


Abstract
In higher eukaryotes, reactive oxygen species (ROS) are generated during respiration in mitochondria in the course of reduction of molecular oxygen as well as by distinct enzyme systems. ROS have been implicated in the regulation of diverse cellular functions including defense against pathogens, intracellular signaling, transcriptional activation, proliferation, and apoptosis. The reduction-oxidation (redox) state of the cell is primarily a consequence of the precise balance between the levels of ROS and endogenous thiol buffers present in the cell, such as glutathione and thioredoxin, which protect cells from oxidative damage. Dramatic elevation of ROS, exceeding compensatory changes in the level of the endogenous thiol buffers, may result in the sustained activation of signaling pathways and expression of genes that induce apoptosis in affected cells. Many cytotoxic drugs function selectively to kill cancer cells by the abrogation of proliferative signals, leading to cell death, and numerous reports have demonstrated that ROS are generated following treatment with these drugs. In this review, we will summarize recent contributions to our understanding of the importance of cytotoxic drug-induced modulation of cellular redox status for signaling and transcription leading to activation of apoptotic effector mechanisms.
In the course of normal metabolism, oxidizing equivalents or reactive oxygen species (ROS) are generated when oxygen is partially reduced as electrons leak out of the electron transport chain during respiration in mitochondria. These “activated” oxygen molecules can readily react with organic substances by noncatalytic means. In addition to the mitochondria, other sources of ROS generation include endogenous enzyme systems, e.g., plasma membrane NADPH-oxidase and cytoplasmic xanthine oxidase as well as organellar sources, e.g., peroxisomal cytochrome P-450 oxidases (Gamaley and Klyubin, 1999). The most common forms of ROS include superoxide anion (O⨪2), hydrogen peroxide (H2O2), and the highly reactive hydroxyl radical (OH⋅). ROS can also give rise to secondary reactive products such as lipid peroxides.
The reduction-oxidation (redox) state of the cell is a consequence of the balance between the levels of oxidizing (ROS) and reducing equivalents. Elevation of ROS in excess of the buffering capacity and enzymatic activities designed to modulate ROS levels result in potentially cytotoxic “oxidative stress”. Under these pro-oxidant conditions, highly reactive radicals can damage DNA, RNA, proteins, and lipid components, which may lead to cell death. To counteract the effects of oxidative stress, cells have developed two important defense mechanisms: a thiol reducing buffer consisting of small proteins with redox-active sulfhydryl moieties [e.g., glutathione (GSH) and thioredoxin (TRX)] and enzymatic systems (e.g., superoxide dismutase, catalase, and glutathione peroxidase) (Nakamura et al., 1997).
Thiol Redox Buffer
The glycine, glutamic acid, cysteine tripeptide, GSH, is the most abundant nonprotein sulfhydryl-containing compound and constitutes the largest component of the endogenous thiol buffer, at cellular concentrations that range between 0.1 to 10 mM (Schroeder et al., 1996). GSH has diverse cellular functions in addition to its antioxidant properties including enzymatic conjugation through the glutathione S-transferase family of proteins and nonenzymatic conjugation to cytotoxic compounds. It is kept in its reduced state by the NADPH-dependent enzyme, glutathione disulfide reductase, and GSH may react with hydrogen peroxides and lipid peroxides by the action of GSH peroxidase to reduce their toxicity.
Investigations into the role of GSH in modulating apoptotic signaling suggest that cellular redox changes following environmental stress induced by cytotoxic agents may not only be modulated by the generation of ROS but also by the extrusion of reduced glutathione from cells (Ghibelli et al., 1995). Treatment of HepG2 hepatoma cells with bleomycin induced the production of reactive oxygen intermediates as well as GSH depletion (Hug et al., 1997). The pro-apoptotic Bcl-2 protein also has an effect on GSH metabolism, as its overexpression leads to redistribution of GSH from cytosol to nucleus (Voehringer et al., 1998). The Bcl-2-mediated nuclear sequestration of GSH alters nuclear redox and blocks caspase activity. In this way, GSH compartmentalization within the cell has consequences for the activity of proteins that promote cell survival (see Fig.1).

Thiol buffer modulation of apoptotic signaling pathways. Arrows represent activation within a pathway; T bars represent inhibition within a pathway. Dashed arrows represent thiol translocation from cytosol into nucleus, or for GSH, extrusion from the cell. GSSG, glutathione disulfide; IL1β, interleukin 1β.
Thioredoxin (Mr 12,000) is a multifunctional and ubiquitous protein characterized by having a redox-active disulfide/dithiol within the conserved active site sequence: -Trp-Cys-Gly-Pro-Cys-Lys- (Holmgren and Bjornstedt, 1995). Thioredoxin reductase specifically reduces Trx-S2to Trx-(SH)2 using NADPH. The Trx-(SH)2 form is a powerful protein-disulfide reductase. Thus TRX, thioredoxin reductase, and NADPH, collectively called the thioredoxin system, operate as a powerful NADPH-dependent protein-disulfide reductase system. Thioredoxin exerts a protective effect on cells exposed to cis-diamminedichloroplatinum (II) (CDDP), and its overexpression is implicated in the mechanism of resistance to this drug (Sasada et al., 1996). The expression and activity of human thioredoxin (hTRX) in Jurkat T cells was dose dependently enhanced by exposure to CDDP, mediated through the generation of intracellular ROS (see Fig. 1). Overexpression of human thioredoxin (hTRX) in Jurkat T cells, which constitutively expressed the exogenous hTRX, displayed increased resistance to CDDP-induced cytotoxicity, compared with control T cell clones. Elevated levels of thioredoxin have also been observed in several human bladder and prostatic cancer cell lines resistant to CDDP (Yokomizo et al., 1995). Introduction of thioredoxin antisense transfectants showed increased sensitivity to cisplatin and also to other agents whose cellular reactions result in increased ROS—doxorubicin, mitomycin C, etoposide, and hydrogen peroxide—as well as to UV irradiation, but not to the tubulin-targeting agents, vincristine and colchicine. These observations underscore the importance of thioredoxin as a component of the thiol redox buffer system in modulating the toxicity of anticancer drugs that generate ROS.
Stress Kinase Signaling and Apoptosis
Many antineoplastic agents eliminate tumor cells by inducing programmed cell death or apoptosis (Thompson, 1995; reviewed inJacobson, 1996), and numerous investigations have documented the cellular changes resulting from oxidative stress induced in cells following exposure to cytotoxic drugs and UV and γ irradiation. Although these agents are structurally dissimilar and act on different cellular targets, (e.g., DNA, cytoskeleton), nevertheless, they may elevate levels of ROS. Correlated with the increase in ROS production by these agents is the activation of the redox-sensitive c-Jun N-terminal kinase/stress activated protein kinase (JNK/SAPK) (reviewed in Powis et al., 1998; and Table1).
Role of JNK/SAPK in apoptosis
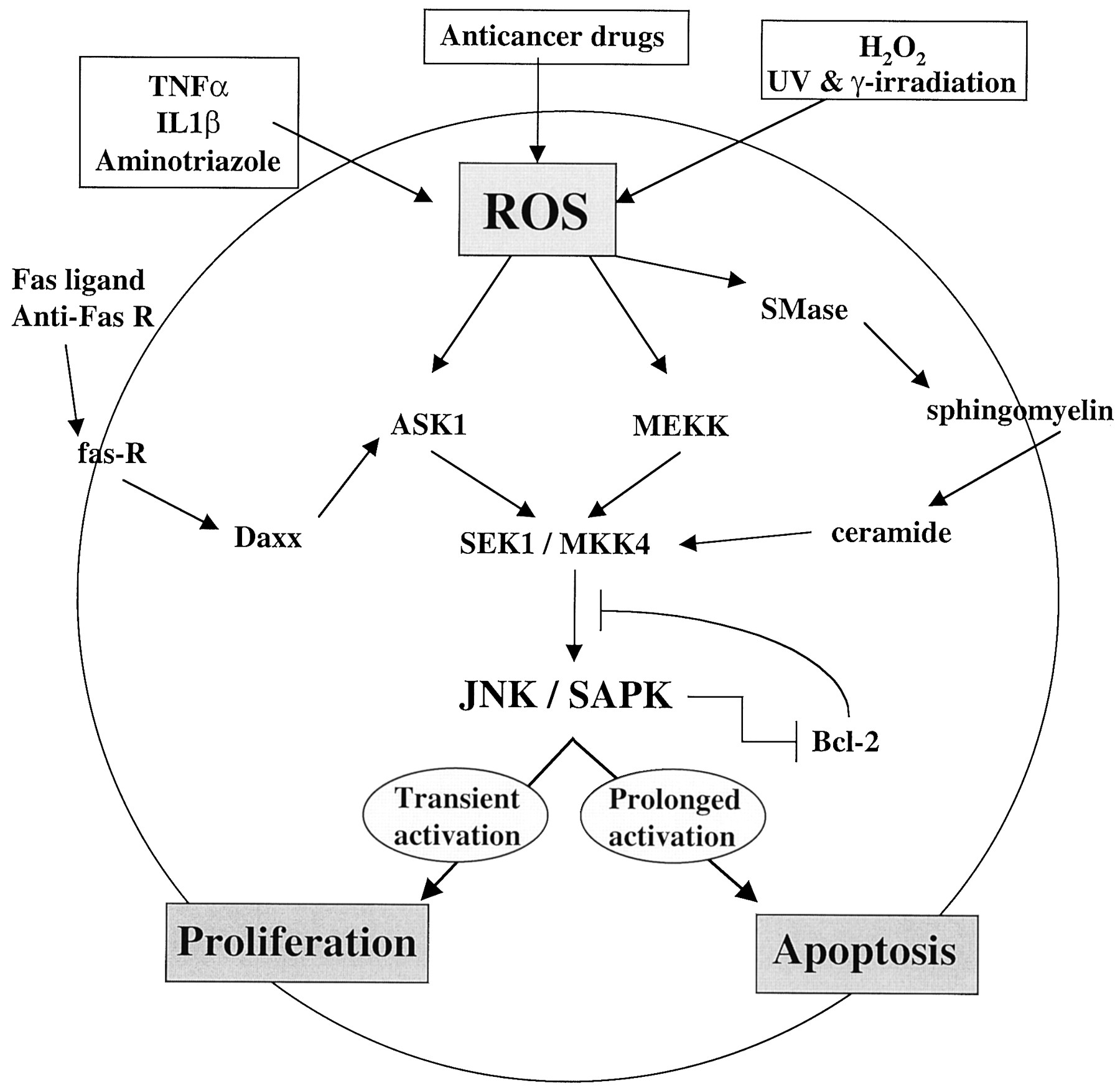
Activation of JNK/SAPK occurs in response to a multitude of inductive stimuli, including X-rays, UV irradiation, cytokines, and chemotherapeutic drugs that induce cellular oxidative stress (reviewed in Fanger et al., 1997; Ip and Davis, 1998). Since in many instances JNK/SAPK activation is necessary for the transcriptional activation of genes and post-translational modification of proteins necessary for the induction of apoptosis, a common ROS-induced pathway, independent of cytotoxic stimulus, may be implied (see Fig.2).

Multiple inputs for ROS generation, JNK/SAPK activation, and apoptosis. Arrows represent activation within a pathway; T bars represent inhibition within a pathway. IL1β, interleukin 1β.
The intracellular second messenger ceramide, generated by the activity of sphingomyelinases on membrane phospholipids, is a key regulator of the activation of JNK/SAPK in response to a number of agents, including tumor necrosis factor (TNF), Fas ligand, and chemotherapeutic agents (Hannun, 1996). Anthracyclines, such as daunorubicin, generate ROS that activate the neutral sphingomyelinase enzyme and elevate intracellular ceramide (Mansat-De Mas et al., 1999). Treatment of U937 human monoblastic leukemia cells with cell-permeant ceramides also induce both an increase in ROS, JNK/SAPK activation, and apoptosis, and these effects are inhibitable by the antioxidants N-acetylcysteine (NAC) and pyrrolidine dithiocarbamate (PDTC).
Ceramide can induce activation of JNK/SAPK and apoptosis by the same pathways as those activated by cellular stress, and the action of ceramide is upstream of JNK/SAPK (Verheij et al., 1996). In U937 human monoblastic leukemia cells and bovine aortic endothelial (BAE) cells, immunoprecipitated JUN/SAPK activity phosphorylated its target c-Jun polypeptide in vitro. Addition of C2 ceramide or the enzyme sphingomyelinase induced a robust dose-dependent JNK/SAPK activation in vivo. Expression of dominant-negative c-Jun by stable transfection inhibited both stress- and C2 ceramide-induced apoptosis. In addition to activating the JNK/SAPK pathway leading to apoptosis in cells under environmental stress, in human leukemia cell lines, ceramide also functions to rapidly translocate protein kinase C (PKC)-δ and -ε isozymes from the plasma membrane to the cytosol, resulting in its inactivation (Sawai et al., 1997). Moreover, elevation in intracellular ceramide following treatment with pro-apoptotic agents sphingomyelinase, TNF-α, or anti-Fas antibody all directed the translocation of the PKC isozymes from the plasma membrane to the cytosol. Treatment of cells with either a specific PKC activator, phorbol 12-myristate 13-acetate (PMA), or a nonspecific kinase inhibitor, staurosporine, prevented ceramide-induced apoptosis by inhibiting cytosolic translocation of PKC-δ and -ε. These results suggest that cytosolic translocation and inactivation of PKC-δ and -ε play an important role in ceramide-mediated apoptosis.
The importance of ROS in cytokine-mediated hydrolysis of sphingomyelin (SM) to ceramide was demonstrated by treatment of primary rat astrocytes with TNF-α or interleukin 1β (Singh et al., 1998). A dramatic change to a pro-oxidant cellular redox state was distinguished by a decrease in cellular GSH and degradation of SM to ceramide. Furthermore, activation of SM hydrolysis and ceramide generation were observed by direct addition of hydrogen peroxide or a pro-oxidant, aminotriazole. Conversely, the antioxidants NAC, a GSH precursor, and PDTC, were found to be potent inhibitors of cytokine-induced degradation of SM to ceramide. These results indicate that cytokine-induced hydrolysis of sphingomyelin to ceramide is redox-sensitive and that high levels of the endogenous thiol buffer negatively modulate the magnitude of ceramide production.
Oxygen radicals have a direct role in the induction of the CD95 (APO-1/Fas) ligand, a specific mediator of apoptosis (Nagata, 1997), and recent findings have elucidated additional components of the signaling cascade to JNK/SAPK and their regulation. A signaling protein termed Daxx binds to the Fas receptor “death domain” by a C-terminal region (Yang et al., 1997). A second component of this cascade, the apoptosis signal-regulating kinase (ASK1) activates two subgroups of MAP kinase kinase (MAPKK), SEK1 (or MKK4), and MKK3/MAPKK6 (or MKK6), which in turn activate JNK/SAPK and p38 subgroups of MAP kinases, respectively. The importance of ASK1 in mediating apoptotic signaling is evident by the fact that overexpression of ASK1 induces programmed cell death (Ichijo et al., 1997). In the model mechanism, the stimulation of the Fas receptor induces associated Daxx to interact with ASK1, thereby activating the kinase and signaling events through JNK/SAPK, leading to apoptosis.
The N-terminal portion of ASK1 physically associates both in vitro and in vivo with thioredoxin, and its activity is negatively regulated in this fashion by protein-protein interaction (Saitoh et al., 1998; and Fig. 1). Expression of thioredoxin inhibited ASK1 kinase activity and subsequent ASK1-dependent apoptosis. The redox status of the cell was the determining factor for the interaction between thioredoxin and ASK1. Experiments using dominant-negative redox-insensitive thioredoxin mutants or antisense oligonucleotides to thioredoxin gene expression resulted in the activation of endogenous ASK1 activity in vivo. These results provide further evidence that thioredoxin is a redox-sensitive physiological regulator of ASK1 activity and the response to environmental stress regulating apoptosis.
It appears paradoxical that the activation of JNK/SAPK can result in both proliferation in response to growth factor stimulation and apoptosis following stimulation by agents that induce oxidative stress. Part of the clue to understanding these distinct cellular effects following JNK/SAPK stimulation is revealed in experiments that suggest that the duration of JNK/SAPK activity determines cell fate. Apoptosis induced by γ irradiation, UV-C, or anti-Fas treatment resulted in persistent activation of JNK/SAPK but not p38 MAPK or extracellular signal-regulated kinase-2 (ERK2), and only dominant-negative forms of JNK1 (but not p38 or c-Raf) inhibited γ and UV irradiation-induced cell death (Chen et al., 1996). The induction of JNK/SAPK in T-cell activation and apoptosis were distinguished by the different activation patterns, transient versus persistent, respectively (see Fig. 2). Cotreatment with a tyrosine phosphatase inhibitor (sodium orthovanadate) and T-cell activation signals (PMA plus ionomycin) prolonged JNK/SAPK induction, followed by T-cell apoptosis. These results suggest that the determination of cell fate, proliferation or cell death, following activation of JNK/SAPK, is correlated with the duration of its activity. Activation of JNK/SAPK has also been observed following treatment of cells with Adriamycin, vinblastine, or etoposide, while in contrast, no significant change in activity was apparent for ERK. These drugs are transport substrates for the multiple drug resistance 1 (MDR-1/P-glycoprotein) gene product, and a 4- and 7-fold elevation in the level of JNK/SAPK activity was measured in two multidrug resistant cell lines selected for resistance to Adriamycin and vinblastine, respectively (Osborn and Chambers, 1996). These findings suggest that JNK/SAPK activation is an important component of the cellular response to several structurally related and functionally distinct anticancer drugs and may also be involved in the multidrug resistance phenotype.
In experiments using HeLa cells and H2O2, the relative influences of mitogen-activated protein kinase (MAPK) subfamilies reveals that the ERK, JNK/SAPK, and p38 are reciprocally activated (Wang et al., 1998). Treatment of HeLa cells with H2O2 resulted in a time- and dose-dependent induction of apoptosis accompanied by sustained activation of all three MAPK subfamilies: ERK, JNK/SAPK, and p38. This H2O2-induced apoptosis was significantly enhanced when ERK2 activation was selectively inhibited by PD098059. Apoptosis decreased when JNK/SAPK activation was inhibited by expression of a dominant-negative mutant form of SAPK/ERK kinase-1. Inhibition of the p38 kinase activity with p38-specific inhibitors SB202190 and SB203580 had no effect on cell survival. Whereas the ERKs are normally activated in response to growth factor stimulation and the JNK/SAPK and p38 MAPK in response to cellular stress, these experiments suggest that there are opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Mitogen-activated extracellular response kinase kinase kinase (MAPKKK/MEKK) is a serine/threonine kinase that regulates the activation of MAPKs, including members of the JNK/SAPK family. Using Swiss 3T3 and REF53 fibroblasts, forced expression of activated MEKK increased the sensitivity of these cells to apoptotic stimuli (Johnson et al., 1996). Whereas proto-oncogenes c-Myc and Elk-1 were induced by MEKK overexpression, activated Raf-1, which signals through the ERK pathway, activated Elk-1 but not c-Myc and did not induce cell death. These experiments provide additional evidence that the JNK/SAPK module is selectively activated to effect the apoptotic response.
The activator protein-1 transcription factor (AP-1), consisting of c-Jun homodimers or c-Jun/c-Fos heterodimers, has been implicated in the response to oxidative stress (Sen and Packer, 1996). Whether AP-1 is an oxidant- or antioxidant-responsive transcription factor is open to interpretation, since both stimuli seem to be capable of activation of AP-1. However, examination of the inducing activities of antioxidants like PDTC or butylated hydroxyanisole using electron paramagnetic resonance spin trapping spectroscopy to detect semiquinone radicals revealed the autoxidation of these compounds and the generation of OH⋅ radicals when measured in hepatoma HepG2 cells (Pinkus et al., 1996). That catalase, which lowers H2O2 levels and reduces production of OH⋅ radicals, inhibited induction of AP-1-dependent glutathione S-transferase (GST) Ya gene expression indicates that activation of AP-1 is due to oxidation and quinone-mediated generation of oxygen radicals. Further confirmation that the induction of AP-1 activity and GST Ya gene expression by butylated hydroxyanisole and tert-butylhydroquinone is due to a pro-oxidant activity was shown by its inhibition using the antioxidant thiol compounds NAC and GSH. These results suggest that AP-1 is an oxidant-responsive factor, and this finding is consistent with activation of c-Jun of AP-1 by JNK/SAPK under conditions of oxidative stress.
JNK/SAPK is a bifunctional protein in that it modulates the activity of its target substrates by phosphorylation in stressed cells, but it also regulates the stability of its substrate proteins (Fuchs et al., 1997). For example, in nonstressed cells, association of JNK/SAPK with c-Jun by the δ domain impairs this protein's ability to undergo transactivation. This association recruits the enzymes of the ubiquitination machinery to c-Jun, thereby marking it for proteosome-dependent degradation. However, phosphorylation of c-Jun by JNK/SAPK in cells under oxidative stress leads to dissociation of JNK/SAPK from c-Jun, which is permissive both for stability and transcriptional activity.
The DNA-binding activity of Jun and Fos requires protein redox factor 1 (Ref-1), which is identical to the DNA repair enzyme, apurinic/apyrimidinic endonuclease (Xanthoudakis et al., 1992). Ref-1 mediates the DNA binding of Fos and Jun heterodimers as well as Jun homodimers by reduction of a conserved cysteine residue in the DNA binding domain of these proteins. Thioredoxin is capable of regulating AP-1 transcriptional activity through its direct association with Ref-1, serving as a proton donor to Ref-1 (Hirota et al., 1997). Cysteine 32 and cysteine 35 of thioredoxin, which constitute the catalytic center, are critical to the protein-protein interaction between thioredoxin and Ref-1. Site-directed mutagenesis studies showed that two cysteines in the redox domain of Ref-1, Cys-63 and Cys-95, are redox-sensitive and can be targets of thioredoxin. Treatment of HeLa cells with PMA mediated the translocation of thioredoxin to the nucleus where Ref-1 is localized. This cytosolic to nuclear translocation is necessary for AP-1-dependent transcriptional potentiation. These data collectively suggest that PMA causes a translocation of thioredoxin into the nucleus, allowing its interaction with Ref-1, and that this association promotes the direct activation of AP-1 by Ref-1. The thioredoxin/Ref-1/AP-1 cascade represents an example of regulation of AP-1 activity by the action of intracellular redox-sensitive thiol compounds (see Fig. 1).
The involvement of the tumor suppressor gene p53 in modulating the cytotoxicity of anticancer agents is well documented. Surprisingly, similar mechanisms are used for stress-mediated activation and stabilization of p53 as for AP-1. For example, the redox/repair protein Ref-1 has been shown to be a potent activator of p53 (Jayaraman et al., 1997); JNK/SAPK can directly phosphorylate p53 in vivo (Milne et al., 1995); MEEK1/JNK signaling stabilizes and activates p53 (Fuchs et al., 1998b); and JNK/SAPK targets p53 ubiquitination and degradation in nonstressed cells (Fuchs et al., 1998a).
JNK/SAPK signaling also regulates the activity of the Bcl-2 family of proteins, which includes both repressors (e.g., Bcl-2, Bcl-XL, Mcl-1, and A1) and activators (e.g., Bax, Bcl-XS, Bak, and Bad) of apoptosis, characterized by their ability to form homo- and heterodimeric complexes. The relative abundance of the pro-apoptotic versus anti-apoptotic members appears to play a crucial role in determining cell fate in response to apoptotic signals (Oltavi and Korsmeyer, 1994). Interestingly, activation of the JNK/SAPK pathway antagonizes the anti-apoptotic action of Bcl-2 (Park et al., 1997). When Bcl-2 was overexpressed in N18TG neuroglioma cells, it suppressed apoptosis induced by etoposide, staurosporine, anisomycin, and UV irradiation, agents that induce the JNK/SAPK pathway. Conversely, overexpression of JNK/SAPK antagonized the death-protective function of Bcl-2, promoting apoptosis. Further evidence that Bcl-2 inhibits JNK/SAPK signaling is provided by data showing inhibition of etoposide-induced stimulation of MEKK1, an upstream activator of JNK/SAPK (Yamamato et al., 1999). Two-dimensional peptide mapping and sequencing have identified Ser-70 in the unstructured loop of Bcl-2 as the target for phosphorylation by ASK1/JNK1 and that mutation of this domain restores resistance to apoptosis. Thus, phosphorylation of Bcl-2 at Ser-70 may contribute to the inactivation of the protective effect of Bcl-2, thereby promoting the apoptotic cascade. These results suggest that the two effectors may be reciprocally regulated. Therefore, suppression of JNK/SAPK activity may be necessary for the survival effect of Bcl-2. These experiments provide further evidence of a convergence of signaling inputs through the JNK/SAPK pathway modulating apoptotic effector molecule function.
In addition to regulation of JNK/SAPK activity by post-translational modification, i.e., phosphorylation/dephosphorylation mediated by kinases and phosphatases (Fanger et al., 1997; Keyse, 2000), recent evidence suggests that GSTπ functions as a negative regulator of JNK/SAPK signaling in nonstressed cells (see Fig. 1). The glutathioneS-transferase family of enzymes functions in the glutathionylation of target substrates using GSH as a cofactor (Tew, 1994). They also have been demonstrated to be functional in the catalytic detoxification of xenobiotics. High expression of GST isozymes has been correlated with acquired drug resistance and tumorigenesis. It is presumed that the dimeric form of GSTπ is responsible for the regulatory control of JNK/SAPK. UV irradiation or H2O2 treatment, which induce oxidative stress, caused GSTπ oligomerization and dissociation of the GSTπ-JNK/SAPK complex resulting in JNK/SAPK activation in cells (Adler et al., 1999). Addition of the glutathione peptidomimetic compound γ-glutamyl-S-(benzyl) cysteinyl-R-phenyl glycine (TER117) and its diethyl ester (TER199), a specific inhibitor of the GSTπ isozyme, caused a dose-dependent activation of JNK/SAPK activity both in vitro and in vivo, respectively. Forced expression of GSTπ decreased MKK4/SEK1 and JNK/SAPK phosphorylation, which coincided with decreased JNK/SAPK activity. Cotransfection of MEKK1 and GSTπ restored MKK4 phosphorylation but did not affect GSTπ inhibition of JNK/SAPK activity, suggesting that the effect of GSTπ on JNK/SAPK is independent of MEKK1-MKK4. These experiments demonstrate a novel mechanism for modulating the activity of stress kinases through the nonenzymatic association of GSTπ, an integral component of the GSH redox buffer system.
Conclusion
This review has focused on regulation of cellular redox by the endogenous thiol buffer systems in response to cytotoxic agents that induce oxidative stress and programmed cell death. Cellular response to these cytotoxic agents may generate ROS in excess of levels of the thiol buffer, and in these cases, the duration of signaling through the stress kinase cascade can lead to the activation of apoptotic effector molecules. Alternatively, cytotoxic agents may modulate the compartmentalization of GSH such that although ROS are not generated directly, the loss of GSH from the nucleus may activate downstream proteolytic caspases that effect cell death. Knowledge of the signaling pathways and physiological responses to cytotoxic agents is essential to understanding the mechanisms of drug toxicity and chemoresistance. Since drug-induced programmed cell death utilizes physiological signaling pathways, examination of the functional status of elements in stress response may provide important new drug targets.
Footnotes
-
Send reprint requests to: Dr. Kenneth D. Tew, Department of Pharmacology, Fox Chase Cancer Center, 7701 Burholme Ave., Philadelphia, PA 19111-2412. E-mail: kd_tew{at}fccc.edu
-
This work was supported in part by National Institutes of Health Grants CA06927 and RR05539, by National Institutes of Health Grant CA85660 to K.D.T., and by an appropriation from the Commonwealth of Pennsylvania.
- Abbreviations:
- ROS
- reactive oxygen species
- GSH
- glutathione
- TRX
- thioredoxin
- h
- human
- CDDP
- cis-diamminedichloroplatinum (II)
- JNK/SAPK
- c-Jun N-terminal kinase/stress activated protein kinase
- TNF
- tumor necrosis factor
- NAC
- N-acetylcysteine
- PDTC
- pyrrolidine dithiocarbamate
- PKC
- protein kinase C
- PMA
- phorbol 12-myristate 13-acetate
- SM
- sphingomyelin
- ASK1
- apoptosis signal-regulating kinase
- ERK
- extracellular signal-regulated kinase
- MAPK
- mitogen-activated protein kinase
- AP-1
- activator protein-1
- Ref-1
- protein redox factor-1
- MEKK1
- mitogen-activated protein kinase kinase kinase 1
- GST
- glutathione S-transferase
- MKK3/4/6
- mitogen-activated protein kinase kinase
- SEK1
- stress-activated protein kinase kinase
- redox
- reduction-oxidation
- Received April 27, 2000.
- Accepted July 31, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}