


Abstract
The benzamide derivative amisulpride shows a unique therapeutic profile being antipsychotic, at high doses, and disinhibitory, at low doses, while giving rise to only a low incidence of extrapyramidal side effects. In vitro, amisulpride has high affinity and selectivity for the human dopamine D2(Ki = 2.8 nM) and D3(Ki = 3.2 nM) receptors. Amisulpride shows antagonist properties toward D3 and both pre- and postsynaptic D2-like dopamine receptors of the rat striatum or nucleus accumbens in vitro. At low doses (≤10 mg/kg) amisulpride preferentially blocks presynaptic dopamine autoreceptors that control dopamine synthesis and release in the rat, whereas at higher doses (40–80 mg/kg) postsynaptic dopamine D2receptor occupancy and antagonism is apparent. In contrast, haloperidol is active in all of these paradigms within the same dose range. Amisulpride preferentially inhibits in vivo binding of the D2/D3 antagonist [3H]raclopride to the limbic system (ID50 = 17 mg/kg) in comparison to the striatum (ID50 = 44 mg/kg) of the rat, increases striatal and limbic tissue 3,4-dihydroxyphenylacetic acid levels with similar potency and efficacy, and preferentially increases extracellular 3,4-dihydroxyphenylacetic acid levels in the nucleus accumbens when compared to the striatum. Haloperidol shows similar potency for the displacement of in vivo [3H]raclopride binding in striatal and limbic regions and preferentially increases striatal tissue 3,4-dihydroxyphenylacetic acid levels. The present data characterize amisulpride as a specific dopamine receptor antagonist with high and similar affinity for the dopamine D2 and D3 receptor. In vivo, it displays a degree of limbic selectivity and a preferential effect, at low doses, on dopamine D2/D3 autoreceptors. This atypical profile may explain the therapeutic efficacy of amisulpride in the treatment of both positive and negative symptoms of schizophrenia.
Clinically, atypical neuroleptics are defined as drugs active in the treatment of schizophrenia but with a lesser propensity than conventional neuroleptics to induce extrapyramidal side effects. Furthermore, some neuroleptics, such as clozapine, are considered atypical because of their therapeutic efficacy in the treatment of schizophenic patients resistant to conventional neuroleptics.
Most neuroleptics display high affinity for the dopamine D2receptor subtype in direct relation to their therapeutic potency or plasma concentration at therapeutically active doses (Seeman, 1992). It has thus been suggested that the atypical characteristics of certain neuroleptics necessarily derive from additional pharmacological properties, such as antagonism toward 5-HT2A or 5-HT2C, muscarinic cholinergic or alpha-1 adrenergic receptors (Meltzer, 1991; Schmidt et al., 1995), or an interaction with ς recognition sites (Ferris et al., 1991).
Molecular biological techniques have recently provided evidence that the dopamine D1 receptor comprises a class of receptors, positively coupled to adenylate cyclase, that includes, besides the classical D1 receptor (also termed D1A), the D5 or D1B receptor (Sunahara et al., 1991) and possibly the mammalian equivalents of the D1C(Sugamori et al., 1994) and D1D (Demchyshynet al., 1995) receptors. Similarly, the dopamine D2 receptor family is now thought to include the D2, D3 (Sokoloff et al., 1990) and D4 (Van Tol et al., 1991) subtypes. In particular, the D3 and D4 receptor subtypes have generated recent interest as potential therapeutic targets in the treatment of schizophrenia because of their preferential limbic localization (Sokoloff et al., 1990; Van Tol et al., 1991). The D3 subtype is selectively recognized by several agonists and antagonists known for their selectivity for the dopamine autoreceptor (Sokoloff et al., 1992a, c), although it is present mainly on dopaminoceptive cells, in particular in the limbic system (Sokoloff et al., 1990, 1992c). Thus it has been suggested that stimulation of a postsynaptic dopamine D3 receptor might be inhibitory to rat locomotor activity (Svensson et al., 1994; Waters et al., 1993b). The D4 receptor represents the subtype of D2receptors for which clozapine shows the highest affinity (Van Tolet al., 1991).
Amisulpride (fig. 1) is a substituted benzamide derivative with dopamine receptor antagonist properties in vitro and in vivo (Chivers et al., 1988;Perrault et al., 1997; Scatton et al., 1994;Sokoloff et al., 1990). Although in clinical studies its efficacy in the treatment of schizophrenia has been clearly demonstrated, its most notable characteristic is its atypical profile. Thus, although amisulpride is efficacious in treating the positive symptoms of schizophrenia, it does so at doses (400–1200 mg/day) that, according to present experience, have only a low propensity to induce extrapyramidal side effects. In addition, amisulpride possesses disinhibitory effects at lower doses (50–300 mg/day) and is successfully used, at these doses, in treatment of the negative symptoms of schizophrenia and of dysthymia, a form of chronic depression (Boyer et al., 1990; Boyer et al., 1995; Delcker et al., 1990; Paillere Martinot et al., 1995; Saletu et al., 1994). These atypical therapeutic characteristics of amisulpride may be a reflection of its atypical neuropharmacological profile. Thus, amisulpride, similarly to classical neuroleptics, antagonizes the hyperactivity or stereotypies that result from the direct or indirect activation of postsynaptic dopaminergic receptors by (high doses of) dopaminomimetics such as apomorphine or amphetamine in the rat (Perrault et al., 1997). Nevertheless, amisulpride does not provoke catalepsy in the rat, which is characteristic of postsynaptic D2 blockade, even at doses maximally effective in these experimental paradigms (Perraultet al., 1997). At lower doses, amisulpride potentiates apomorphine- and amphetamine-induced stereotyped behavior in mice, particularly favoring a transition from sniffing to gnawing behavior (Vasse et al., 1985). Furthermore, low doses of amisulpride inhibit hypokinesia induced by the administration of low doses of apomorphine, 7-OH-DPAT or quinpirole in rats (Perrault et al., 1997). Classical neuroleptics such as haloperidol are devoid of such prostereotypic effects and inhibit the diverse behavioral effects of dopamine agonists within the same dose range.
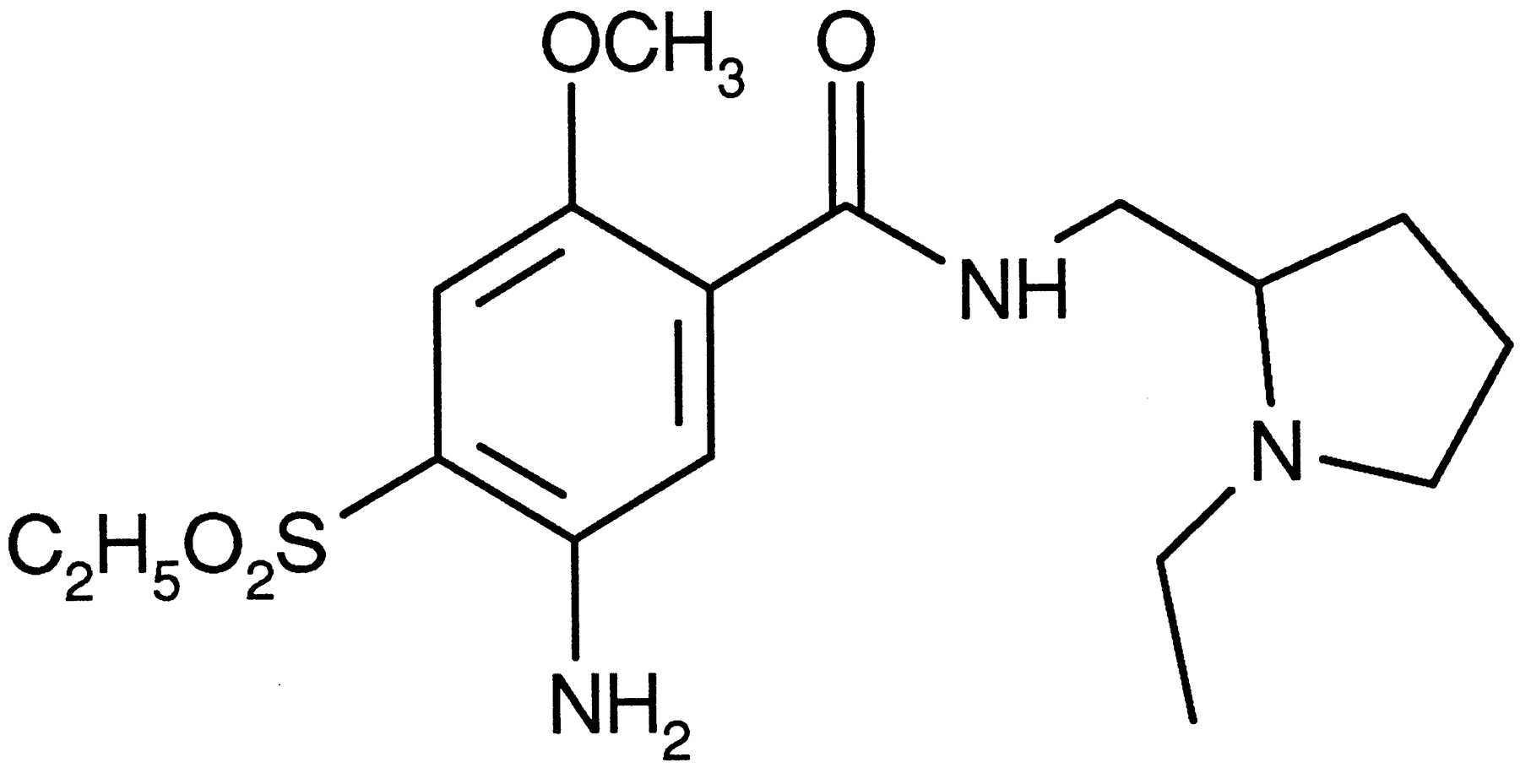
Amisulpride, (±)-4-amino-N-1((1-ethyl-2-pyrrolidinyl)methyl)-5-(ethylsulphonyl)-2-methoxybenzamide.
Various hypotheses have been put forward to explain the spectrum of animal behaviors provoked by dopaminomimetics and the dichotomy in their antagonism by atypical neuroleptics. These include the presence of behaviorally suppressant dopamine receptors in the nucleus accumbens (Waters et al., 1993b) or frontal cortex (Carter and Pycock, 1980) and the presence of inhibitory dopamine autoreceptors (Di Chiaraet al., 1986). Thus, in addition to postsynaptic dopamine receptors, D2-like autoreceptors have been amply demonstrated and are thought to be involved in the regulation of neuronal firing (Lejeune and Millan, 1995), dopamine release (Suaud-Chagny et al., 1991) and tyrosine hydroxylase activation (Claustre et al., 1985; Walters and Roth, 1976). Their molecular identity, however, remains subject to debate. Alternatively, drug activity at nondopaminergic receptors, such asalpha-1 adrenoceptors (Wiszniowska-Szafraniec et al., 1983), 5-HT receptor subtypes (Carter and Pycock, 1981;Mogilnicka et al., 1977), beta adrenergic receptors (Costall et al., 1978), muscarinic cholinergic receptors (Christensen et al., 1976) and histamine H1 receptors (Dadkar et al., 1976), may modulate the expression of dopamine receptor activation or blockade.
In view of the established atypical profile of amisulpride in the treatment of schizophrenia and dysthymia, and in view of current hypotheses with respect to dopamine receptor subtypes and the possible contribution of nondopaminergic effects to neuroleptic atypicity, we have sought to define its neurochemical characteristics and mechanism of action, largely in comparison with that of the typical neuroleptic haloperidol. To this end, we studied the interaction of amisulpride with a variety of drug receptor and recognition sites in vitro, which demonstrated that amisulpride is highly specific and selective for both the dopamine D2 and D3receptor subtypes. Its antagonist character was demonstrated in vitro by its inhibition of dopamine D3receptor-mediated mitogenesis and by its pre- and postsynaptic effects on neurotransmitter release and was demonstrated in vivo by its effects on indices of dopamine synthesis, release and metabolism and on ACh levels. These studies show that amisulpride displays a degree of limbic selectivity and a preferential effect, at low doses, on D2/D3 autoreceptors as compared with the classical neuroleptic, haloperidol.
The behavioral characteristics of amisulpride are described in a companion paper (Perrault et al., 1997).
Some of the data presented here have previously been reported in abstract form (Scatton et al., 1994; Schoemaker et al., 1995).
Materials and Methods
Animals.
Unless otherwise indicated, adult male Sprague-Dawley rats (OFA or COBS, Iffa Credo, St. Germain sur l’arbresle, France, or Charles River, St. Aubin-les-Elbeus, France), Dunkin Hartley guinea pigs (Iffa Credo) and Fauves de Bourgogne rabbits (ESD, Romans, France) were used.
Throughout these studies, the phrase the limbic systemrefers to the nucleus accumbens and the olfactory tubercle.
Materials and drugs.
Pig choroid plexi and bovine caudate nuclei were obtained from CollectOrgane (Paris, France). Cell lines expressing the human dopamine receptor subtypes or membrane preparations thereof were obtained from: D1 and D5 (New England Nuclear, Boston, MA), D2S and D3 (Dr. J.-C. Schwartz, INSERM, Paris, France), D4.4 (Receptor Biology, Baltimore, MD).
Radioligands were obtained from the following sources: [3H]spiperone, [3H]SCH 23390, [3H]GBR12935, [3H]prazosin, [3H]clonidine, [3H]dihydroalprenolol, [3H]desipramine, [3H]serotonin, [3H]quipazine, [3H]quinuclidinyl benzylate, [3H]pyrilamine, [3H]flumazenil, [3H]TBOB, [3H]nipecotic acid, [3H]strychnine, [3H]CGP 39653, [3H]glycine, [3H]MK801, [3H]AMPA, [3H]kainate, [3H]angiotensin II, [5-methyl-3H]-nitrendipine, [benzoyl-2,5-3H]-batrachotoxinin A 20-α-benzoate, [3H]Ro5-4864, [3H]raclopride, [3H]dopamine, [14C]choline, [125I]angiotensin II (New England Nulear/DuPont de Nemours, Boston, MA); [125I]iodosulpride, [3H]7-OH-DPAT, [3H]mesulergine, [3H]GR 113808, ((−)N6-R[G-3H]-phenylisopropyladenosine, 5′-N-ethylcarboxamido [8(n)-3H]-adenosine, [3H]idazoxan, [3H]thymidine (Amersham, Little Chalfont, UK); [3H]8-OH-DPAT (CEA, Saclay, France); [3H]GABA (Dositek, Orsay, France). [3H]ifenprodil was custom-synthesized by Amersham. Amisulpride, sulpride, haloperidol, remoxipride, clozapine and 7-OH-DPAT were synthesized at Synthélabo Recherche (Bagneux, France). All other chemicals were obtained commercially at the highest purity available.
Drugs were administered through the i.p. route, except when indicated otherwise; control groups received an equal volume of the corresponding vehicle. Amisulpride was administered as a hydrochloride salt. Doses refer to the free base equivalent.
Radioligand binding studies in vitro.
Studies of radioligand binding to various receptor and/or drug recognition sites were performed essentially as described by the authors indicated: the human dopamine D2 receptor expressed in CHO cells (1.0 nM [125I]iodosulpride; Sokoloff et al., 1992a), the human dopamine D3 receptor expressed in CHO cells (0.2 nM [125I]iodosulpride; Sokoloff et al., 1992a), the human dopamine D4.4 receptor expressed in CHO cells (0.5 nM [3H]spiperone; Van Tol et al., 1991), the human dopamine D1 receptor in Sf9 cells (1.6 nM [3H]SCH23390; Sunahara et al., 1991), the human dopamine D5 receptor expressed in Sf9 cells (1.8 nM [3H]SCH23390; Sunahara et al., 1991), the dopamine D1 receptor in rat striatum (0.3 nM [3H]SCH23390; Billard et al., 1984), the dopamine D2 receptor in rat striatum (0.3 nM [3H]spiperone; Briley and Langer, 1978), the dopamine D3 receptor in bovine caudate nucleus (0.8 nM [3H]7-OH-DPAT in the presence of 0.2 μM eliprodil;Schoemaker, 1993), the plasma membrane dopamine transporter from the rat striatum (1 nM [3H]GBR12935; Janowsky et al., 1986), the alpha-1A adrenoceptor in the rat salivary gland (0.2 nM [3H]prazosin; Faure et al., 1994), the alpha-1B adrenoceptor in the rat liver (0.1 nM [3H]prazosin; Faure et al., 1994), thealpha-2 adrenoceptor in the rat cerebral cortex (5 nM [3H]clonidine; Pimoule and Langer, 1982), thebeta adrenoceptor in the rat cerebral cortex (2 nM [3H]dihydroalprenolol; Mogilnicka et al., 1980), the plasma membrane noradrenaline transporter from the rat vas deferens (2 nM [3H]desipramine; Raisman et al., 1982), the 5-HT1A receptor in rat hippocampus (1 nM [3H]8-OH-DPAT in the presence of 3 μM paroxetine;Sanger and Schoemaker, 1992; Schoemaker and Langer, 1986), the 5-HT1B receptor in rat striatum (5 nM [3H]5-HT in the presence of 100 nM 8-OH-DPAT and 100 nM mesulergine; Herrick-Davis et al., 1988; Heuring and Peroutka, 1987; Peroutka, 1986), the 5-HT1D receptor in bovine caudate nucleus (2 nM [3H]5-HT in the presence of 100 nM 8-OH-DPAT and 100 nM mesulergine; Herrick-Davis et al., 1988; Heuring and Peroutka, 1987), the 5-HT2Areceptor in rat cerebral cortex (0.4 nM [3H]spiperone;Hicks et al., 1984), the 5-HT2C receptor in the pig choroid plexus (1 nM [3H]mesulergine; Pazos et al., 1984; Yagaloff and Hartig, 1986), the 5-HT3receptor in the rat cerebral cortex (0.8–0.9 nM [3H]quipazine in the presence of 10 nM ketanserin and 100 nM paroxetine; Angel et al., 1993), the 5-HT4receptor in the guinea pig striatum (0.1 nM [3H]GR 113808; Grossman et al., 1993), the muscarinic cholinergic receptor in the rat cortex (0.3 nM [3H]quinuclidinyl benzylate; Yamamura and Snyder, 1974), the histamine H1receptor in the guinea pig cerebellum (1 nM [3H]pyrilamine; Tran et al., 1978), the GABAA receptor in rat brain (4 nM [3H]GABA;Langer et al., 1985), the ω1/benzodiazepine receptor in the rat cerebellum (1 nM [3H]flumazenil;Arbilla et al., 1985), the ω2/benzodiazepine receptor in the rat spinal cord (1 nM [3H]flumazenil;Arbilla et al., 1985), the ω5/benzodiazepine receptor in the rat hippocampus (1 nM [3H]flumazenil in the presence of 5 μM zolpidem; Tan and Schoemaker, 1993), the picrotoxin site of the GABAA receptor channel in the rat cerebral cortex (2 nM [3H]TBOB; Van Rijn et al., 1990), the GABAB receptor in rat brain (10 nM [3H]GABA; Hill and Bowery, 1981), the presynaptic GABA transporter in the rat cerebral cortex (5 μM [3H]nipecotic acid; Vargas et al., 1993), the strychnine-sensitive glycine receptor in the rat spinal cord (2 nM [3H]strychnine; Young and Snyder, 1974; Young and Snyder, 1973), the glutamate recognition site of the NMDA receptor in whole rat brain (1.5 nM [3H]CGP 39653; Sills et al., 1991), the strychnine-insensitive glycine recognition site of the NMDA receptor in the whole rat brain (14–17 nM [3H]glycine;Kishimoto et al., 1981; Zukin et al., 1974), the polyamine-sensitive modulatory site of the NMDA receptor complex in the rat cerebral cortex (1 nM [3H]ifenprodil in the presence of 3 μM GBR 12909; Schoemaker et al., 1991), the NMDA receptor-ion channel site in well-washed membranes from the whole rat brain (2 nM [3H]MK801; Reynolds et al., 1987;Wong et al., 1988), the quisqualate/AMPA subtype of glutamate receptors in the rat brain (4 nM [3H]AMPA;Honoré et al., 1982; Honoré and Drejer, 1988), the kainate subtype of glutamate receptors in the rat brain (2 nM [3H]kainate; Simon et al., 1976), the adenosine A1 receptor in rat hippocampal membranes (3 nM (−)N6-R[G-3H]-phenylisopropyladenosine;Schwabe and Trost, 1980), the adenosine A2 receptor in rat striatal membranes (4 nM 5′-N-ethylcarboxamido[8(n)-3H]-adenosine in the presence of 50 nM N6-cyclopentyladenosine; Bruns et al., 1986), the angiotensin II AT1 receptor in rabbit adrenocortical membranes (2 nM [3H]angiotensin II;Glossmann et al., 1974), the angiotensin II AT2receptor in membranes from the rat adrenal medulla (0.1 nM [125I]angiotensin II; Glossmann et al., 1974), the L-type Ca++ channel in rat cerebral cortex (0.1 nM [5-methyl-3H]-nitrendipine; Schoemaker and Langer, 1989), the Na+ channel in rat cerebral cortex (2 nM [benzoyl-2,5-3H]-batrachotoxinin A 20-α-benzoate in the presence of 1 μM tetrodotoxin and scorpion toxin; Pauwels et al., 1986), ς recognition sites in rat cortex (0.5 nM [3H]ifenprodil; Schoemaker et al., 1991), the p-site (peripheral benzodiazepine receptor) in the rat kidney (0.5 nM [3H]Ro 5-4864; Schoemaker et al., 1983), the imidazoline I2 recognition site in the whole rat brain (1 nM [3H]idazoxan in the presence of 10 μM (−)adrenaline; Le Rouzic et al., 1995).
Radioactivity was quantified using liquid scintillation spectometry. Data from radioligand inhibition experiments were evaluated by graphical methods or by linear or nonlinear regression analysis when appropriate and, unless indicated otherwise, are presented as the drug concentration required to inhibit 50% of specific radioligand binding (IC50) or are converted to Ki values as described by Cheng and Prusoff (1973).
Dopamine D3 receptor-stimulated mitogenic activityin vitro.
The functional effects of amisulpride at the dopamine D3 receptor subtype were assessed as described byPilon et al. (1994) and Sautel et al. (1995). Briefly, the mitogenic response elicited in NG108-15 neuroblastoma-glioma cells stably transfected with human dopamine D3 receptor cDNA by the addition of 10 nM quinpirole in the presence of 1 μM forskolin was quantified by the incorporation of [3H]thymidine. Antagonism of quinpirole-induced mitogenesis was measured in the presence of increasing (0.1–100 nM) concentrations of amisulpride.
Radioligand binding studies in vivo.
In vivo [3H]raclopride binding to rat brain structures was measured according to a previously described procedure (Köhler et al., 1985). The radioligand (9 μCi/200 μl) was injected into the tail vein of male Sprague-Dawley rats 45 min before sacrifice. Test drug or vehicle was administered in a final volume of 1 ml 75 min before [3H]raclopride. Brain structures (striatum, limbic system and cerebellum) were dissected by hand, and the incorporated radioactivity was measured after overnight digestion in 0.5 ml of Soluene. The radioactivity incorporated into the cerebellum was taken as nonspecific binding.
[3H]Spiperone binding in vivo was studied according to a similar protocol. The radioligand (5 μCi/200 μl) was administered into the tail vein 60 min before sacrifice. Drug or vehicle was given 60 min before the radioligand.
Neurotransmitter release in vitro.
The modulation of electrically evoked [3H]dopamine and [14C]ACh release from slices (1 × 1 × 0.3 mm) of the rat striatum and nucleus accumbens was studied essentially as described by Arbilla and Langer (1984). Briefly, slices were incubated with 0.1 μM [3H]dopamine and 19 μM [14C]choline for 30 min at 37°C in Krebs buffer (mM: NaCl, 118; KCl, 4.7; CaCl2, 1.3; MgCl2, 1.2; NaH2PO4, 1.0; NaHCO3, 25.0; glucose, 11.0; EDTA, 0.04, equilibrated with 5% CO2/95% O2). Slices were then washed, transferred to superfusion chambers (1 slice/chamber) and superfused with Krebs buffer supplemented with 10 μM hemicholinium-3 for 60 min at a flow rate of 0.7 or 1 ml/min. After this period, fractions (7 and 6 min, respectively) were collected until the end of the experiment. Slices were initially stimulated electrically (2 min, 3 Hz, 2 ms, 16 mA) in the absence of amisulpride or 7-OH-DPAT, 74 min after the beginning of the superfusion, and were stimulated 40 min thereafter in their presence. Amisulpride or 7-OH-DPAT was added to the superfusion buffer 20 min before the second stimulation period. When the interaction between amisulpride and 7-OH-DPAT was studied, amisulpride was present in the superfusion buffer as of 20 min before the first stimulation period. In each case, the stimulation-evoked [3H] and [14C] overflow (S1 and S2, respectively) was calculated with respect to the spontaneous outflow (sp1 and sp2, respectively) in the fractions immediately before stimulation. Although both the neurotransmitter and derived metabolites probably contribute to stimulation-evoked [3H] and [14C] overflow (see, for instance,Parker and Cubeddu, 1985), the terms [3H]dopamine and [14C]ACh are used for convenience.
Microdialysis studies.
Adult male Sprague-Dawley rats were anesthetized with chloral hydrate (400 mg/kg), and guide cannulae were stereotaxically implanted onto the dura mater above the striatum and the nucleus accumbens (+0.7 mm anterior, +3 mm lateral and +1.7 mm anterior, +1.0 mm lateral from bregma, respectively) (Paxinos and Watson, 1982). At least 5 days after surgery, microdialysis probes (Carnegy Medicine) 250 μm in diameter with an exposed membrane length of 4 (striatum) and 2 (nucleus accumbens) mm were positioned within the guide cannulae (vertical coordinates: −7 mm and −8 mm, respectively, from the dura surface) and perfused with artificial CSF (mM: NaCl, 147; KCl, 4; CaCl2, 1.2; MgCl2, 1.0) using a CMA/100 pump (BAS) at a flow rate of 2 μl/min. Twenty-minute dialysate fractions were collected in a valve loop and analyzed online using HPLC with electrochemical detection. The average concentration of five stable fractions immediately preceding drug administration was defined as the 100% control value.
Electrically evoked dopamine release in vivo.
Presynaptic autoregulation of electrically evoked dopamine release in the rat olfactory tubercle was studied in vivo as described by Suaud-Chagny et al. (1991). Briefly, dopamine release was monitored every 1 s with electrochemically treated 12-μm-diameter carbon-fiber electrodes (1.6 mm lateral, 1.7 mm anterior to bregma and 9.0 mm below the cortical surface) combined with differential pulse amperometry with the final potential adjusted to +85 mV. Bipolar stimulating electrodes were implanted in the ascending dopaminergic pathway at 1.2 mm lateral and 4.0 mm posterior to bregma, at a depth adjusted for each experiment so that the stimulation-evoked dopamine response was maximal. Electrical stimulation was by twenty 1-s trains of six square wave form current pulses (pulse interval 70 ms, 250 μA, 0.5 ms) and was repeated every 10 min.
Monoamines and metabolites in vivo.
Two hours after drug administration, animals were sacrificed by decapitation, and the brain structures (frontal cortex, striatum and limbic system) were dissected on ice. Samples were then weighed, homogenized in 20 volumes of 0.1 M HClO4 and centrifuged at 10,000 ×g for 10 min. Dopamine and DOPAC levels were measured in the supernatant by HPLC with electrochemical detection as described previously (Sermerdjian-Rouquier et al., 1981). ACh and choline levels were measured in aliquots of a buffered supernatant (0.5 M Tris-citrate, pH 4) by HPLC with electrochemical detection using platinum electrodes (Asano et al., 1986).
Measurement of dopamine and 5-HT synthesis rates.
The rate of dopamine and 5-HT synthesis was estimated by measuring the accumulation of dopa and 5-HTP, respectively, 30 min after the administration of NSD-1015 (100 mg/kg) (Claustre et al., 1985). The presynaptic modulation of dopamine synthesis (Walters and Roth, 1976) was studied by co-administration of γ-hydroxy-butyric acid (750 mg/kg) 45 min before sacrifice. Amisulpride and 7-OH-DPAT were given 75 and 15 min, respectively, before γ-hydroxy-butyric acid. Dopa and 5-HTP levels were measured by HPLC with electrochemical detection according to Sermerdjian-Rouquier et al. (1981).
Statistical analyses.
Statistical differences between groups were assessed using the ANOVA test, followed by Dunnett’s or Duncan’s test where appropriate. Simple statistical comparisons were done using a two-tailed Student’s t test. ED50 values for drug inhibition of in vivo radioligand binding in the striatum and limbic system were tested for statistical identity using the partial F test.
Results
In vitro radioligand binding studies.
Amisulpride shows high affinity toward the cloned and stably transfected human dopamine D2 (Ki = 2.8 ± 0.4 nM; n = 7) and D3(Ki = 3.2 ± 0.3 nM; n = 7) receptor subtypes labeled with [125I]iodosulpiride and fails to recognize the D1, D4 and D5 receptor subtypes. Both haloperidol and clozapine recognize all human dopamine receptor subtypes (table1).
Affinity of amisulpride for molecularly identified human dopamine receptor subtypes in vitro
In agreement with its high affinity for the human D2 and D3 receptor subtypes, amisulpride potently inhibits radioligand binding to the native dopamine D2 receptor in membranes from the rat striatum (IC50 = 21 nM, table2). Similarly, amisulpride recognizes the native bovine dopamine D3 receptor labeled with [3H]7-OH-DPAT, at low concentrations (IC50 = 2.9 nM). The selectivity of amisulpride for dopamine receptors of the D2 family was studied using [3H]SCH23390 to label the D1 receptor in the rat striatum; amisulpride fails to affect the binding of this radioligand at concentrations up to 10 μM. In contrast, haloperidol inhibits radioligand binding to the native dopamine D2 receptor at lower concentrations than in the case of the D3 receptor and, in addition, recognizes the native D1 receptor subtype (table 2). Clozapine recognizes these three dopamine receptor subtypes with similar affinity. The dopamine D4 and D5 receptors are currently not accessible to radioligand binding studies in their native state.
Inhibitory effects of amisulpride against radioligand binding to native receptor and drug recognition sites in vitro
Amisulpride was devoid of significant affinity (IC50 > 1 μM) for radioligand binding to a variety of other neurotransmitter receptors and/or drug recognition sites (table 2), which attested to its selectivity. Both haloperidol and clozapine recognize with high affinity the 5-HT2A serotonergic and the alpha-1 adrenergic receptor subtypes. In addition, haloperidol inhibits radioligand binding to ς-sites, whereas clozapine affects binding to 5-HT2C, 5-HT3 and H1 histamine receptors.
In vivo radioligand binding studies.
[3H]Raclopride selectively recognizes D2 and D3 receptors in vitro and may be used as a label for D2-like receptors in vivo (Köhleret al., 1985). Amisulpride displaces [3H]raclopride binding in vivo (fig.2) with an ED50 value of 17.3 ± 1.86 mg/kg in the rat limbic system but is significantly (P < .05) less active in displacing binding in the striatum (ED50 = 43.6 ± 6.2 mg/kg; table 3). Like amisulpride, sulpiride is more potent in displacing [3H]raclopride binding in the limbic system, whereas remoxipride and haloperidol show similar activity in both brain regions.

Displacement by amisulpride of [3H]raclopride binding to the rat striatum and limbic system in vivo. Data represent specific [3H]raclopride binding, the cerebellum being used as a measure of nonspecific binding, and are the mean with S.E.M. of results obtained from six rats per group. Test drugs were administered 75 min before the radioligand.
Comparative potencies of several neuroleptics at inhibiting in vivo[3H]raclopride-specific binding in the rat striatum and limbic structures
Spiperone similarly recognizes dopamine D2 and D3 receptors (Sokoloff et al., 1990) and, in addition, labels the 5-HT2 receptor in vivo(Laduron et al., 1978). [3H]Spiperone binding to D2-like receptors in vivo is significantly (P < .05) more sensitive to inhibition by amisulpride in the limbic system than in the striatum (ED50 = 29 ± 5 and 87 ± 9 mg/kg, respectively; n = 5–14/group). [3H]Spiperone binding in the frontal cortex, which mainly represents 5-HT2 receptors (Laduron et al., 1978), is not affected by amisulpride at doses up to 100 mg/kg.
Dopamine D3 receptor-mediated mitogenesis in vitro.
Quinpirole (10 nM) stimulated [3H]thymidine incorporation, a measure of mitogenesis, in NG108-15 cells stably transfected with the human D3 dopamine receptor, to 171.9 ± 5.0% of controls (100.0 ± 4.6%; n= 3; P < .01). Although amisulpride (100 nM) failed to stimulate [3H]thymidine incorporation (95.8 ± 7.1%;n = 3; P > .05 vs. control), it inhibited quinpirole-elicited [3H]thymidine incorporation with an IC50 value of 22 ± 3 nM (n = 3).
Neurotransmitter release studies in vitro.
Dopamine D2/D3 receptor antagonism can be demonstratedin vitro by studying the modulation of neurotransmitter release. Thus the electrically stimulated [3H]dopamine release from slices of the striatum or the nucleus accumbens is subject to inhibitory modulation through a D2-like (D2/D3) terminal autoreceptor and is inhibited by the D2/D3 agonist 7-OH-DPAT with EC50 values of 13.9 ± 0.7 and 4.7 ± 0.6 nM, respectively. At maximally effective concentrations, 7-OH-DPAT reduces the evoked release to 3.7% and 13.8% of controls in the striatum and nucleus accumbens, respectively.
The effects of amisulpride were studied alone and against a concentration of 7-OH-DPAT that produces approximately 70% of its maximal inhibitory effect on evoked [3H]dopamine overflow (30 nM in the striatum, 10 nM in the nucleus accumbens). Amisulpride slightly but significantly increased [3H]dopamine release from slices of the rat striatum (S2/S1 = 0.88 ± 0.04 under control conditions, n = 6; 1.04 ± 0.08 in the presence of 100 nM amisulpride,n = 4; P < .05) and opposed the inhibitory effects of 7-OH-DPAT in both brain areas (fig. 3).

Effect of amisulpride on electrically evoked [3H]dopamine release from the rat striatum and nucleus accumbens in vitro. The effect of amisulpride on electrically evoked [3H]dopamine release was studied using slices prepared from the rat striatum and nucleus accumbens. Slices were initially stimulated electrically (2 min, 3 Hz, 16 mA) in the absence of amisulpride or 7-OH-DPAT and, 40 min thereafter, in their presence. Amisulpride or 7-OH-DPAT (30 nM for the striatum, 10 nM for the nucleus accumbens) was added to the superfusion buffer 20 min before the second stimulation period. When the interaction between amisulpride and 7-OH-DPAT was studied, amisulpride was present in the superfusion buffer as of 20 min before the first stimulation period. In each case, the stimulation-evoked [3H]-overflow (S1 and S2, respectively) was calculated with respect to the spontaneous outflow in the fractions immediately before stimulation (sp1 and sp2, respectively). Data are shown as the mean and S.E.M. of 3 to 11 observations. * and †: P < .05 compared with control and 7-OH-DPAT, respectively.
Electrically stimulated [14C]ACh (formed after preincubation with [14C]choline) release from slices of the rat striatum is inhibited through the stimulation of a postsynaptic D2 receptor (Arbilla and Langer, 1984). The dopamine D2/D3 agonist 7-OH-DPAT inhibited electrical stimulation-evoked [14C]ACh release from rat striatal slices with an IC50 value of 19.5 ± 4.7 nM to a maximum of 23% of control values. Amisulpride opposed the effects of 7-OH-DPAT, thus attesting to postsynaptic dopamine receptor blockade. Amisulpride concentration-response curves against the effects of 30 nM 7-OH-DPAT on [3H]dopamine release (EC50 = 2.2 ± 0.3 nM) and [14C]ACh (EC50 = 1.2 ± 0.3 nM) release are compared in figure 4. Amisulpride did not affect basal [3H]dopamine or [14C]ACh efflux at any concentration tested (data not shown).

Effect of amisulpride on electrically evoked [3H]dopamine and [14C]ACh release from the rat striatum in vitro. The effects of amisulpride (▪) on the 7-OH-DPAT (30 nM)-induced (○) inhibition of electrically evoked [3H]dopamine and [14C]ACh release were studied using slices prepared from the rat striatum. Data are shown as a percentage of the corresponding S2/S1 control (•) ratio ([3H]dopamine: 0.92 ± 0.02,n = 32; [14C]ACh: 0.90 ± 0.01,n = 37).
Effects on dopaminergic neurotransmission in vivo.
Tissue dopamine and DOPAC levels. The effects of amisulpride, haloperidol, sulpiride and clozapine on regional dopamine and DOPAC tissue levels are shown in table 4. The doses of the latter three reference compounds were chosen as those previously shown to have maximal effects on dopamine turnover in the striatum (Scatton et al., 1977).
Effects of amisulpride on dopamine and DOPAC levels in the rat brain
Only the highest dose of amisulpride (100 mg/kg) significantly reduced dopamine levels in the striatum or limbic system. No other neuroleptic significantly decreased dopamine levels in the striatum at the doses used. Sulpiride significantly decreased limbic dopamine levels by approximately 10%.
Amisulpride increased tissue DOPAC levels in a dose-dependent manner in all brain regions. This effect was significant (P < .05) from 2.5 to 100 mg/kg in the limbic system and from 10 to 100 mg/kg in the striatum. Apart from this difference in threshold sensitivity, no differences in the potency of amisulpride were observed between the two regions studied. The maximal effects of amisulpride (100 mg/kg) were similar in the limbic system and striatum (319% and 285% of controls, respectively).
Haloperidol significantly increased tissue DOPAC levels in both regions (P < .001). At the dose tested, its effects were more marked in the striatum (344%) than in the limbic (265%) system. The effects of sulpiride, like those of amisulpride, were of a similar order of magnitude in each region (striatum = 290%, limbic = 272%). Clozapine had a relatively minor effect on striatal (159%) and limbic (174%) tissue DOPAC levels.
Dopamine synthesis. The effects of amisulpride on tyrosine hydroxylase activity, the rate-limiting step in dopamine synthesis that is subject to negative-feedback modulation through dopamine autoreceptors (Claustre et al., 1985; Walters and Roth, 1976), were specifically addressed after inhibition of L-dopa decarboxylase by pretreatment with NSD-1015 (100 mg/kg).
Amisulpride significantly increased the synthesis of dopamine, as measured by the accumulation of dopa, in the rat striatum and limbic system at doses of 20 and 100 mg/kg (table 5). It shows a relative selectivity for the limbic system (ED50 = 18.6 ± 4.7 mg/kg) as compared with the striatum (ED50= 43.7 ± 6.5 mg/kg). At the dose of 100 mg/kg, its maximal effects appear similar to those of haloperidol (0.3 mg/kg, table 5).
Effects of amisulpride on dopamine synthesis in the rat brain
In order to isolate presynaptic regulatory mechanisms, pharmacologically, animals were additionally pretreated with γ-hydroxy-butyrate to block neuronal impulse flow (Claustre et al., 1985). Under these conditions, dopa accumulation is increased to approximately 210% and 115% of controls in the striatum and limbic system, respectively. Amisulpride (0.5–75 mg/kg, table6) fails to provoke an additional increase in dopa accumulation in the striatum but slightly accelerates, at 75 mg/kg, dopamine synthesis in the limbic system. In both brain regions, haloperidol modestly but significantly stimulates the accumulation of dopa.
Effects of amisulpride on dopamine synthesis in the rat brain after blockade of impulse flow
Activation of terminal dopamine autoreceptors by the administration of 7-OH-DPAT (0.082 mg/kg s.c.) to γ-hydroxy-butyrate-pretreated animals results in a decrease in dopa accumulation in both regions (table7). Amisulpride opposes the effects of 7-OH-DPAT with ED50 values of 10.6 ± 5.2 and 10.4 ± 6.4 mg/kg in the striatum and limbic system, respectively.
Effects of 7-OH-DPAT and amisulpride on dopamine synthesis in the rat brain after blockade of impulse flow
Effects on extracellular dopamine and DOPAC as measured by microdialysis. In comparison with vehicle-treated controls, amisulpride (10 mg/kg) increases extracellular dopamine levels, measured by the technique of microdialysis coupled to online HPLC with electrochemical detection, in both the striatum and nucleus accumbens (fig. 5). Maximal levels, approximately 150% of controls, are reached within 60 min of drug administration. Amisulpride, administered at the dose of 30 mg/kg, produced similar effects (data not shown). A concomitant but slightly delayed increase in extracellular DOPAC levels in the striatum and the nucleus accumbens reached, 140 min after drug administration, 137 ± 7% and 200 ± 22%, respectively. The effect of amisulpride on dialysate DOPAC levels appeared greater in the nucleus accumbens than in the striatum.

Effects of amisulpride on extracellular dopamine and DOPAC levels, as measured by microdialysis, in the rat striatum and nucleus accumbens. Extracellular dopamine and DOPAC levels in the rat striatum (n = 4) and nucleus accumbens (n = 6) were measured after the administration of amisulpride (10 mg/kg i.p.), using the microdialysis technique. Dialysates were collected as 20-min fractions and analyzed by online HPLC coupled to electrochemical detection. Data are shown as a percentage of basal outflow, taken over five stable fractions immediately before drug administration, and represent the mean and S.E.M.
In comparison, the effects of haloperidol were studied at a dose (0.03 mg/kg) that yields an occupancy (21%–30%) of [3H]raclopride-labeled D2-like receptorsin vivo similar to that produced by 10 mg/kg of amisulpride (19%–37%). Compared with vehicle-treated controls, haloperidol increased extracellular dopamine concentrations to 140% to 150% of basal values in both brain regions (fig. 6). Like amisulpride, haloperidol appeared to increase dialysate DOPAC levels to a greater extent in the nucleus accumbens than in the striatum.
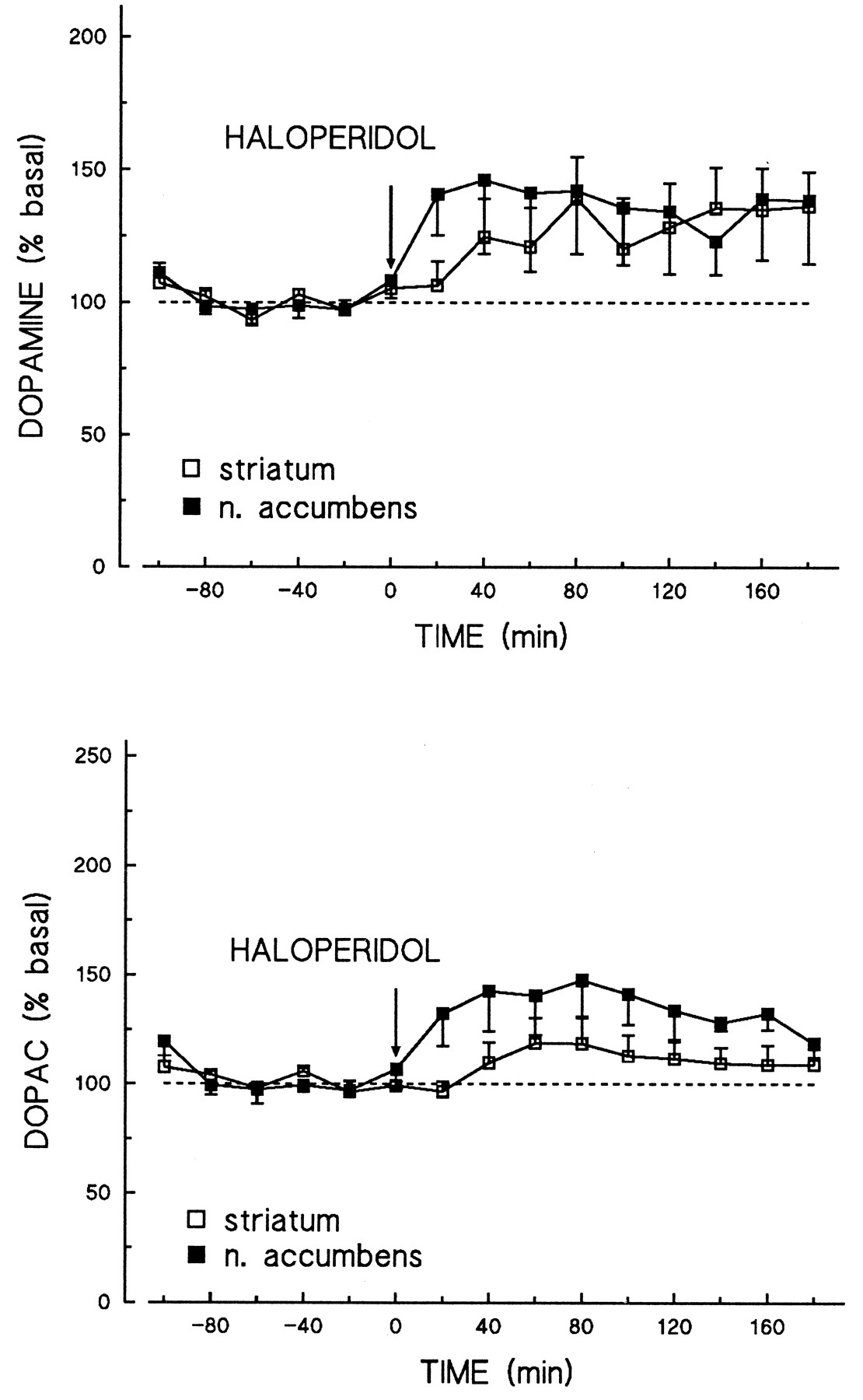
Effects of haloperidol on extracellular dopamine and DOPAC levels, as measured by microdialysis, in the rat striatum and nucleus accumbens. Extracellular dopamine and DOPAC levels in the rat striatum (n = 6) and nucleus accumbens (n = 6) were measured after the administration of haloperidol (0.03 mg/kg i.p.), using the microdialysis technique. Dialysates were collected as 20-min fractions and analyzed by on-line HPLC coupled to electrochemical detection. Data are shown as a percentage of basal outflow, taken over five stable fractions immediately before drug administration, and represent the mean and S.E.M.
Effects on stimulation-evoked dopamine release in the olfactory tubercle. The administration of amisulpride (0.5–15 mg/kg s.c.) provokes a time- and dose-dependent increase in the stimulation-evoked dopamine release, measured by differential pulse amperometry in the rat olfactory tubercle (fig. 7). Its maximal effect, obtained at a dose of 10 mg/kg, is similar to that previously seen after haloperidol (0.5 mg/kg s.c.; Suaud-Chagny et al., 1991).

Effects of amisulpride on the amplitude of evoked dopamine release in the rat olfactory tubercle in vivo. The effect of amisulpride was studied on dopamine release evoked by electrical stimulations of the ascending dopaminergic pathway, which were repeated every 10 min. Dopamine release was measured using an electrochemically treated carbon-fiber electrode implanted in the olfactory tubercle and was recorded every 1 s. Data are expressed as a percentage of the mean evoked dopamine release during the three stimulations immediately before s.c. drug administration, and represent the mean and S.E.M. of four (amisulpride) or six (vehicle) rats per group.
Evoked dopamine release at different doses of amisulpride, measured 90 min after drug administration and expressed as a percentage of vehicle-treated controls, is shown in figure 8. The ED50 value of amisulpride for its enhancement of stimulation-evoked dopamine release may be estimated as 3.7 mg/kg s.c.

Dose-response curve of the effect of amisulpride on stimulation-evoked dopamine release in the rat olfactory tuberclein vivo. The effect of amisulpride, administered at the dose of 0.5, 1, 3, 5, 10 and 15 mg/kg s.c., on dopamine release was studied as described in fig. 7. Shown is the effect, expressed as a percentage of the mean evoked dopamine release in vehicle-treated controls, of amisulpride 90 min after drug treatment (mean ± S.E.M.;n = 4/group).
Effects on striatal choline and ACh levels.
Amisulpride, sulpiride, haloperidol and clozapine did not affect striatal choline levels at the doses tested. Amisulpride decreased striatal ACh levels significantly at 30 and 100 mg/kg (87.5% and 56.3% of control levels, respectively). Haloperidol (2 mg/kg) and sulpiride (100 mg/kg) decreased striatal ACh levels to a similar extent (68% and 62% of controls, respectively), whereas clozapine (10 mg/kg) was without significant effect (table 8).
The effects of amisulpride, haloperidol, sulpiride and clozapine on rat striatal ACh and choline levels
Discussion
The present studies indicate that amisulpride, in comparison with other neuroleptics both typical and atypical, possesses complex neurochemical characteristics and can largely be defined as a specific dopamine receptor antagonist with a high degree of selectivity for the D2 and D3 receptor subtypes that it recognizesin vitro with similar affinity. In vivo, these properties translate into a degree of selectivity for the presynaptic dopamine autoreceptors that control the dopaminergic system and for its limbic projections.
Selectivity for the dopaminergic system.
The present data confirm and extend previous reports (Chivers et al., 1988) in showing that amisulpride is highly selective for the D2receptor family. Within this class of receptors, it recognizes the cloned human and native D2 (rat) and D3(bovine) subtypes with similar and low-nanomolar (Ki = 3 nM) affinity in vitro. In this respect, amisulpride is similar to drugs such as sulpiride, AJ76 and UH232 that show a D2/D3 affinity ratio close to unity and is different from the classical neuroleptics, such as haloperidol, which generally show higher affinity for the D2 than for the D3 receptor in vitro(Sokoloff et al., 1990, 1992a). Previous studies have shown that amisulpride recognizes the two isoforms of the human D2 receptor (D2S and D2L) with equal affinity (Malmberg et al., 1993).
The mitogenic response to dopamine agonists of NG108-15 cells stably transfected with human dopamine D3 receptor cDNA is one of the few functional effects unequivocally associated with this receptor subtype (Pilon et al., 1994; Sautel et al., 1995). In this test, amisulpride inhibited the stimulation of mitogenesis induced by quinpirole with a potency (IC50 = 22 nM) compatible with its affinity for the dopamine D3receptor but failed to stimulate mitogenesis when added alone. Thus amisulpride behaves as a full antagonist at the human dopamine D3 receptor.
Like other neuroleptics of the benzamide structure, amisulpride does not recognize the dopamine D4 receptor subtype (Van Tolet al., 1991), subtypes of the D1 receptor family (Sunahara et al., 1991) or the plasma membrane dopamine transporter.
Among the different 5-HT receptors studied, amisulpride recognizes only the 5-HT2A subtype, albeit with low affinity (IC50 = 2.0 μM). This observation agrees well with the IC50 (5.6 μM) reported by Chivers et al.(1988) against the 5-HT2A receptor labeled using [3H]ketanserin and with the observation that amisulpride fails to inhibit the binding of [3H]spiperone to the 5-HT2A receptor of the rat frontal cortex in vivo (ID50 > 100 mg/kg). In this respect, amisulpride thus differs from the majority of dopamine receptor antagonists,i.e., neuroleptics, that often do possess high affinity for this receptor (Meltzer et al., 1989; Stockmeier et al., 1993). On the basis of an analysis of the affinity of neuroleptics for the 5-HT2 receptor and their degree of atypicity, Meltzer et al. (1989) suggested that both properties are closely associated. Clearly, this conclusion does not apply to amisulpride.
Drug affinity for muscarinic cholinergic and alpha-1 adrenergic receptors or ς recognition sites has been suggested to contribute to antipsychotic therapeutic activity or atypicity (Ferriset al., 1991; Meltzer, 1991). Amisulpride fails to display significant affinity (IC50 > 1 μM) for any of these receptors, so they are not expected to contribute to its atypical profile as a neuroleptic.
Amisulpride does not display significant activity against radioligand binding to the alpha-2 adrenoceptor or the norepinephrine transporter, or to receptors involved in GABAergic or glutamatergic neurotransmission, the histamine H1 receptor, the strychnine-sensitive glycine receptor, adenosine receptor subtypes, the angiotensin AT1 and AT2 receptors, the Na+ or L-type Ca++ channel, p-sites (i.e., peripheral-type benzodiazepine) or I2imidazoline recognition sites.
Thus, much like other neuroleptics derived from the benzamide structure, such as sulpiride, remoxipride and raclopride (Chiverset al., 1988; Leysen et al., 1993), amisulpride is highly selective for the dopamine D2 receptor family. Within the D2 receptor family, amisulpride specifically recognizes its D2 and D3 subtypes with high and equal affinity in vitro. It seems reasonable to suggest that its neuropharmacological and clinical profile would derive from these characteristics.
Presynaptic dopamine autoreceptor selectivity.
As stated in the Introduction, amisulpride, like most neuroleptics, antagonizes the hyperactivity and stereotypies that result from the activation of postsynaptic dopaminergic receptors by (high doses of) direct- or indirect-acting dopaminomimetics such as apomorphine or amphetamine (Perrault et al., 1997). However, a characteristic feature of amisulpride is that at lower doses, it potentiates apomorphine- and amphetamine-induced stereotyped behavior (Vasse et al., 1985) and inhibits hypokinesia induced by the administration of low (presynaptic) doses of apomorphine, 7-OH-DPAT or quinpirole (Perraultet al., 1997). The aim of the present study was to examine whether the effects of low and high doses of amisulpride could be neurochemically discriminated at the level of dopaminergic neurotransmission.
The IC50 values of 7-OH-DPAT for inhibiting electrically evoked [3H]dopamine and [14C]ACh release from slices of the rat striatum in vitro are similar within experimental limits. Similarly, the EC50 values of amisulpride in opposing the effects of 7-OH-DPAT (30 nM) in this model were indistinguishable (2.2 and 1.2 nM, respectively). Together, these observations suggest a close pharmacological similarity between these populations of pre- and postsynaptic dopamine receptors. Gifford and Johnson (1993), using quinpirole as the agonist in an otherwise similar experimental approach, came to an identical conclusion using AJ-76 and UH-232, dopamine antagonists for which a selectivity for the dopamine autoreceptor has been amply demonstrated in vivo (Johanssonet al., 1985; Svensson et al., 1986; Waterset al., 1993a, 1994). Thus, for reasons that remain to be explored, this in vitro model may not be fully representative of in vivo drug effects.
As a second approach to characterizing the interaction of amisulpride with presynaptic dopaminergic systems, we studied its effects on extracellular dopamine levels, using the microdialysis technique coupled to online HPLC analysis and electrochemical detection. Amisulpride at low doses (10 mg/kg) increased dialysate dopamine levels in both striatum and nucleus accumbens. Its effects at this dose were similar to those of haloperidol at 0.03 mg/kg, a dose expected to yield an occupancy of D2-like receptors in vivosimilar to that produced by amisulpride at 10 mg/kg.
The effects of amisulpride on terminal dopamine autoreceptors that modulate impulse flow-dependent release in the rat olfactory tubercle were studied by differential pulse amperometry in vivo. Because of the experimental design, where the impulse flow is imposed by electrical stimulation of the ascending pathways, the modulation of dopamine release as measured does not depend on postsynaptic or somatodendritic autoreceptors but takes place at the terminal level (Suaud-Chagny et al., 1991). The design is thus different from that in the microdialysis technique as employed, where extracellular dopamine levels depend on the modulation of spontaneous neuronal firing and dopamine release through somatodendritic as well as terminal autoreceptors. Dopamine release evoked by electrical stimulation of the ascending pathways was inhibited by the mixed dopamine agonist apomorphine with an ED50 value of 70 μg/kg, as well as by the D2/D3 receptor agonist quinpirole, whereas maximally effective doses of haloperidol (0.5 mg/kg) and sulpiride (50 mg/kg) increased stimulation-evoked dopamine release 4- to 5-fold (Suaud-Chagny et al., 1991). Amisulpride similarly increased the amplitude of dopamine release, its maximal effect (a 5-fold increase) being observed at 10 mg/kg. Amisulpride was approximately 3 times as active as sulpiride in this respect and showed an ED50 value of 3.7 mg/kg. It should be noted that the absolute potencies of both agonists (apomorphine) and antagonists (amisulpride, haloperidol) are fully within the range for behavioral effects thought to be mediated by presynaptic dopamine autoreceptors (Perrault et al., 1997), which further attests to the validity of the model.
Thus it is clear that amisulpride behaves as an antagonist toward presynaptic dopamine receptors that modulate [3H]dopamine release in vitro as well as in vivo. At higher doses, amisulpride possesses characteristics found in classical neuroleptics. Thus amisulpride occupies postsynaptic D2-like receptors in the rat striatum, labeled using [3H]raclopride or [3H]spiperone (ED50 = 44 and 87 mg/kg, respectively), and stimulates dopamine turnover, increasing tissue DOPAC levels with similar potency and effectiveness in the striatum and limbic system. Its maximal effects were similar to those of haloperidol and sulpiride and greater than those of clozapine. Although striatal ACh levels were significantly decreased only from 30 mg/kg amisulpride, inspection of the relative overall dose-response curves for the effects of amisulpride on tissue DOPAC and ACh levels does not suggest any critical differences in potency. Both phenomena thus are likely to be related.
In a result consistent with its stimulation of dopamine turnover, amisulpride increased the rate of dopamine synthesis (ED50= 20–40 mg/kg), as reflected by the increase in dopa accumulation after the administration of NSD-1015. It is also likely that in this model, its effects were not mediated through presynaptic receptor occupancy at the level of the dopamine nerve terminal because they were largely eliminated by pretreatment with γ-hydroxy-butyrate (table 6), although we cannot rule out the possibility that low synaptic concentrations of dopamine after impulse-flow blockade prevented expression of a presynaptic component that might contribute under control conditions. In contrast, haloperidol increased dopa accumulation in the absence as well as in the presence of γ-hydroxy-butyrate, which suggests that its effects on dopamine synthesis are mediated by postsynaptic (as evidenced in the absence of γ-hydroxy-butyrate) as well as presynaptic (as evidenced in the presence of γ-hydroxy-butyrate) mechanisms.
7-OH-DPAT decreased the rate of dopa accumulation after pretreatment with NSD-1015 and γ-hydroxy-butyrate. Under those conditions where impulse flow was interrupted, amisulpride opposed the effects of 7-OH-DPAT with an ED50 value of approximately 10 mg/kg. The fact that, particularly in the striatum, amisulpride failed to increase dopa accumulation after NSD-1015 and γ-hydroxy-butyrate pretreatment yet opposed the effects of 7-OH-DPAT needs to be explored further. It may be that synaptic concentrations of dopamine are too low after blockade of neuronal activity to allow dopamine autoreceptor antagonism to be expressed. A low endogenous DA tonus may similarly explain why γ-hydroxy-butyrate administration caused only a small, nonsignificant increase in dopa accumulation in the limbic system, even though the effects of 7-OH-DPAT clearly demonstrated the presence of synthesis-modulating DA autoreceptors in this area. Alternatively, the modulation of dopamine synthesis by endogenous dopamine and the exogenous agonist 7-OH-DPAT may be mediated at least in part through different (extrajunctional?) dopamine receptor subtypes, differentially recognized by amisulpride in vivo. Recent evidence suggests that the molecularly defined D3 receptor might be a candidate for this dopamine autoreceptor subtype (Meller et al., 1993; Nissbrandt et al., 1995). Pharmacological studies in vitro (Aretha and Galloway, 1996) and in vivo (Aretha et al., 1995) support this hypothesis. In particular, the selective dopamine D3 receptor antagonist (+)-S 14297 fails to affect dopamine synthesis in vivo, as assessed by its effects on tissue DOPAC/dopamine ratios, when studied alone, but opposes the inhibitory effects of 7-OH-DPAT on this parameter (Gobert et al., 1995).
Thus all available data suggest that, in vivo, amisulpride affects presynaptic parameters of dopaminergic neurotransmission at doses lower than those that block postsynaptic dopamine receptors. These data are in full agreement with the observations that amisulpride preferentially blocks behavioral effects thought to be associated with the stimulation of presynaptic dopamine receptors (Perrault et al., 1997).
Selectivity for limbic D2/D3receptors.
It has been suggested that the atypical character of certain neuroleptics arises from a greater effect on the limbic system, which is thought to be involved in emotional and cognitive processes, than on the extrapyramidal system, which is intimately related to the control of motor behavior (Bischoff, 1992; Meltzer, 1993; Scatton and Zivkovic, 1985).
By using [3H]raclopride, a high-affinity radioligand for dopamine D2 and D3 receptors, we have shown that amisulpride as well as its analog sulpiride, but not haloperidol or remoxipride, displayed a preferential affinity for limbic dopamine D2 (+ D3) receptors. Using the nonbenzamide dopamine antagonist [3H]spiperone as the radioligand, we confirmed the limbic selectivity of amisulpride. These observations are consistent with previous in vivo binding studies using [3H]spiperone (Bischoff, 1992). At first, the regional differences in displacing potencies of amisulpride would not seem to be related to a selective inhibition of [3H]raclopride binding of a particular dopamine receptor subtype, because in vitro, amisulpride displays a similar affinity for D2and D3 dopamine receptors. Moreover, the low density and restricted distribution of the mRNA coding for the D3subtype (Sokoloff et al., 1990, 1992b) suggests that the presence of dopamine D3 receptors cannot account for thein vivo pharmacological differences between limbic and striatal [3H]raclopride binding sites. Nevertheless, the dose-ratio of amisulpride for the occupation of D2 and D3 receptors in vivo remains to be established. In addition, and even though D3 mRNA is expressed at low levels, the D3 receptor density may be equal to that of the D2 receptor in the nucleus accumbens (Booze and Wallace, 1995).
Microdialysis studies point to a second regionally specific effect of amisulpride. Thus, although amisulpride at low doses (10 mg/kg) produced an essentially similar increase in striatal and limbic dialysate dopamine levels, dialysate DOPAC levels were increased to a greater extent in the limbic system. Although the origin of such differential effects remains unknown, it has been reported that the dopamine receptors that control extracellular dopamine and DOPAC levels may be localized at the terminal and somatodendritic levels, respectively. Thus the local infusion of dopamine agonists or antagonists, including haloperidol, raclopride, AJ76 or UH232, into the striatum or nucleus accumbens only minimally affects extracellular DOPAC levels (Imperato and Di Chiara, 1988; Timmerman et al., 1990; Waters et al., 1994). However, significant increases and decreases, respectively, are observed after their systemic administraiton, which suggests that extracellular DOPAC levels in the projection fields are a function of impulse flow and thus are modulated through postsynaptic receptors or somatodendritic autoreceptors (Waters et al., 1994). On the basis of this hypothesis, it would follow that amisulpride increases dopaminergic neuronal activity to a greater extent in the limbic than in the striatal projections. Electrophysiological studies are in progress to test this hypothesis.
A further index of a potential limbic selectivity in the interaction of amisulpride with dopaminergic neurotransmission is its stimulation of tyrosine hydroxylase activity. Thus amisulpride increased dopa accumulation in the limbic system with higher potency (ED50= 18.6 mg/kg) than in the striatum (ED50 = 43.7 mg/kg). Also in this case, it appears that the origin of limbic selectivity is not at the level of the terminal autoreceptor, because amisulpride opposed the effects of 7-OH-DPAT, after the administration of γ-hydroxy-butyrate, with similar potency in both regions.
Within the higher dose ranges, amisulpride increased limbic and striatal DOPAC tissue levels to a similar degree in each region. In this respect, amisulpride differs markedly from classical neuroleptics such as haloperidol, whose effects on tissue DOPAC levels are considerably more pronounced in striatal regions (Scatton et al., 1977), and can be grouped together with the atypical neuroleptics clozapine and sulpiride.
Taken together, the present results demonstrate that after systemic administration, amisulpride preferentially interacts with limbic dopamine D2-like receptors. Regional differences in pharmacological effects may be related to the regional brain distribution of systemically administered amisulpride (Köhleret al., 1992). Alternatively, a regional selectively may arise from the differential involvement of dopamine D2 and D3 receptor subtypes in the striatal and limbic systems.
Conclusions.
With reference to other neuroleptics, amisulpride displays several characteristic features. Thus its specificity and selectivity for D2-like (D2 and D3) receptors are uncommon and are largely confined to neuroleptics from the substituted benzamide series. Although average clinical doses and plasma concentrations of neuroleptics appear to correlate with D2 receptor affinity (Seeman, 1992), which suggests that D2 receptor occupancy constitutes a major molecular target for therapeutic efficacy, additional pharmacological mechanisms are thought to participate in, or to contribute to, an atypical therapeutic spectrum. Among these are an antagonism toward 5-HT2A or 5-HT2C receptors, muscarinic cholinergic receptors or alpha adrenoceptors or an affinity toward ς recognition sites (Ferris et al., 1991; Meltzer, 1991; Schmidt et al., 1995). The present data suggest that selective dopamine D2 antagonism can also lead to therapeutically efficacious atypical neuroleptics. Within the dopamine D2 receptor family, amisulpride selectivity recognizes the D2 and D3 subtypes. The observation that amisulpride shows only low affinity for the D4 subtype would support the suggestion (Seeman, 1992) that drug affinity for the dopamine D4 receptor is not an absolute requirement for therapeutic efficacy in the treatment of schizophrenia. Amisulpride selectively recognizes the D2 and D3 subtypes with equal affinity, unlike most conventional neuroleptics, which display higher affinity for the D2 than for the D3 subtype. Although the functional significance of the D3 dopamine receptor subtype remains to be firmly established, it is tempting to speculate that a causal relationship exists with the selectivity of amisulpride for dopamine autoreceptors. Thus, in vivo, amisulpride selectively affects presynaptic dopamine receptors that control dopamine synthesis and release, at doses below those that produce significant postsynaptic D2receptor occupancy, decrease striatal ACh levels, provoke an increase in dopamine turnover or block apomorphine-induced gnawing. The involvement of both D2 and D3 receptor subtypes in [3H]raclopride/[3H]spiperone bindingin vivo, as well as dopamine synthesis and release modulation, taken together with a greater relative abundance of D3 vs. D2 receptors in the limbic than in the striatal regions, may be the basis of the selectivity of amisulpride for the limbic dopaminergic system.
In summary, amisulpride has complex neurochemical effects on central dopaminergic neurotransmission but appears to show a selectivity, at low doses, for dopamine autoreceptors involved in the presynaptic regulation of dopaminergic neurotransmission, as well as a selectivity for its limbic projections. These characteristics probably contribute to its therapeutic activity in the treatment of dysthymia and negative symptoms of schizophrenia at low doses, and in the treatment of the positive symptoms of schizophrenia at high doses, because low doses selectively increase dopaminergic activity whereas high doses block postsynaptic D2 receptors.
Acknowledgments
The authors gratefully acknowledge Drs. S. Tan, A.-M. Galzin and D. Graham for their contribution to in vitro receptor binding studies as well as the excellent technical assistance of N. Allouard, M. Bas, N. Brunel, D. Chautrel, G. Danielou, G. Darles, C. Delatte, F. Demange, P. Gaubert, C. Lemaire, B. Peny, C. Sellier, A. Thiola, F. Thuret, M. Vasseur, and M. Vinay.
Footnotes
-
Send reprint requests to: Dr. Hans Schoemaker, Synthélabo Recherche, CNS Research Department, 31 ave Paul Vaillant-Couturier, BP 110, 92225 Bagneux, France.
- Abbreviations:
- L-dopa
- L-3,4-dihydroxyphenylalanine
- DOPAC
- 3,4-dihydroxyphenylacetic acid
- HVA
- homovanillic acid
- DOPEG
- 3,4-dihydroxyphenylethyleneglycol
- 5-HTP
- 5-hydroxytryptophane
- 5-HT
- serotonin
- 5-HIAA
- 5-hydroxyindoleacetic acid
- NSD-1015
- m-hydroxybenzylhydrazine dihydrochloride
- 7-OH-DPAT
- (±)7-hydroxy-N,N-di-n-propyl-2-aminotetralin
- ANOVA
- analysis of variance
- HPLC
- high-performance liquid chromatography
- QNB
- quinuclidinyl benzylate
- TBOB
- t-butylbicycloorthobenzoate
- PEA
- phenylisopropyladenosine
- NECA
- N-ethylcarboxamidoadenosine
- CHO cells
- Chinese hamster ovary cells
- Received April 16, 1996.
- Accepted October 1, 1996.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}