Article Text

Abstract
Considerable advances made in defining the aetiology, pathogenesis, and pathology of Parkinson’s disease (PD) have resulted in the development and rapid expansion of the pharmacopoeia available for treatment. Anticholinergics were used before the introduction of levodopa which is now the drug most commonly used. Dopamine agonists are effective when used alone or as an adjunct to levodopa, while monoamine oxidase B inhibitors improve motor function in early and advanced PD. However, treatment mainly addresses the dopaminergic features of the disease and leaves its progressive course unaffected; the drug treatment available for the management of non-motor symptoms is limited. This article seeks to set current treatment options in context, review emerging and novel drug treatments for PD, and assess the prospects for disease modification. Surgical therapies are not considered.
- COMT, catechol-O-methyltransferase
- GDNF, glial cell derived nerve growth factor
- MAO, monoamine oxidase
- MPP, 1-methyl 4-phenylpyridinium
- MPTP, 1-methyl, 4-phenyl 1,2,3,6 tetrahydropyridine
- PD, Parkinson’s disease
- PET, positron emission tomography
- UPDRS, Unified Parkinson Disease Rating Scale
- SPECT, single photon emission computerised tomography
- dopamine agonists
- levodopa
- MAO-B inhibitors
- Parkinson’s disease
Statistics from Altmetric.com
- COMT, catechol-O-methyltransferase
- GDNF, glial cell derived nerve growth factor
- MAO, monoamine oxidase
- MPP, 1-methyl 4-phenylpyridinium
- MPTP, 1-methyl, 4-phenyl 1,2,3,6 tetrahydropyridine
- PD, Parkinson’s disease
- PET, positron emission tomography
- UPDRS, Unified Parkinson Disease Rating Scale
- SPECT, single photon emission computerised tomography
First described formally almost 200 years ago, Parkinson’s disease (PD) is one of the most common human degenerative disorders and has a major socio-economic impact. Direct medical costs in the UK were estimated at £383 million in 1992 and are almost certainly a gross under-estimate. Over 75% of these costs are related to institutionalised care, but indirect costs and the burden of disease on patients and carers are inestimable. With an aging population, the management of PD is likely to prove an increasingly important and challenging aspect of medical practice. The cost of the development and use of novel therapies will, therefore, have to be set in this context.
Considerable advances have been made in defining the aetiology, pathogenesis, and pathology of PD and these advances in turn have resulted in the development and rapid expansion of the pharmacopoeia available for treatment. However, treatment remains unsatisfactory, as it mainly addresses only the dopaminergic features of the disease and leaves its progressive course unaffected. The future of PD drug treatment will need to focus on the symptomatic management of the non-dopaminergic and non-motor features of the disease and on the need for disease modification in terms of the delay or prevention of progression. This article seeks to set current treatment options in context, review emerging and novel drug treatments for PD, and assess the prospects for disease modification. Surgical therapies will not be considered.
PATHOLOGY
PD is a multicentric neurodegenerative disease in which the development of pathological abnormalities follows a specific sequence. Recent studies suggest that the earliest changes are seen in the dorsal motor nucleus and in the olfactory bulbs and nucleus – Braak stage 1.1 In this context, it is noteworthy that loss of olfactory function can occur before the onset of dopaminergic symptoms or signs and may serve to define an at risk population.2 Cell loss and Lewy body formation then develop in the locus coeruleus and progress in the medulla and pons – Braak stage 2.3 The appearance of cell loss and inclusion formation in the substantia nigra pars compacta defines the onset of Braak stage 3. At this stage there is also degeneration in the pedunculopontine nucleus, the dorsal raphe nuclei, and the hypothalamus. Stages 5 and 6 involve progressive involvement of the cerebral cortex and neurodegeneration in those regions already affected.
According to Braak’s staging, clinical features diagnostic of Parkinson’s disease manifest during stage 3. The implication is that successful therapeutic disease modifying intervention at/or prior to stage 3 would prevent progression to the point where dopaminergic features became severe. Importantly, such treatment if effective on non-dopaminergic systems, would prevent the clinical evolution of the non-motor complications that characterise the more advanced stages of PD.
DOPAMINERGIC FEATURES AND THEIR TREATMENT
Patients with PD usually present with features indicative of degeneration of nigrostriatal pathways. A useful clinical definition for PD is “asymmetric onset of an akinetic (bradykinetic) rigid syndrome with resting tremor and a good response to levodopa”. When applied by neurologists with an interest in movement disorders, this definition has a pathological correlation exceeding 98%.4 When treatment is considered appropriate, and this is a topic discussed in detail below, a variety of options is available. The use of dopaminergic drugs improves motor function, significantly reduces both the morbidity and mortality of PD, and improves quality of life.5–7
Levodopa remains the drug most commonly used in PD. It is very effective in improving bradykinesia and rigidity, and in practice remains the gold standard against which other drugs are judged. Some studies, predominantly in vitro, have suggested that levodopa may be toxic.8,9,10 However, such data are conflicting,11,12 and some laboratory studies have suggested a growth factor-like effect for levodopa.13 Overall, the pre-clinical evidence for levodopa toxicity is not convincing and there are no data to indicate that any toxic action is of clinical relevance.14,15
Levodopa remains efficacious throughout the course of PD, but its effects are modified as a consequence of disease progression and the loss of the dopaminergic cells required to metabolise the drug and store and release dopamine. In addition, levodopa use does result in motor complications including dyskinesias and “wearing off”. The mechanisms by which these effects develop are not completely understood but include pulsatile stimulation of dopamine receptors by short acting agents such as levodopa, and the degree of striatal denervation.16 Motor complications develop at a rate of approximately 10% per annum in those diagnosed over 60 years of age, but faster in younger onset patients, 70% of whom have motor complications 3 years after diagnosis.17 The efficacy of levodopa can be enhanced by co-administration of a catechol-O-methyltransferase (COMT) inhibitor, which reduces O-methylation in the gut, increases levodopa absorption, and prolongs half-life.18 Tolcapone was the first COMT inhibitor to be licensed but was withdrawn because of hepatic toxicity. However, it has recently been re-introduced with a restricted license and requires appropriate monitoring. It should only be used for those who cannot tolerate or fail to benefit from entacapone. Entacapone is the most widely used COMT inhibitor and, when combined with levodopa, increases “on” time and reduces “off” time.19 There is evidence from the 1-methyl, 4-phenyl 1,2,3,6 tetrahydropyridine (MPTP) primate model of PD that levodopa combined with entacapone and given four times a day results in significantly less dyskinesia than without entacapone or if given less frequently.20 Stalevo is a preparation combining levodopa with a dopa decarboxylase inhibitor and entacapone. A study is currently underway to determine whether the effect seen in the MPTP model translates to a lower dyskinesia rate in patients initiated on Stalevo rather than levodopa.
Dopamine agonists are effective when used alone or as an adjunct to levodopa.21 Agonist monotherapy can effectively control dopaminergic symptoms for a while22,23 (table 1), but inevitably patients will require levodopa supplementation at some point during their disease. Perhaps one of the commonest reasons for agonist failure during early disease is that too often they are used at too low a dose and abandoned before an appropriate level is reached. If used correctly, they can produce symptom control comparable to that achieved with levodopa. Although the two monotherapy studies22,23 showed the use of levodopa increased Unified Parkinson Disease Rating Scale (UPDRS) scores by five points, patients in the agonist arms did not require further additional medication to reach an equivalent clinical benefit, that is patients and physicians perceived the agonist and levodopa arms to be clinically the same in the context of a blinded study. Quality of life scores were also equivalent for the 4 year period.22 This implies that the patients were equally well controlled on agonist (with levodopa supplementation when required) or levodopa alone. Of course the levodopa group had more dyskinesias, but at 4 years these did not intrude significantly into patient quality of life nor yet start to limit treatment options for motor control.
Percentage of patients remaining on dopamine agonist monotherapy at years 1–4 (CALM-PD) and years 1–5 (ropinirole 056) during treatment trials
One open label study demonstrated that the addition of cabergoline, a long acting ergot, to pramipexole or ropinirole, both non-ergots, resulted in improved motor control.24 Long acting agonists are useful in the management of dyskinesias in a proportion of patients, presumably due to their ability to provide more continuous dopaminergic stimulation while avoiding rapid fluctuations in receptor stimulation.25 Rotigitine, a new dopamine agonist, will be available for use as a skin patch and offers another opportunity for sustained dopaminergic effect. A similar effect may be obtained with levodopa or apomorphine infusions,26 and a preparation for the duodenal infusion of levodopa will be available shortly. The common side effects of dopamine agonists are those of dopaminergic agents in general in addition to a higher rate of cognitive disturbances (confusion, hallucinations), sleepiness, and leg oedema. A recent report has linked pergolide with fibrotic cardiac valvular disease.27 There are insufficient data at present to establish whether this is associated with ergot agonists alone or all dopamine agonists, and whether the effect is dose and/or time related. Currently the possibility of fibrotic cardiac valvular disease is being managed by vigilance, appropriate investigations (echocardiogram, chest x ray, and ESR), and referral to cardiology departments in specific cases.
Monoamine oxidase (MAO) B inhibitors such as selegiline and rasagiline both improve motor function in early and advanced PD.28–30 Although there was some concern regarding the long term safety of selegiline, several studies have allayed these fears and shown this drug to be both effective and safe for PD patients.31
Any recommendations regarding the nature and sequence of therapy must always be considered with regard to the fact that treatment for PD is always tailored to the specific needs and circumstances of the patient. The following is a personal approach to the initial management of the dopaminergic related features of PD. Traditionally, drug treatment has only been initiated when the patient’s symptoms interfere significantly with their employment or social activities. However, the introduction of drugs that are well tolerated with a low and acceptable side effect profile in the short and long term has led some clinicians to re-evaluate this approach. In my view, the option of early treatment to improve the very symptoms that have brought the patient to medical attention should be discussed with the patient. Those aged 70–75 years or younger, with no cognitive impairment and no significant co-morbidity, should be considered for introduction of a dopamine agonist or a MAO-B inhibitor. The agonist will improve motor dysfunction more than a MAO-B inhibitor, but the latter is probably better tolerated and the choice between these will depend upon the patient’s degree of symptomatic dysfunction. For those patients aged over 70–75 years, with cognitive dysfunction or significant co-morbidity, levodopa would be the drug of choice for the initiation of therapy. At some point all patients whether started on a dopamine agonist or MAO-B inhibitor, will need the addition of levodopa to their regimen. At present, levodopa is initiated without entacapone, but this may change if an on-going study demonstrates that the introduction of levodopa as Stalevo is associated with a lower risk of motor complications.
NON-DOPAMINERGIC FEATURES AND THEIR TREATMENT
The widespread and progressive neurodegeneration in the PD brain leads to the emergence of a variety of features that are collectively grouped under the title of non-motor symptoms. These are predominantly, but not exclusively, the consequence of the loss of non-dopaminergic pathways. The non-motor symptoms of PD range from cognitive problems such as apathy, depression, anxiety disorders, and hallucinations to fatigue, gait and balance disturbances, hypophonia, sleep disorders, sexual dysfunction, bowel problems, drenching sweats, sialorrhoea, and pain. These symptoms are often the most troubling for patients and contribute significantly to morbidity and impaired quality of life.32 The drug treatment available for the management of these problems is limited.
Depression, and to some extent apathy (anhedonia), may respond to tricyclics such as amitriptyline, or to SSRIs. Pramipexole may be useful as an antidepressant, separate from its action to improve the motor features of PD, and has shown some benefit in both depressed non-PD33 and depressed PD patients,34 although further studies are required to confirm this effect. Anxiety and panic attacks can be prominent in PD and while these may sometimes relate to wearing off and so respond to dopaminergic therapy, additional anxiolytic therapy may be needed. Hallucinations can arise as a consequence of both the neurodegeneration in PD and the dopaminergic drugs used in treatment. Studies have shown that dopamine agonists are more associated than levodopa with the development of hallucinations, particularly in the elderly. If troublesome, modification of existing therapy is the easiest strategy to reduce or stop hallucinations, but in some patients this is difficult due to the re-emergence of motor features. Well controlled clinical trials have showed that low dose clozapine can significantly improve PD patients with psychosis35,36; alternatively, some may respond to quetiapine.37 Hallucinations are, of course, an important symptom of diffuse Lewy body disease, and their emergence early in the course of PD is a risk factor for dementia. PD patients who developed dementia at least 2 years after the appearance of motor features, showed a modest but significant benefit for cognitive evaluations with rivastigmine, to a degree similar to that seen with this drug in Alzheimer’s disease.38
Abnormalities of sleep are common in PD and are the result of the natural consequences of aging, the underlying disease pathology,39 motor and non-motor complications,40,41 and drugs.42 Disordered sleep often results in excessive daytime drowsiness and this in turn may be compounded by the sedative effect of dopaminergic drugs.43 Several strategies are available to improve both night time sleep and day time alertness in PD and include improving sleep hygiene, treating nocturnal motor problems, better management of nocturia, modifying medication,44 and the use of modafinil in refractory patients with daytime drowsiness.45
Sexual and bladder dysfunction are common and occur in both sexes. The dopaminergic treatment of PD may lead to increased sex drive, but the effects of the disease often result in impaired sexual performance.46 Not surprisingly, this combination may cause frustration and sildenafil or apomorphine can, in selected cases, usefully manage this.47–49 Bladder abnormalities particularly cause problems at night but can be successfully managed by a range of options that include non-pharmacological as well as pharmacological strategies.50 The latter include the use of oxybutinin or detrusitol, or amitriptyline in patients with concomitant depression. Sialorrhoea and drooling is often the result of reduced frequency of swallowing and may be helped by simple things such chewing gum or sucking sweets. Anti-cholinergic drugs may sometimes help, but often cause unwanted side effects. Botulinum toxin can be used for refractory cases.51
Pain is a frequent symptom in PD and some patients manifest especially with shoulder pain. Pain, anxiety, akathisia, respiratory distress, depressive mood swings, and slowed and impaired thought are symptoms which may be experienced during “off” periods and which will respond, at least in part, to dopaminergic therapy.52–54 The recognition that a number of non-motor features may improve with dopaminergic drugs is important in improving the quality of life of PD patients.55
NON-DOPAMINERGIC TREATMENTS FOR PD
Anticholinergic use in PD pre-dated the introduction of levodopa and for some physicians still remains first line therapy. This is particularly so for patients with tremor dominant PD, although there is no convincing evidence to indicate that anticholinergics are better than dopaminergics in this respect,56 and both are often found to be of limited value. However, anticholinergic use is often associated with significant side effects including dry mouth, cognitive disturbance, and, in men, urinary retention. Amantadine has also been used for PD for several years and can still offer some useful symptomatic control of motor features in a proportion of patients, although it is substantially less potent than dopaminergics.57 Amantadine may be useful in improving dyskinesias, particularly at peak dose, although the data are limited and the benefit is usually restricted to 6–12 months.58
Alpha 2-adrenergic α-2a and 2c receptors are distributed widely within the basal ganglia, including the substantia nigra. Idazoxan and fipamezole, α-2 antagonists, reduce levodopa induced dyskinesia and extend the duration of action of levodopa in MPTP treated primates.59–62 Idazoxan has shown variable benefit in clinical trials.63,64 Interestingly, mirtazapine, an anti-depressant whose principal action is α-2 receptor antagonism, decreased levodopa induced dyskinesias in PD patients.65
The basal ganglia receive a significant serotonergic input from the raphe nuclei and many types of 5HT receptors are present in the striatum and other areas of the basal ganglia. Fluoxetine, a 5HT reuptake inhibitor, can suppress levodopa induced dyskinesias in PD patients,66,67 although there does not appear to be any difference in the occurrence of dyskinesias in those patients with PD who do and do not receive SSRI treatment.68 SSRI use can sometimes be associated with worsening of the motor features of PD.
The adenosine A2a receptor antagonists have attracted interest as potential symptomatic drugs for PD. A2a receptors are localised on striatal medium spiny neurones and modulate the release of GABA.69–71 A2a antagonists also affect the release of acetylcholine from striatal cholinergic interneurones and release dopamine from the nigro-striatal tract.71 The A2a antagonist, KW 6002, increases locomotor activity in MPTP treated mice and potentiates rotational behaviour produced by levodopa or dopamine agonist drugs in the unilateral 6-OHDA lesioned rat.72–78 Such drugs might be important not only in controlling the symptoms of PD but also in preventing the wearing off seen with chronic treatment. In the MPTP primate model, KW 6002 produced long lasting improvement in motor activity without the development of dyskinesias.79 The benefits of KW 6002 were additive to levodopa or a dopamine agonist even when given 24–48 h later and did not result in any increase in dyskinesias.80 As A2a receptors are present in the limbic areas, hippocampus and amygdala, A2a antagonists may also be effective in treating anxiety and depression in PD.
DISEASE MODIFICATION
The potential benefits of successful disease modifying therapy in PD are enormous. However, the true effectiveness of such therapy might well depend on whether the putative agent is able to protect only dopaminergic neurones or, preferably, all neuronal types that degenerate in this disorder. If only the former, the patient would not be prevented from suffering the non-dopaminergic features of PD that come to dominate the disease in its more advanced stages. If the latter, however, the advantages would be profound and delay or prevent disability due to both motor and non-motor symptoms.
There have been considerable advances in our understanding of the aetiology and pathogenesis of PD.81 Several gene mutations have been identified and other loci await refinement and characterisation.82 Environmental risk factors and exposure to potential toxins may influence PD onset either alone or in association with genetic susceptibility.83 Biochemical abnormalities including mitochondrial dysfunction, free radical mediated damage, excitotoxicity, inflammatory change, and proteosomal dysfunction have all been identified in the PD brain. Importantly, these abnormalities have provided targets for potential disease modifying drugs (fig 1). Several of these drugs have been investigated in laboratory models of PD and some of these in turn have been studied in clinical trials (table 2).84–86
Putative neuroprotective agents that have been investigated or are currently in trial for disease modification in PD

Main factors currently identified as contributing to aetiology and pathogenesis in PD.
Several compounds have been studied and many more are under evaluation for disease modification in PD. The DATATOP trial demonstrated that selegiline (deprenyl) could delay the introduction of levodopa in early PD by 9–12 months.87 The interpretation of this result was confounded by selegiline’s symptomatic effect. In an effort to avoid this, selegiline was compared to placebo using, as the primary endpoint, the change in motor score between an untreated baseline visit and an untreated final visit performed after 12 months of treatment and 2 months of study drug withdrawal.88 Patients treated with selegiline had less deterioration from baseline than placebo patients, again suggesting that selegiline might be neuroprotective. However, this study may also have been confounded by the potential of selegiline to have long lasting symptomatic effects. Nevertheless, in a long term follow up study of levodopa, patients who had been taking selegiline for 7 years compared to those who were changed to placebo after 5 years, there was a significantly slower decline, less wearing off, on-off, and freezing, but more dyskinesias in those on selegiline.89
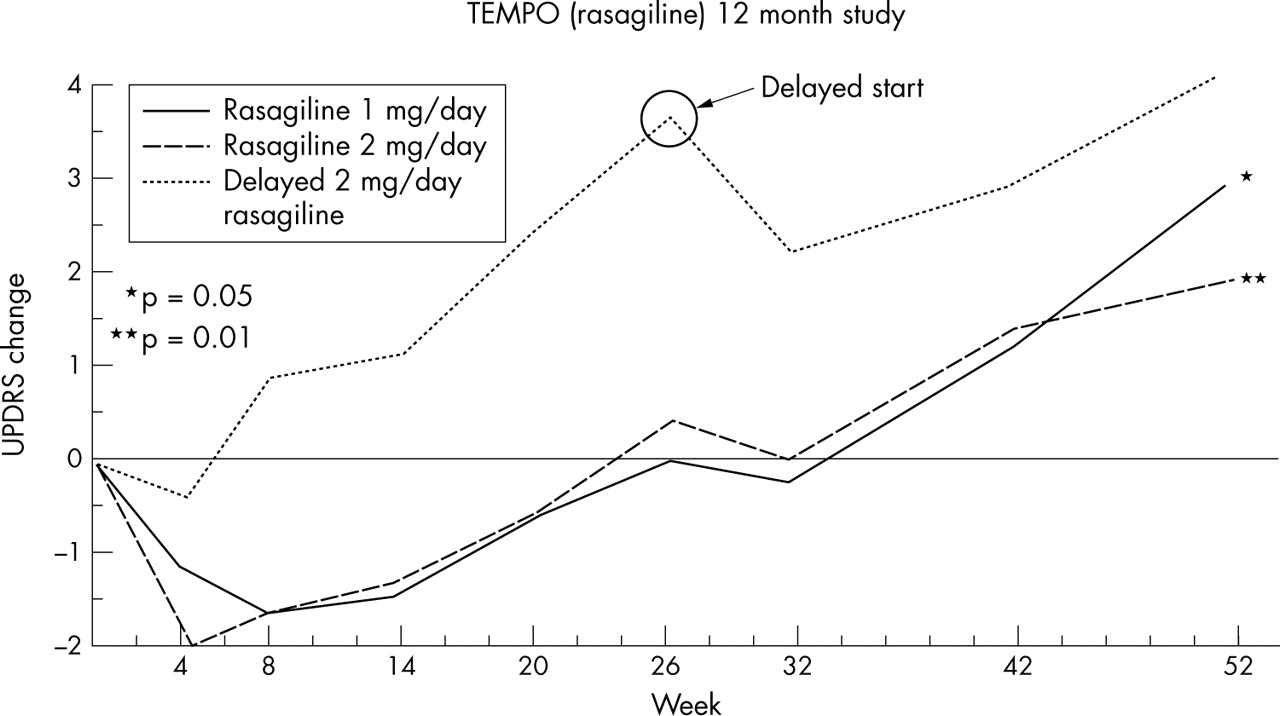
Rasagiline is a more potent MAO-B inhibitor than selegiline and has been shown to provide symptomatic benefit to PD patients with either early or late disease.28–30 This drug has also shown neuroprotective effects in in vitro and in vivo models of PD. For instance, it protects against MPTP/MPP and 6-hydroxydopamine toxicity and excitotoxic mediated damage, and stabilises the mitochondrial membrane potential to reduce apoptotic cell death.90–93 These effects are independent of its MAO-B inhibition. The TEMPO study randomised patients with early PD to placebo or rasagiline 1 or 2 mg and evaluated progression with change from baseline in UPDRS scores over 6 months. At this point, the placebo group was placed on 2 mg rasagiline and all groups were followed for a further 6 months. At the end of the study, the rate of progression in the patients on rasagiline and those switched to the drug was slower than in the placebo group during the first 6 months, the slopes of the two treated populations running in parallel during the second half of the study (fig 2).94 The interpretation of the TEMPO result is complex.95 The results cannot be explained by a symptomatic effect alone and, at face value, they represent an early disease modifying effect. However, there are potential confounding effects, including lasting benefit from treating earlier.

{kind=link}
{kind=link}
Comparison between rasagiline for 1 year and delayed rasagiline. The effect size comparing rasagiline 1 mg/day for 1 year with delayed rasagiline was −1.8 U (95% CI: −3.64 to 0.01, p = 0.05), and the effect size comparing rasagiline 2 mg/day for 1 year with delayed rasagiline was −2.3 U (95% CI: −4.11 to −0.48, p = 0.01).94
Coenzyme Q10 has been evaluated in a pilot study of early PD patients to determine whether it might have disease modifying capabilities.96 The rationale for the use of coenzyme Q10 in PD was based upon the observation that mitochondrial complex I activity is decreased in PD substantia nigra, PD patients have reduced levels of coenzyme Q10, and this compound protects against MPTP toxicity. Coenzyme Q10 is both an anti-oxidant and an integral component of oxidative phosphorylation which has been shown to enhance electron transport. Patients were randomised to either a placebo arm or one of three doses of coenzyme Q10 (300, 600, or 1200 mg) and followed for 16 months. There was a significant benefit for coenzyme Q10 1200 mg in terms of change from baseline in total UPDRS scores compared to placebo at 16 months, and a non-significant trend to benefit for lower doses. This interesting and important result is sufficient to support further study of coenzyme Q10, but insufficient at present to advocate that PD patients should use this compound.
Dopamine agonists have demonstrated neuroprotective actions in the laboratory in vitro and in vivo. They can protect cells in tissue culture against the toxic actions of 1-methyl-4-phenylpyridinium (MPP+), 6-hydroxydopamine, rotenone, and 3-nitropropionic acid and in vivo, pramipexole protects against dopaminergic cell loss in the substantia nigra in the MPTP primate model.97 Interestingly, studies using pramipexole have shown that the protective action of dopamine agonists may not be dependent on the presence of dopamine receptors.98 Pre-incubation of the tissue culture cells or prior exposure to the agonist appears to be an important factor in the agonist’s effect and may imply that the drug has to reach specific intracellular compartments or initiate protective cellular responses before toxin exposure. Such a requirement does not preclude a clinical application but rather emphasises the need to give a protective agent as early in the course of the disease as possible.
Two studies in PD patients with early disease have sought to determine whether agonists can modify the course of the disease. The CALM-PD study used beta-CIT single photon emission computerised tomography (SPECT) to follow the rate of loss of dopamine transporter as a marker of dopaminergic nigrostriatal cell density.99 PD patients were randomised to pramipexole or levodopa and followed for a total of 4 years; levodopa supplementation was allowed in both arms. At 2, 3, and 4 years there was a significant reduction in the rate of transporter loss in the pramipexole group, averaging out at approximately 40%. A similar result was seen in the REAL-PET ropinirole study which used a similar trial design but utilised positron emission tomography (PET) to follow loss of nigrostriatal cell density with fluorodopa.100 This demonstrated an approximate 34% reduction in the ropinirole group compared to those on levodopa. As in previous neuroprotection studies, interpretation of the results is complex. Unfortunately, a placebo group was not included in either study and so it is not known whether the agonist was protective or levodopa toxic.
The ELLDOPA trial randomised early PD patients to a placebo arm or one of three levodopa treatment groups (150, 300, or 600 mg daily) and performed a SPECT analysis before and at 40 weeks.101 The levodopa arms showed a greater rate of transporter signal loss than did the placebo group over the period of the trial. The levodopa treated patients had a better UPDRS score than placebo patients after washout, although this was only for 2 weeks and a prolonged symptomatic effect could not be excluded. Patients in the high dose levodopa arm had significantly more dyskinesias than other groups.
Interpretation of the imaging studies cited above is complicated by the possibility that the agonists or levodopa might themselves have modified the respective signal expression in SPECT or PET. For instance, exposure to levodopa and/or the agonists could regulate dopamine metabolism or transporter expression. However, explanation of the results on this basis alone requires an intricate model that at present has no support from laboratory data.86
Glial cell derived nerve growth factor (GDNF) is important in dopaminergic cell growth and maintenance. Intrastriatal or intraventricular administration of GDNF to MPTP treated non-human primates increased dopaminergic neuronal density and improved motor function without dyskinesias.102,103 Intraventricular injection of GDNF in patients did not result in benefit.104 A pilot study in five PD patients who received intraputaminal infusions of GDNF demonstrated significant clinical improvement in both motor deficits and dyskinesias and a parallel increase in fluorodopa uptake on PET.105 A larger and blinded clinical trial of intraputaminal GDNF in PD was recently terminated and although the results have not yet been published, informal presentations suggest an adverse event profile.
CONCLUSIONS
Current therapy options for PD remain focussed on the symptomatic improvement of motor features related predominantly to loss of dopaminergic neurones in the substantia nigra. Such treatment is effective in improving morbidity and mortality. Recent insights into the aetiology, pathology, and pathogenesis of PD are providing important opportunities to develop disease modifying therapies that will have an even greater impact on the disease than did the introduction of levodopa. Importantly, current attempts to define an at risk population on genetic or clinical grounds should provide the ideal group in which to study potential protective therapies. Improvements in trial design are required to evaluate candidate drugs more appropriately, perhaps with the introduction of validated clinical markers. Despite all the caveats in the interpretation of existing disease modifying trials, treating physicians need practical guidance both to help patients make a judgement on what drug to use and when to initiate it. This remains very much an individual decision and will need to take account of a number of factors including the patient’s age and co-morbidity and the physician’s own interpretation of the data available and the information presented here.
REFERENCES
Footnotes
-
Competing interests: none declared