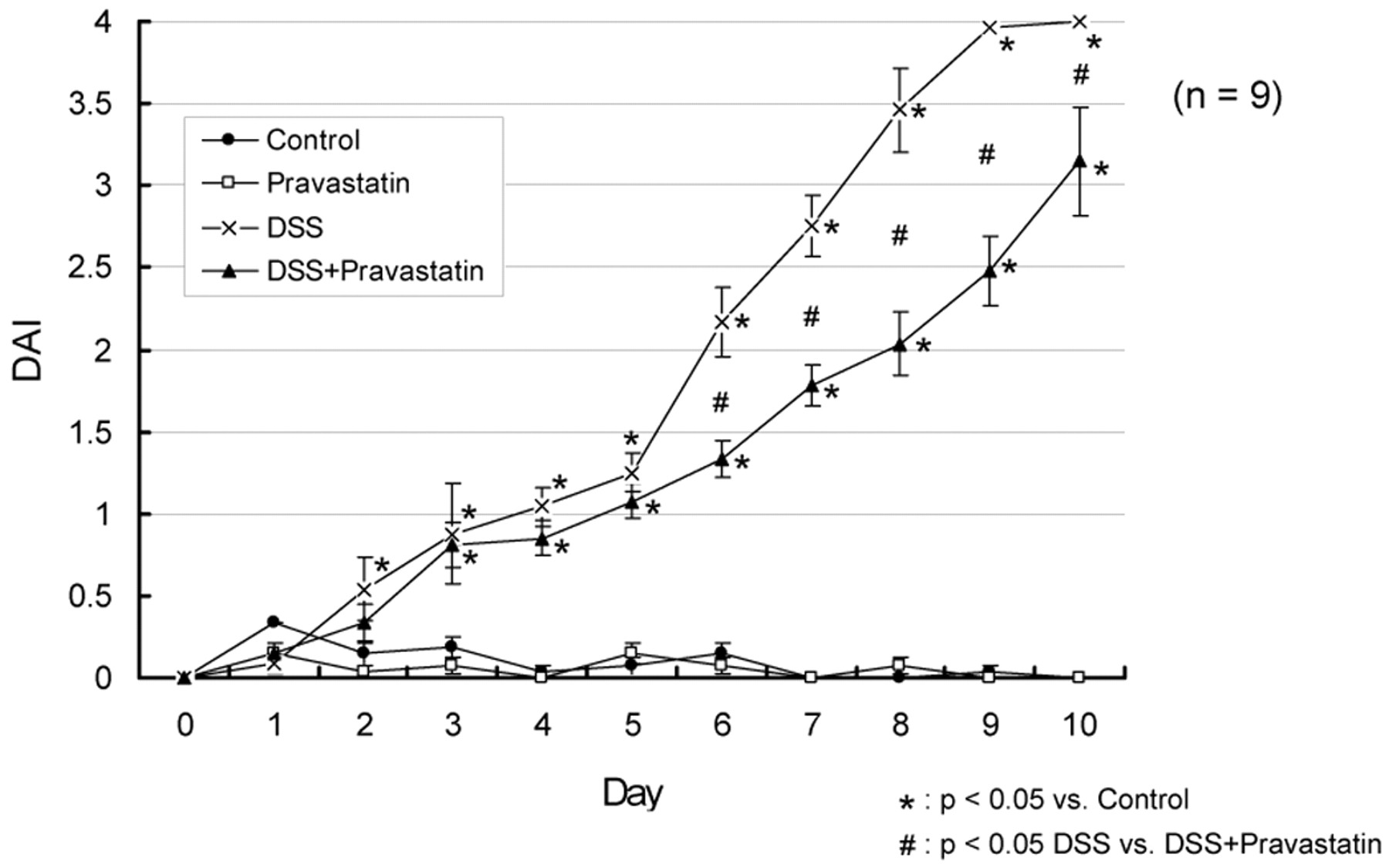
DAI in mice treated with DSS/pravastatin. The cumulative disease activity index included body weight, stool characteristic, and fecal blood on a scale of 0 to 4. Compared with control (untreated) mice, mice receiving DSS showed a significant increase in disease activity (★, p < 0.05), which was significantly but not completely attenuated by a treatment with pravastatin (#, p < 0.05). Pravastatin alone did not promote disease. One-way ANOVA plus Fisher's PLSD test.
Colonic Epithelial Mucosal Permeability.
The colonic permeability to EB albumin was used as an index of colon epithelial permeability. Permeability was expressed as the measured absorbance 620 nm/g of DMF-extracted distal colon tissue. Colonic epithelial permeability to EB was significantly increased in DSS-treated mice (11.0 ± 3.3) compared with untreated controls. Importantly, the DSS-induced increase in epithelial permeability associated with induction of colitis was blocked (4.6 ± 2.9) by treatment with pravastatin (1 mg/kg) (Fig. 3).

Distal colon protein permeability. Distal colon epithelial permeability to Evans blue was significantly increased in mice treated with DSS (★, p < 0.05 from control) that was attenuated by treatment with pravastatin (#,p < 0.05 from DSS). Pravastatin alone did not alter permeability. One-way ANOVA plus Fisher's PLSD test.
Measurement of Colon Length.
Decreased colon length in DSS-treated mice was observed, which was significantly different between DSS and control mice. Colon length in controls was 79.1 ± 2.8 mm (measured from rectum to jejunum). The length of the colon in the DSS group (46.0 ± 1.3 mm) was markedly shorter than that in the DSS + pravastatin group (51.3 ± 0.8 mm) (Fig.4) and shows that pravastatin partially reduces the colon shortening associated with induction of DSS colitis.

Pravastatin prevents DSS-induced colon shortening. Treatment with DSS induced significant shortening of the colon (★,p < 0.05 versus control), which was partially prevented by treatment with pravastatin (#, p < 0.05 versus DSS). One-way ANOVA plus Fisher's PLSD test.
Histological Analysis.
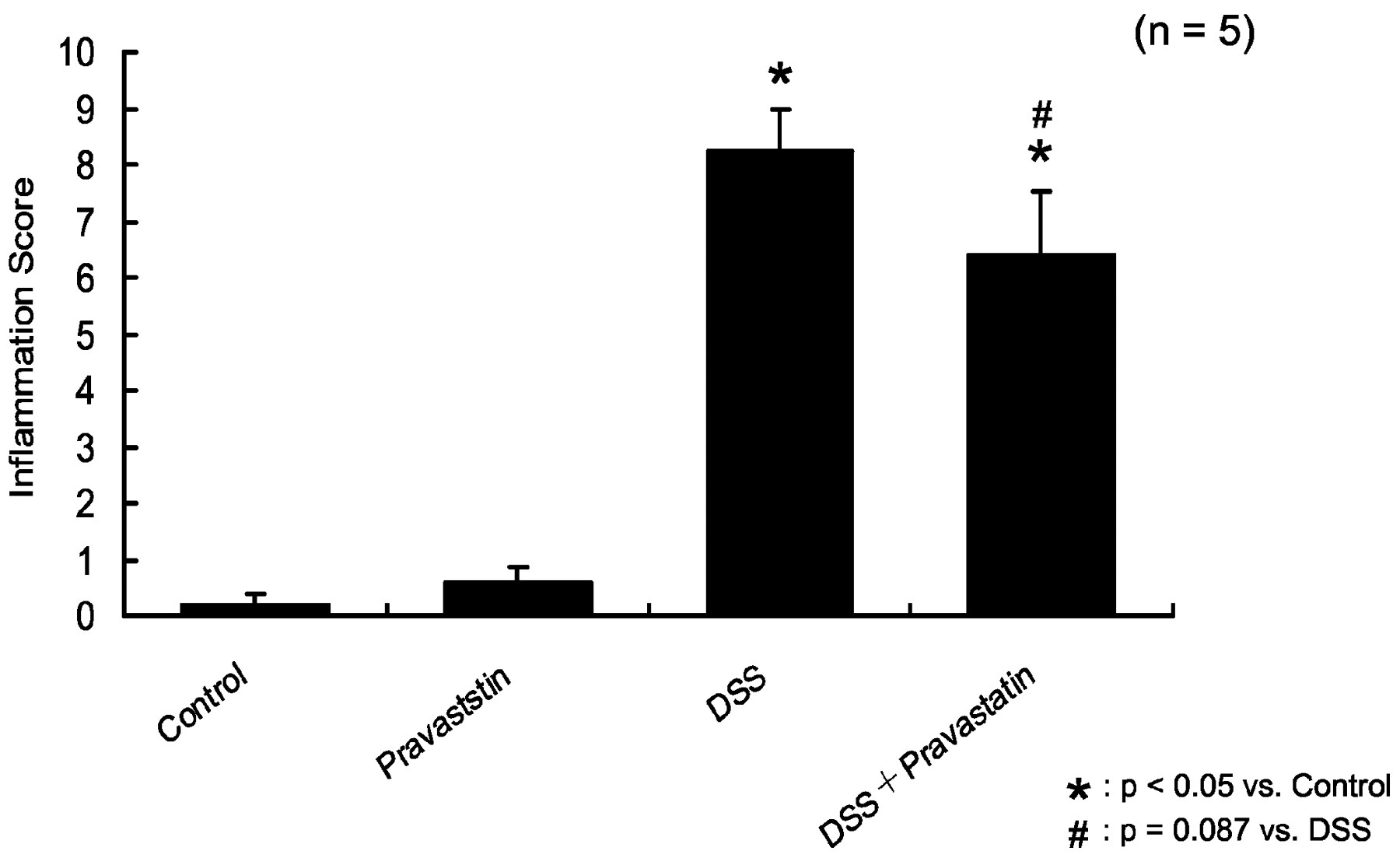
Figure 5, a and b, show histologically normal colon structure in control (5a) and pravastatin-treated mice (5b) shows similar, normal colonic architecture. Colon structure following DSS was characterized by severe disintegration of tissue architecture, edema, and a massive, mixed immune cell infiltrate (mononuclear cells, neutrophils, and eosinophils) with ulcerations and large areas of complete epithelial denudation, and muscular thickening (Fig. 5c). Conversely, animals cotreated with pravastatin showed an improvement in colonic histology (Fig. 5d). The inflammation score (Fig.6) and crypt damage score (Fig.7) were marginally improved in pravastatin-treated DSS mice. Inflammation was significantly increased by DSS (8.3 ± 0.8; *p < 0.05 versus control) was marginally reduced by pravastatin (6.4 ± 1.1; #p= 0.087 versus DSS; Fig. 6). DSS also induced significant crypt damage (*p < 0.05 versus control; Fig. 7). Crypt damage was also marginally reduced by pravastatin (12.5 ± 2.4 versus 7.4 ± 2.6) (*p < 0.05 versus control;#p = 0.056 versus DSS).

Pravastatin blocks DSS-induced changes in colon histology. Control colon histology (a) is dramatically altered by DSS and leads to mucosal thickening, leukocyte infiltration, crypt damage, and loss of goblet cells (c). Pravastatin alone (b) does not significantly affect colon histology but protects against the DSS induced destruction of the colon and maintains epithelial integrity (d); however, pravastatin does not reduce mucosal thickening.

Pravastatin reduces inflammation score. Although pravastatin does not alter the inflammation score compared with controls, it marginally reduced the inflammation produced by DSS treatment. ★, p < 0.05 versus control; #,p = 0.087 versus DSS. One-way ANOVA plus Fisher's PLSD test.

Pravastatin reduces crypt damage score. Although pravastatin does not alter the crypt damage score compared with controls, it marginally reduced the crypt damage produced by DSS treatment. ★, p < 0.05 versus control; #,p < 0.056 versus DSS. One-way ANOVA plus Fisher's PLSD test.
MAdCAM-1 Immunohistochemistry.
Figure8 shows the immunohistochemical staining of MAdCAM-1 in distal colon sections collected on day 10. Figure 8c (DSS group) shows strong MAdCAM-1 staining on several enlarged vessels in the mucosa; this staining pattern was eliminated by coadministration of pravastatin (Fig. 8d). Much less staining was observed in controls and in pravastatin-treated mice (Fig. 8, a and b). Figure9 shows the average number of MAdCAM-1-possitive vessels in the lamina propria of the distal colon. DSS administration dramatically increased the number of MAdCAM-1-positive vessels in the colonic lamina propria (50.4 ± 4.7) compared with controls (7.3 ± 1.0). Cotreatment with pravastatin significantly reduced the number of MAdCAM-1-positive vessels (26.2 ± 1.8) compared with DSS treatment. There were no differences in the number of MAdCAM-1-positive vessels between pravastatin-treated and control mice (Fig. 9).

Pravastatin reduces MAdCAM-1 induction in the inflamed colon. Obvious MAdCAM-1 staining was not seen in controls or in pravastatin treated samples (a and b, respectively) but was remarkably enhanced in the mucosa of DSS-treated animals (c). The increase in MAdCAM-1 staining was reduced but not eliminated by pravastatin treatment (d).
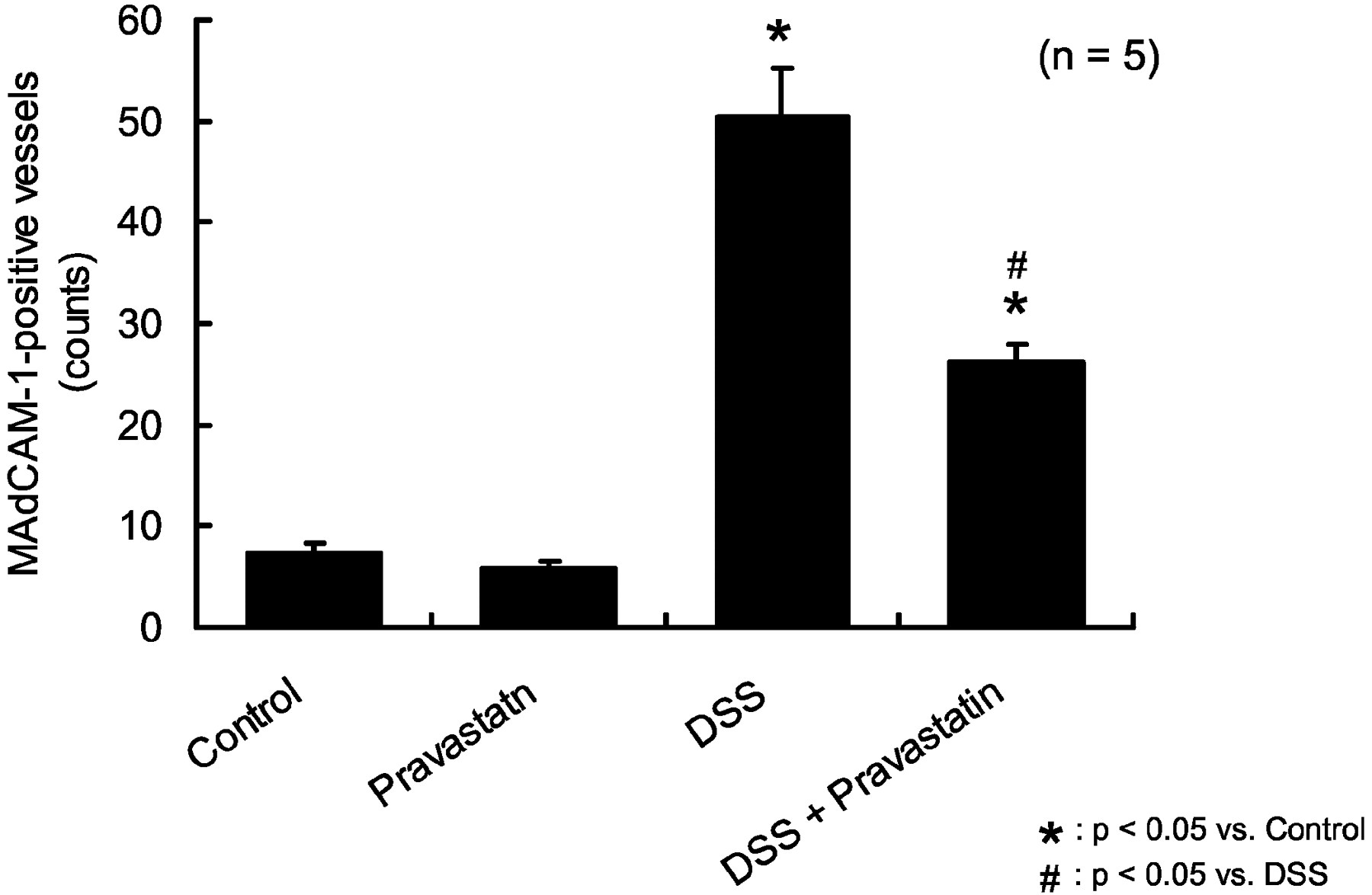
Pravastatin blocks the DSS-induced increase in MAdCAM-1-positive blood vessel staining. The number of MAdCAM-1-positive vessels in the lamina propria of DSS-treated colon dramatically increased (50.4 ± 4.7) compared with untreated controls (7.3 ± 1.0). Cotreatment with pravastatin significantly attenuated this increase in the number of MAdCAM-1-positive vessels (26.2 ± 1.8). There were no differences in the number of MAdCAM-1-positive vessels between pravastatin-treated and control-treated mice. ★, p < 0.05 versus control; #, p < 0.05 versus DSS. One-way ANOVA plus Fisher's PLSD test.
Serum Cholesterol.
Pravastatin administration had no effect of serum cholesterol (79.5 ± 2.9 mg/ml) compared with nontreated control mice (83.0 ± 4.1 mg/ml). DSS-treated mice showed significantly higher serum cholesterol (102.3 ± 4.6 mg/ml) compared with control, and those cotreated with pravastatin showed no remarkable increase compared with control.
Colonic eNOS mRNA Expression.
To assess the role of eNOS in DSS-induced colitis and/or pravastatin administration, RT-PCR for eNOS was performed in gut tissue samples. Pravastatin administration increased eNOS mRNA expression 17.7% over control levels. The DSS-treated group showed a 65.1% decrease in eNOS mRNA expression, whereas DSS mice cotreated with pravastatin showed only a small reduction (−11.7%) in eNOS mRNA.
The Effect of Pravastatin on DSS—Induced Colonic Injury on eNOS Knockout Mice.
To confirm that the mechanism of pravastatin in the reduced injury in DSS-induced colitis was eNOS-dependent, eNOS knockout mice were administered DSS and/or pravastatin, as described above. The DSS-treated eNOS knockout mice showed identical features of clinical colitis as control mice. Body weight in eNOS knockouts was significantly reduced (81.4 ± 1.1%), and DAI significantly increased (3.85 ± 0.17) at day 10. Decreased colon length in DSS-treated eNOS-knockout mice was also observed. Nevertheless, wild-type eNOS mice showed significant protection against DSS, and the administration of pravastatin to eNOS-knockout mice provided no measurable protection against the loss of body weight, DAI, or colon shortening (Table 1), demonstrating the dependence of this effect on functional eNOS.
The eNOS−/− model
Discussion
HMG-CoA reductase inhibitors are used mainly to treat hypercholesteremia. The HMG-CoA reductase blockers known as statins prevent the synthesis of cholesterol at the mevalonate and provide significant protection against coronary artery disease (Shepherd et al., 1995; Treasure et al., 1995) stroke (Delanty and Vaughan, 1997) and ischemic injury (Lefer et al., 1999). In addition to the effects of statins on cholesterol, statins also preserve endothelial function in animal models (Osborne et al., 1989) and in humans (Levine et al., 1995; Treasure et al., 1995; Kinlay et al., 1996).
High levels of serum cholesterol precede the clinical atherosclerosis and have been suggested to contribute to endothelial dysfunction (Verbeuren et al., 1986; Osborne et al., 1989). Hypercholesteremia exacerbates tissue injury after ischemia before the existence of atherosclerosis (Tilton et al., 1987). Cholesterol-mediated endothelial dysfunction is correlated with the decreased bioavailability of endothelial-derived NO, which could reflect high levels of oxidant generation by endothelium (which would consume NO), or decreased endothelial production of NO or both. Therefore, the statins ability to block cholesterol accumulation within endothelial cells alone logically was initially thought responsible for preserving functions like NO synthetic capacity.
NO regulates multiple events in chronic inflammation that may be important in both experimental and human forms of colitis. NO blocks leukocyte-endothelial adhesion (Kubes et al., 1991) by preventing the synthesis of endothelial cell adhesion molecules, e.g., MAdCAM-1 (Oshima et al., 2001) VCAM-1, ICAM-1, E-selectin (De Caterina et al., 1995), and P-selectin (Davenpeck et al., 1994). These endothelial adhesion molecules are mobilized in colitis in response to the cytokines found in the gut during active human and animal IBD models (Shigematsu, 1998; Connor et al., 1999; Kawachi et al., 2000a,b,c;Krieglstein and Granger, 2001; Shigematsu et al., 2001). Therefore, therapies to increase NO bioavailability may limit injury in inflammatory conditions at least in part by blocking expression of these adhesive determinants.
In our colitis model, increased mRNA for eNOS in the colons of pravastatin-treated mice suggests that the prevention of MAdCAM-1 induction following DSS, and the accompanying infiltration of the mucosa by leukocytes could in fact be limited by NO derived from eNOS.
The role of NO in IBD is still unsettled. NO both enhances, and blocks some types of chronic inflammation depending on the synthase and location (Binion et al., 1998, 2000; Krieglstein et al., 2001). For example, endothelial iNOS produce protective levels of NO within the gut to reduce inflammation (Binion et al., 1998, 2000). In several articles, Binion et al. (1998, 2000) argue that after repeated rounds of injury/repair, the gut microvasculature sustains a loss of “differentiated” functions, including iNOS expression, which in these cells, reduces leukocyte adhesion.
Nevertheless, in experimental animal models of colitis, particularly the DSS model, NO synthesized by high levels of cytokine-induced endothelial iNOS or iNOS in leukocytes does not appear to protect but rather contributes to development of disease. Krieglstein et al. (2001)showed that transfer of wild-type or iNOS−/− bone marrow to wild-type or iNOS-deficient mice (producing chimera with iNOS deficient in blood cells) or only in tissues (e.g., endothelium) produced protection in all models where iNOS was deficient (Lefer et al., 1999). Although these results are compelling, since knockout animals may have unknown mechanisms that compensate iNOS deficiency, these studies may need further investigation, possibly using animals treated with highly selective iNOS blockers (e.g., 1400ω).
We did not evaluate iNOS expression in our model of colitis; however, pravastatin did increase eNOS mRNA levels and also prevented DSS-induced colonic injury. Therefore, maintenance or enhancement of at least one form of nitric-oxide synthase appears to reduce tissue injury. In any case, it is doubtful that statins, like pravastatin, work through iNOS; statins have not been described as modifiers of either iNOS synthesis or activity and is therefore unlikely that pravastatin effects reflect mostly changes in iNOS metabolism.
Currently, the protective effects of statins in chronic inflammation are now mainly attributed to effects on eNOS. Lefer et al. (1999)suggest that statins (e.g., pravastatin) augment the endothelial NO synthetic capacity (limiting leukocyte adhesion and dependent tissue injury). Fluvastatin (another statin) also decreased human monocyte CD11b expression and adhesion to the endothelium, independent of effects on cholesterol (Weber et al., 1997). CD11b, the α-chain of the β2-integrins, helps firmly adhere leukocytes to the endothelium and was also blocked by statins. This group (Lefer et al., 1999) also demonstrated that simvastatin attenuated neutrophil CD18 mobilization following coronary ischemia. Statins also inhibit neutrophil and monocyte chemotaxis (Dunzendorfer et al., 1997). Therefore, the decreased leukocyte infiltration observed in our present study could as well reflect protective effects on the colon microvasculature and possibly effects of statins on leukocyte functions. Still, in this study, pravastatin only slightly reduced neutrophil infiltration. It is possible that at higher doses of pravastatin a greater reduction of neutrophil infiltration might be observed.
Initially, we carried out pilot experiments in mice with the objective of showing that relatively low doses of pravastatin would be nontoxic to the strain of mice chosen for this study. In those pilot experiments, animals were administered only 0.2 mg/kg pravastatin per day. This low dose of pravastatin produced no overt signs of toxicity or organ injury at this relatively low dose (which is equivalent to 20% of the dose typically given to treat hypercholesteremia in humans). Importantly, pravastatin at this low dose did appear to provide some protection against DSS induced colitis. At day 15, body weight loss was 16.0 ± 3.5% in the DSS group but only 8.1 ± 3.6% in the DSS + pravastatin group, which was a significant reduction (7.9%). Similarly, disease activity was 3.3 ± 0.3 in the DSS group but only 2.2 ± 0.4 in the DSS + pravastatin group, again significant prevention (by 1.1 in the disease activity). Based on these preliminary data (at 0.2 mg/kg), we performed the full current study at 1 mg/kg dose, which is roughly equivalent to the human regimen for this statin when used as anticholesteremic.
In the endothelium, statins stabilize endothelial NO synthase mRNA, increase levels of eNOS, and promote NO bioavailability (Mital et al., 2000). Statins like pravastatin also lead to a net activation eNOS independent of eNOS levels. Statins will activate the Akt/protein kinase B system (Luo et al., 2000) to phosphorylate the eNOS serine 1179 residue stimulating NO formation (Fulton et al., 1999) seen in shear stress/PKA activation (Boo et al., 2002). RT-PCR found a slightly increased colon tissue eNOS expression in pravastatin-treated mice (pravastatin and DSS + pravastatin) compared with untreated animals. In these animals, there was also reduced staining for MAdCAM-1 in the colitic bowel (DSS + pravastatin-treated animals versus DSS alone), consistent with lower leukocyte entrapment promoted by statins. Since statins reduce hypoxic inhibition of NOS activity (Endres et al., 1998), they may preserve blood flow to the ischemic bowel sometimes seen in some forms of IBD, e.g., ulcerative colitis (Guslandi et al., 1998).
Besides effects of statins on endothelium and leukocytes, statins also increase eNOS levels in platelets (Tannous et al., 1999). We did not evaluate platelet contributions, but statin effects on platelets eNOS might further reduce tissue injury by blocking platelet-endothelial adhesion and platelet-leukocyte aggregation, two steps in ischemic tissue injury (Krieglstein and Granger, 2001; Salter et al., 2001).
Statins increase both eNOS and tissue NO levels (Shepherd et al., 1995;Treasure et al., 1995), and our results suggest an increase in eNOS mRNA levels following pravastatin. The dependence of the protective effects of statins in our model are also supported by findings that eNOS-deficient mice (unlike wild-type mice) showed no protection against DSS when given pravastatin (Shepherd et al., 1995). These latter results appear consistent only if pravastatin effects are mediated by enhanced eNOS activity. These data are not entirely conclusive, however, and leave the door open for further study of other NOS isoforms.
Lastly, we must consider one current report that lipophilic statins like fluvastatin and lovastatin increase iNOS in cardiomyocytes (Ikeda et al., 2001). These findings may be cardiac tissue-specific; other groups have reported either no effect (Tannous et al., 1999) or measure a reduction in iNOS with statins (Park et al., 2000). Taken together, our results suggest pravastatin protection is eNOS-dependent and unrelated to cholesterol metabolism. Statins maintain endothelial function and microvascular homeostasis against chronic inflammation, e.g., DSS colitis. Importantly, since statins are relatively safe and well tolerated, it is likely that they could have important future applications in the treatment of IBD and other inflammatory and tissue injury conditions (e.g., arthritis and lupus).
Footnotes
-
DOI: 10.1124/jpet.102.044099
- Abbreviations:
- IBD
- inflammatory bowel disease
- NO
- nitric oxide
- NOS
- nitric-oxide synthase
- eNOS
- endothelial nitric-oxide synthase
- iNOS
- inducible nitric-oxide synthase
- HMG
- 3-hydroxy-3-methylglutaryl
- DSS
- dextran sulfate sodium
- PBS
- phosphate-buffered saline
- DAI
- disease activity index
- EB
- Evans blue
- DMF
- N,N-dimethyl-formamide
- RT-PCR
- reverse transcription-polymerase chain reaction
- PLSD
- protected least significant difference
- MAdCAM-1
- mucosal addressin cell adhesion molecule-1
- ANOVA
- analysis of variance
- Received September 6, 2002.
- Accepted November 19, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}